Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology




Abstract
1. Introduction
2. Results and Discussion
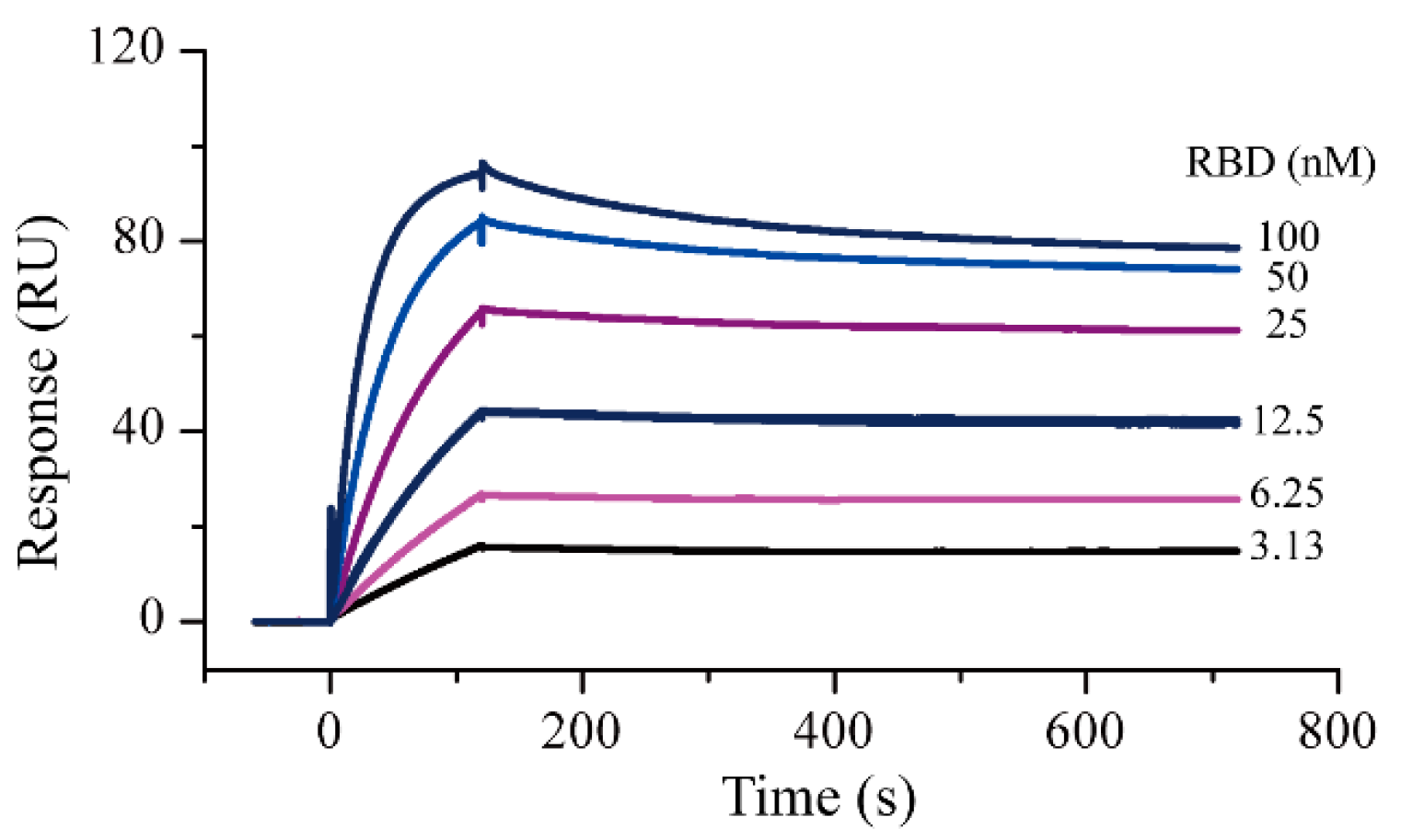
2.1. Surface Plasmon Resonance Assay Development and Protein–Protein Interaction Testing
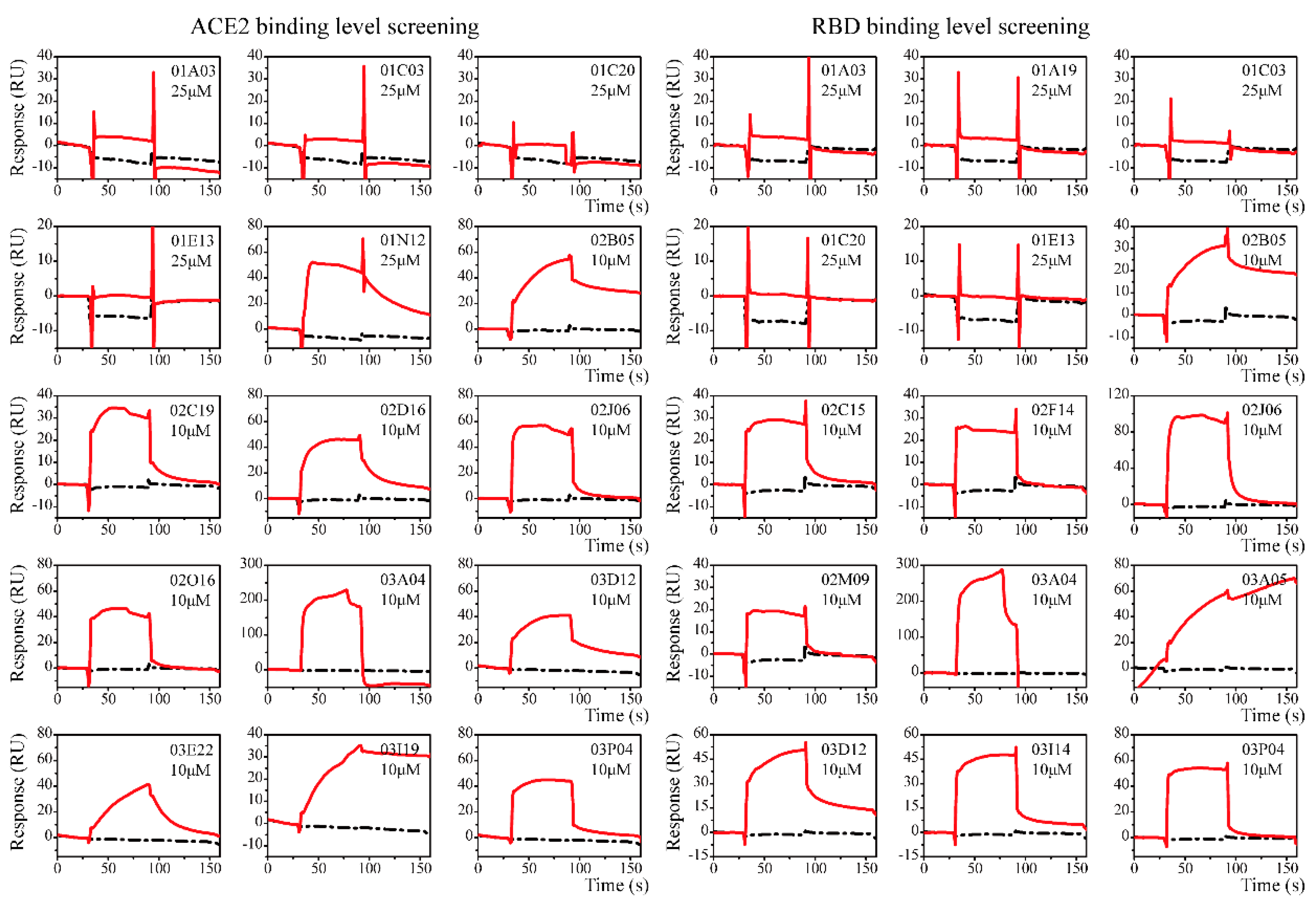
