
Hydrogenation of CO2 Promoted by Silicon-Activated H2S: Origin and Implications


Abstract
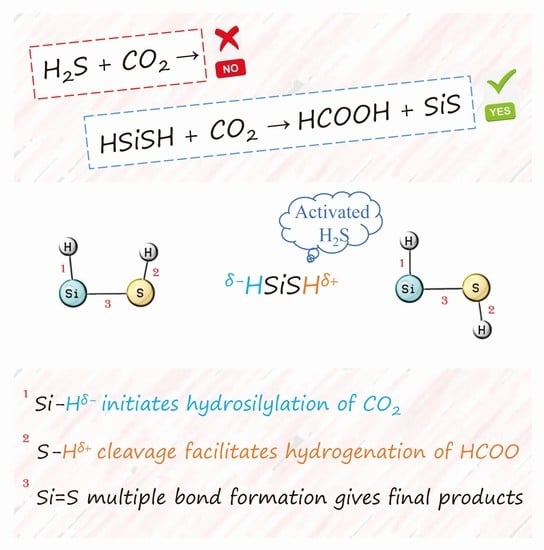
1. Introduction
2. Results
3. Discussion
4. Computational Methods
5. Conclusions
Supplementary Materials
Funding
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Meinshausen, M.; Meinshausen, N.; Hare, W.; Raper, S.C.B.; Frieler, K.; Knutti, R.; Frame, D.J.; Allen, M.R. Greenhouse-gas emission targets for limiting global warming to 2 °C. Nature 2009, 458, 1158–1196. [Google Scholar] [CrossRef] [PubMed]
- Seneviratne, S.I.; Rogelj, J.; Seferian, R.; Wartenburger, R.; Allen, M.R.; Cain, M.; Millar, R.J.; Ebi, K.L.; Ellis, N.; Hoegh-Guldberg, O.; et al. The many possible climates from the Paris Agreement’s aim of 1.5 °C warming. Nature 2018, 558, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Cheng, K.; Kang, J.C.; Zhou, C.; Subramanian, V.; Zhang, Q.H.; Wang, Y. New horizon in C1 chemistry: Breaking the selectivity limitation in transformation of syngas and hydrogenation of CO2 into hydrocarbon chemicals and fuels. Chem. Soc. Rev. 2019, 48, 3193–3228. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.Y.; Zhang, C.; Gao, P.; Wang, H.; Li, X.P.; Zhong, L.S.; Wei, W.; Sun, Y.H. A review of the catalytic hydrogenation of carbon dioxide into value-added hydrocarbons. Catal. Sci. Technol. 2017, 7, 4580–4598. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, T.; Das, S. Catalytic transformation of CO2 into C1 chemicals using hydrosilanes as a reducing agent. Green Chem. 2020, 22, 1800–1820. [Google Scholar] [CrossRef]
- Yant, W.P. Hydrogen sulphide in industry—Occurrence, effects, and treatment. Am. J. Public Health Nations Health 1930, 20, 598–608. [Google Scholar] [CrossRef]
- Shen, L.J.; Cao, Y.N.; Du, Z.J.; Zhao, W.T.; Lin, K.; Jiang, L.L. Illuminate the active sites of γ-FeOOH for low-temperature desulfurization. Appl. Surf. Sci. 2017, 425, 212–219. [Google Scholar] [CrossRef]
- Zhong, J.W.; Yang, X.F.; Wu, Z.L.; Liang, B.L.; Huang, Y.Q.; Zhang, T. State of the art and perspectives in heterogeneous catalysis of CO2 hydrogenation to methanol. Chem. Soc. Rev. 2020, 49, 1385–1413. [Google Scholar] [CrossRef]
- Liu, G.X.; Poths, P.; Zhang, X.X.; Zhu, Z.G.; Marshall, M.; Blankenhorn, M.; Alexandrova, A.N.; Bowen, K.H. CO2 hydrogenation to formate and formic acid by bimetallic palladium-copper hydride clusters. J. Am. Chem. Soc. 2020, 142, 7930–7936. [Google Scholar] [CrossRef]
- Jiang, X.; Nie, X.W.; Guo, X.W.; Song, C.S.; Chen, J.G. Recent advances in carbon dioxide hydrogenation to methanol via heterogeneous catalysis. Chem. Rev. 2020, 120, 7984–8034. [Google Scholar] [CrossRef]
- Baltrusaitis, J.; Patterson, E.V.; Hatch, C. Computational studies of CO2 activation via photochemical reactions with reduced sulfur compounds. J. Phys. Chem. A 2012, 116, 9331–9339. [Google Scholar] [CrossRef] [PubMed]
- He, R.T.; Hu, B.Y.; Zhong, H.; Jin, F.M.; Fan, J.J.; Hu, Y.H.; Jing, Z.Z. Reduction of CO2 with H2S in a simulated deep-sea hydrothermal vent system. Chem. Commun. 2019, 55, 1056–1059. [Google Scholar] [CrossRef] [PubMed]
- Upadhyay, L.R.; Rangarajan, S.; Baltrusaitis, J. Inhibitor, co-catalyst, or co-reactant? Probing the different roles of H2S during CO2 hydrogenation on the MoS2 catalyst. ACS Catal. 2019, 9, 10044–10059. [Google Scholar]
- Kumar, M.; Francisco, J.S. Hydrogen sulfide induced carbon dioxide activation by metal-free dual catalysis. Chem. Eur. J. 2016, 22, 4359–4363. [Google Scholar] [CrossRef] [PubMed]