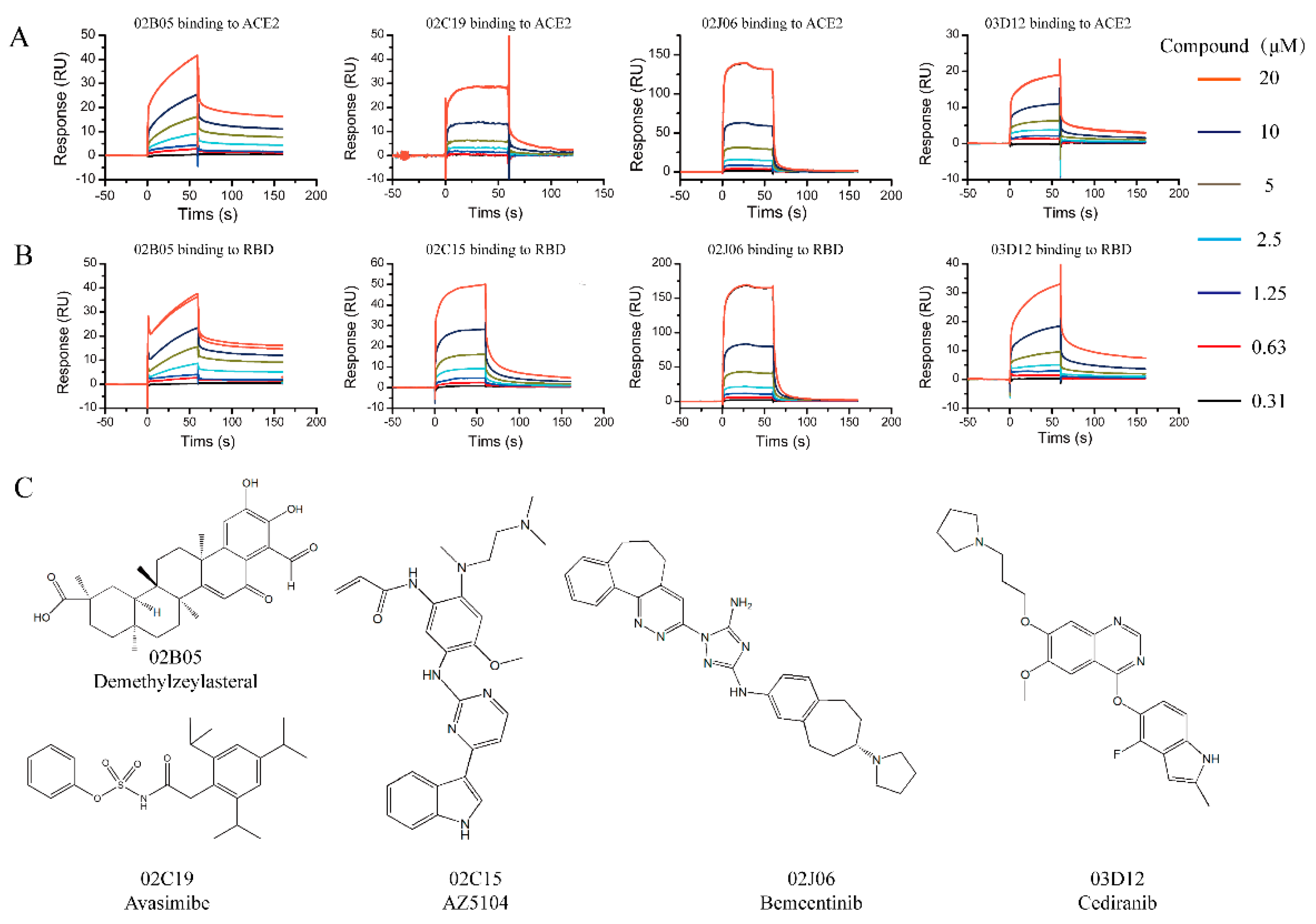
2.2. Screening and Affinity Analysis
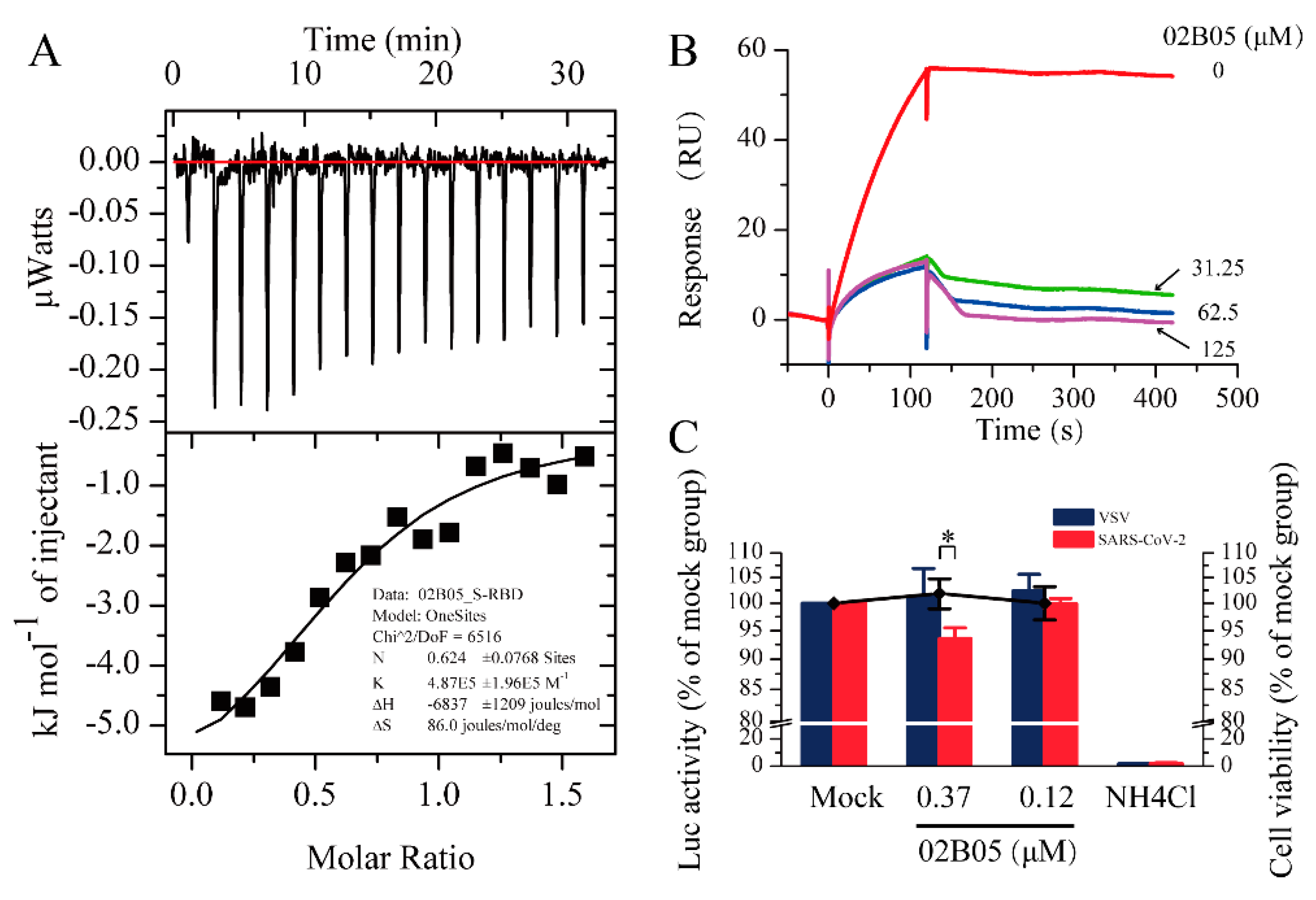
2.3. Validity of Interaction by Isothermal Titration Calorimetry (ITC)
2.4. Surface Plasmon Resonance-Based Competition Assay