- Mul, G.; Wachs, I.E.; Hirschon, A.S. Catalytic synthesis of methanethiol from hydrogen sulfide and carbon monoxide over vanadium-based catalysts. Catal. Today 2003, 78, 327–337. [Google Scholar] [CrossRef]
- Gutierrez, O.Y.; Kaufmann, C.; Hrabar, A.; Zhu, Y.Z.; Lercher, J.A. Synthesis of methyl mercaptan from carbonyl sulfide over sulfide K2MoO4/SiO2. J. Catal. 2011, 280, 264–273. [Google Scholar] [CrossRef]
- Gutierrez, O.Y.; Kaufmann, C.; Lercher, J.A. Synthesis of methanethiol from carbonyl sulfide and carbon disulfide on (Co)K-promoted sulfide Mo/SiO2 catalysts. ACS Catal. 2011, 1, 1595–1603. [Google Scholar] [CrossRef]
- Chen, A.P.; Wang, Q.; Li, Q.L.; Hao, Y.J.; Fang, W.P.; Yang, Y.Q. Direct synthesis of methanethiol from H2S-rich syngas over sulfided Mo-based catalysts. J. Mol. Catal. A Chem. 2008, 283, 69–76. [Google Scholar] [CrossRef]
- Zhang, B.J.; Taylor, S.H.; Hutchings, G.J. Catalytic synthesis of methanethiol from CO/H2/H2S mixtures using α-Al2O3. New J. Chem. 2004, 28, 471–476. [Google Scholar] [CrossRef]
- Liu, X.; Liu, X.R.; Wang, X.F. Splitting of hydrogen sulfide by group 14 elements (Si, Ge, Sn, Pb) in excess argon at cryogenic temperatures. J. Phys. Chem. A 2018, 122, 7023–7032. [Google Scholar] [CrossRef]
- Cockett, M.C.R.; Dyke, J.M.; Morris, A.; Niavaran, M.H.Z. High-temperature photoelectron spectroscopy. A study of SiS(X 1Σ+). J. Chem. Soc. Faraday Trans. 2 Mol. Chem. Phys. 1989, 85, 75–83. [Google Scholar]
- Muller, H.S.P.; McCarthy, M.C.; Bizzocchi, L.; Gupta, H.; Esser, S.; Lichau, H.; Caris, M.; Lewen, F.; Hahn, J.; Esposti, C.D.; et al. Rotational spectroscopy of the isotopic species of silicon monosulfide, SiS. Phys. Chem. Chem. Phys. 2007, 9, 1579–1586. [Google Scholar] [CrossRef] [PubMed]
- Schnockel, H.; Koppe, R. Matrix IR spectrum and ab initio SCF calculations of molecular SiS2. J. Am. Chem. Soc. 1989, 111, 4583–4586. [Google Scholar] [CrossRef]
- Gottle, A.J.; Koper, M.T.M. Determinant role of electrogenerated reactive nucleophilic species on selectivity during reduction of CO2 catalyzed by metalloporphyrins. J. Am. Chem. Soc. 2018, 140, 4826–4834. [Google Scholar] [CrossRef] [PubMed]
- Tang, Q.; Lee, Y.J.; Li, D.Y.; Choi, W.; Liu, C.W.; Lee, D.; Jiang, D.E. Lattice-hydride mechanism in electrocatalytic CO2 reduction by structurally precise copper-hydride nanoclusters. J. Am. Chem. Soc. 2017, 139, 9728–9736. [Google Scholar] [CrossRef]
- Khandelwal, M.; Wehmschulte, R.J. Deoxygenative reduction of carbon dioxide to methane, toluene, and diphenylmethane with [Et2Al]+ as catalyst. Angew. Int. Ed. Chem. 2012, 51, 7323–7326. [Google Scholar] [CrossRef]
- Sun, W.; Qian, C.X.; He, L.; Ghuman, K.K.; Wong, A.P.Y.; Jia, J.; Jelle, A.A.; O’Brien, P.G.; Reyes, L.M.; Wood, T.E.; et al. Heterogeneous reduction of carbon dioxide by hydride-terminated silicon nanocrystals. Nat. Commun. 2016, 7, 1–9. [Google Scholar] [CrossRef]
- Dasog, M.; Kraus, S.; Sinelnikov, R.; Veinot, J.G.C.; Rieger, B. CO2 to methanol conversion using hydride terminated porous silicon nanoparticles. Chem. Commun. 2017, 53, 3114–3117. [Google Scholar] [CrossRef]
- Wong, A.P.Y.; Sun, W.; Qian, C.X.; Jelle, A.A.; Jia, J.; Zheng, Z.Q.; Dong, Y.C.; Ozin, G.A. Tailoring CO2 reduction with doped silicon nanocrystals. Adv. Sustain. Syst. 2017, 1, 1700118. [Google Scholar] [CrossRef]
- Qian, C.X.; Sun, W.; Hung, D.L.H.; Qiu, C.Y.; Makaremi, M.; Kumar, S.G.H.; Wan, L.L.; Ghoussoub, M.; Wood, T.E.; Xia, M.K.; et al. Catalytic CO2 reduction by palladium-decorated silicon-hydride nanosheets. Nat. Catal. 2019, 2, 46–54. [Google Scholar] [CrossRef]