2.5. Pseudovirus Entry Assay
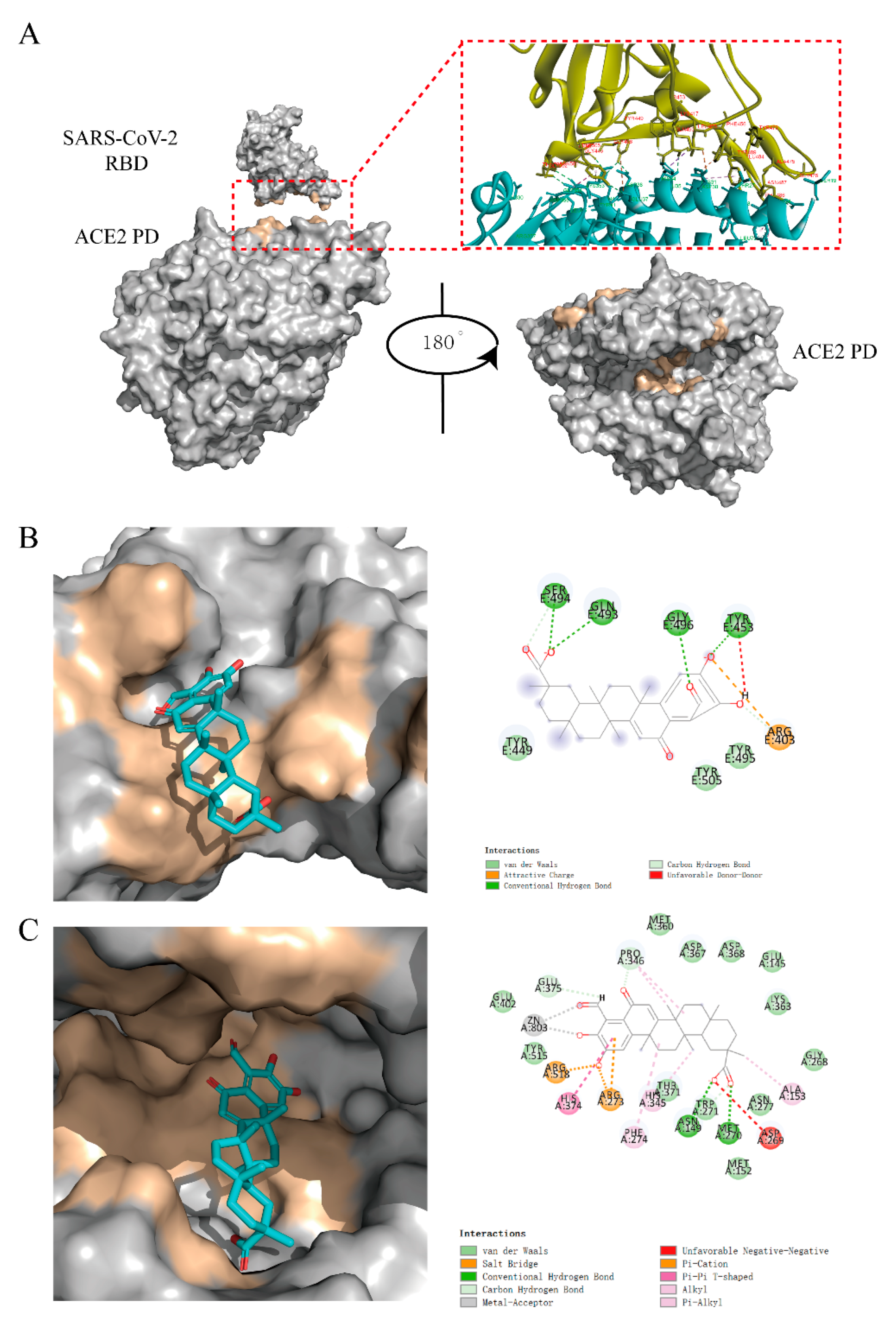
2.6. Virtual Docking
3. Conclusions
4. Materials and Methods
4.1. Chemicals and Reagents
4.2. Protein–Protein Interaction Testing
4.3. Screening and Kinetic Analysis
4.4. Interactions Determined by Isothermal Titration Calorimetry (ITC)
4.5. Surface Plasmon Resonance-Based Competition Assay
4.6. Pseudotyped Lentiviral Particle Entry into Human Cells and the Luciferase Assay
4.7. Docking
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wang, C.; Horby, P.W.; Hayden, F.G.; Gao, G.A. novel coronavirus outbreak of global health concern. Lancet 2020, 395, 470–473. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. WHO Coronavirus Disease (COVID-19) Dashboard. Available online: https://covid19.who.int/overview (accessed on 7 November 2020).
- Lu, R.J.; Zhao, X.; Li, J.; Niu, P.H.; Yang, B.; Wu, H.L.; Wang, W.L.; Song, H.; Huang, B.Y.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.W.; Yu, J.F.; Shan, S.S.; Zhou, H.; Fan, S.L.; Zhang, Q.; Shi, X.L.; Wang, Q.S.; Zhang, L.Q.; et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Wan, Y.; Shang, J.; Graham, R.; Baric, R.S.; Li, F. Receptor Recognition by the Novel Coronavirus from Wuhan: An Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J. Virol. 2020, 94, 1–9. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM Structure of the 2019-nCoV Spike in the Prefusion Conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Yan, R.H.; Zhang, Y.Y.; Li, Y.N.; Xia, L.; Guo, Y.Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef]
- Li, Q.Q.; Wu, J.J.; Nie, J.H.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.Y.; Zhang, Q.; Liu, H.; Nie, L.L.; et al. The impact of mutations in SARS-CoV-2 spike on viral infectivity and antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Chen, J.H.; Wang, R.; Wang, M.L.; Wei, G.W. Mutations strengthened SARS-CoV-2 infectivity. J. Mol. Biol. 2020, 432, 5212–5226. [Google Scholar] [CrossRef] [PubMed]
- Zumla, A.; Chan, J.F.W.; Azhar, E.I.; Hui, D.S.C.; Yuen, K. Coronaviruses-drug discovery and therapeutic options. Nat. Rev. Drug Discov. 2016, 15, 327–347. [Google Scholar] [CrossRef] [PubMed]
- Guy, R.K.; DiPaola, R.S.; Romanelli, F.; Dutch, R.E. Rapid repurposing of drugs for COVID-19. Science 2020, 368, 829–830. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.Y.; Tian, L.; Liu, Y.Z.; Hui, N.; Qiao, G.P.; Li, H.; Shi, Z.F.; Tang, Y.H.; Zhang, D.Z.; Xie, X.L.; et al. A promising antiviral candidate drug for the COVID-19 pandemic: A mini-review of Remdesivir. Eur. J. Med. Chem. 2020, 201, 112527. [Google Scholar] [CrossRef]
- Hall, D.C., Jr.; Ji, H.F. A search for medications to treat COVID-19 via in silico molecular docking models of the SARS-CoV-2 spike glycoprotein and 3CL protease. Travel Med. Infect. Dis. 2020, 35, 101646. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.M.; Du, X.Y.; Xu, Y.C.; Deng, Y.Q.; Liu, M.Q.; Zhao, Y.; Zhang, B.; Li, X.F.; Zhang, L.K.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Robson, B. Computers and viral diseases. Preliminary bioinformatics studies on the design of a synthetic vaccine and a preventative peptidomimetic antagonist against the SARS-CoV-2 (2019-nCoV, COVID-19) coronavirus. Comput. Biol. Med. 2020, 119, 103670. [Google Scholar] [CrossRef]
- Ton, A.T.; Gentile, F.; Hsing, M.; Ban, F.; Cherkasov, A. Rapid Identification of Potential Inhibitors of SARS-CoV-2 Main Protease by Deep Docking of 1.3 Billion Compounds. Mol. Inf. 2020, 39, 2000028. [Google Scholar] [CrossRef]
- Dai, W.H.; Zhang, B.; Jiang, X.M.; Su, H.X.; Li, J.; Zhao, Y.; Xie, X.; Jin, Z.M.; Peng, J.J.; Liu, F.J.; et al. Structure-based design of antiviral drug candidates targeting the SARS-CoV-2 main protease. Science 2020, 368, 1331–1335. [Google Scholar] [CrossRef]
- Du, L.Y.; He, Y.X.; Zhou, Y.S.; Liu, S.W.; Zheng, B.J.; Jiang, S.B. The spike protein of SARS-CoV--a target for vaccine and therapeutic development. Nat. Rev. Microbiol. 2009, 7, 226–236. [Google Scholar] [CrossRef]
- Han, Y.X.; Kral, P. Computational Design of ACE2-Based Peptide Inhibitors of SARS-CoV-2. ACS Nano 2020, 14, 5143–5147. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.R.; Liu, Y.; Yang, Y.Y.; Zhang, P.; Zhong, W.; Wang, Y.L.; Wang, Q.Q.; Xu, Y.; Li, M.X.; Li, X.Z.; et al. Analysis of therapeutic targets for SARS-CoV-2 and discovery of potential drugs by computational methods. Acta Pharm. Sinica B 2020, 10, 766–788. [Google Scholar] [CrossRef]
- Hoa, X.D.; Kirk, A.G.; Tabrizian, M. Towards integrated and sensitive surface plasmon resonance biosensors: A review of recent progress. Biosens. Bioelectron. 2007, 23, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Homola, J. Present and future of surface plasmon resonance biosensors. Anal. Bioanal. Chem. 2003, 377, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Rich, R.L.; Myszka, D.G. Higher-throughput, label-free, real-time molecular interaction analysis. Anal. Biochem. 2007, 361, 1–6. [Google Scholar] [CrossRef]
- Boucher, L.E.; Bosch, J. Development of a multifunctional tool for drug screening against plasmodial protein-protein interactions via surface plasmon resonance. J. Mol. Recognit. 2013, 26, 496–500. [Google Scholar] [CrossRef][Green Version]
- Hoare, S.R.J.; Fleck, B.A.; Williams, J.P.; Grigoriadis, D.E. The importance of target binding kinetics for measuring target binding affinity in drug discovery: A case study from a CRF(1) receptor antagonist program. Drug Discov. Today 2019, 25, 7–14. [Google Scholar] [CrossRef]
- Zhao, G.Y.; Du, L.Y.; Ma, C.Q.; Li, Y.; Li, L.; Poon, V.K.M.; Wang, L.L.; Yu, F.; Zheng, B.J.; Jiang, S.B.; et al. A safe and convenient pseudovirus-based inhibition assay to detect neutralizing antibodies and screen for viral entry inhibitors against the novel human coronavirus MERS-CoV. Virol. J. 2013, 10, 266. [Google Scholar] [CrossRef]
- Bergsdorf, C.; Wright, S.K. A Guide to Run Affinity Screens Using Differential Scanning Fluorimetry and Surface Plasmon Resonance Assays. Methods Enzymol. 2018, 610, 135–165. [Google Scholar] [CrossRef]
- GE Healthcare Life Sciences. Buffer and sample preparation for direct binding assay in 5% DMSO. Lab. Guidel. 2014, 1–4. [Google Scholar]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-ray structures reveal a large hinge-bending motion important for inhibitor binding and catalysis. J. Biol. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Target | kon (1/Ms) | koff (×10−3 1/s) | KD (×10−6 M) |
|---|---|---|---|---|
| 02B05 | ACE2 | 1989 | 3.345 | 1.736 a |
| S-RBD | 2351 | 2.443 | 1.039 a | |
| 02C15 | S-RBD | 2360 | 21.50 | 9.110 a |
| 02C19 | ACE2 | 2344 | 52.05 | 22.21 a |
| 02J06 | ACE2 | 2613 | 635.9 | 243.4 a |
| S-RBD | 5123 | 328.9 | 64.20 a | |
| 02M09 | S-RBD | - | - | 8.368 b |
| 03D12 | ACE2 | 2764 | 9.480 | 3.429 a |
| S-RBD | 2102 | 9.644 | 4.588 a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhu, Z.-L.; Qiu, X.-D.; Wu, S.; Liu, Y.-T.; Zhao, T.; Sun, Z.-H.; Li, Z.-R.; Shan, G.-Z. Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules 2021, 26, 57. https://doi.org/10.3390/molecules26010057
Zhu Z-L, Qiu X-D, Wu S, Liu Y-T, Zhao T, Sun Z-H, Li Z-R, Shan G-Z. Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules. 2021; 26(1):57. https://doi.org/10.3390/molecules26010057
Chicago/Turabian StyleZhu, Zhi-Ling, Xiao-Dan Qiu, Shuo Wu, Yi-Tong Liu, Ting Zhao, Zhong-Hao Sun, Zhuo-Rong Li, and Guang-Zhi Shan. 2021. "Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology" Molecules 26, no. 1: 57. https://doi.org/10.3390/molecules26010057
APA StyleZhu, Z.-L., Qiu, X.-D., Wu, S., Liu, Y.-T., Zhao, T., Sun, Z.-H., Li, Z.-R., & Shan, G.-Z. (2021). Blocking Effect of Demethylzeylasteral on the Interaction between Human ACE2 Protein and SARS-CoV-2 RBD Protein Discovered Using SPR Technology. Molecules, 26(1), 57. https://doi.org/10.3390/molecules26010057