- Grasemann, M.; Laurenczy, G. Formic acid as a hydrogen source—recent developments and future trends. Energy Environ. Sci. 2012, 5, 8171–8181. [Google Scholar] [CrossRef]
- Villegas-Escobar, N.; Ortega, D.E.; Cortés-Arriagada, D.; Durán, R.; Yepes, D.; Gutiérrez-Oliva, S.; Toro-Labbé, A. Why low valent lead (II) hydride complex would be a better catalyst for CO2 activation than its 14 group analogues? J. Phys. Chem. C 2017, 121, 12127–12135. [Google Scholar] [CrossRef]
- Castleman, A.W. From elements to clusters: The periodic table revisited. J. Phys. Chem. Lett. 2011, 2, 1062–1069. [Google Scholar] [CrossRef]
- Jena, P. Beyond the periodic table of elements: The role of superatoms. J. Phys. Chem. Lett. 2013, 4, 1432–1442. [Google Scholar] [CrossRef]
- Luo, Z.X.; Castleman, A.W. Special and general superatoms. Acc. Chem. Res. 2014, 47, 2931–2940. [Google Scholar] [CrossRef]
- Jena, P.; Sun, Q. Super atomic clusters: Design rules and potential for building blocks of materials. Chem. Rev. 2018, 118, 5755–5870. [Google Scholar] [CrossRef]
- Dean, J.A. Lange’s Handbook of Chemistry, 15th ed.; McGraw Hill: New York, NY, USA, 1999. [Google Scholar]
- Chang, X.X.; Wang, T.; Gong, J.L. CO2 photo-reduction: Insights into CO2 activation and reaction on surfaces of photocatalysts. Energy Environ. Sci. 2016, 9, 2177–2196. [Google Scholar] [CrossRef]
- Wang, Y.C.; Lv, J.; Zhu, L.; Ma, Y.M. CALYPSO: A method for crystal structure prediction. Comput. Phys. Commun. 2012, 183, 2063–2070. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09 Revision d 01; Gaussian Inc.: Wallingford Center, CO, USA, 2009. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, Y.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B Condens. Matter Mater. Phys. 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Frisch, M.J.; Pople, J.A.; Binkley, J.S. Self-consistent molecular orbital methods 25. Supplementary functions for Gaussian basis sets. J. Chem. Phys. 1984, 80, 3265–3269. [Google Scholar] [CrossRef]
- Lv, J.; Wang, Y.C.; Zhu, L.; Ma, Y.M. Particle-swarm structure prediction on clusters. J. Chem. Phys. 2012, 137, 084104. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.A.; Dunning, T.H.; Harrison, R.J. Electron affinities of the first-row atoms revisited. Systematic basis sets and wave functions. J. Chem. Phys. 1992, 96, 6796–6806. [Google Scholar] [CrossRef]
- Woon, D.E.; Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. III. The atoms aluminum through argon. J. Chem. Phys. 1993, 98, 1358–1871. [Google Scholar] [CrossRef]
- Peterson, K.A. Systematically convergent basis sets with relativistic pseudopotentials. I. Correlation consistent basis sets for the post-d group 13–15 elements. J. Chem. Phys. 2003, 119, 11099–11112. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Luo, S.J.; Zhao, Y.; Truhlar, D.G. Validation of electronic structure methods for isomerization reactions of large organic molecules. Phys. Chem. Chem. Phys. 2011, 13, 13683–13689. [Google Scholar] [CrossRef]
- Li, X.Y.; Xu, X.F.; You, X.Q.; Truhlar, D.G. Benchmark calculations for bond dissociation enthalpies of unsaturated methyl esters and the bond dissociation enthalpies of methyl linolenate. J. Phys. Chem. A 2016, 120, 4025–4036. [Google Scholar] [CrossRef]
- Hratchian, H.P.; Schlegel, H.B. Using Hessian updating to increase the efficiency of a Hessian based predictor-corrector reaction path following method. J. Chem. Theory Comput. 2005, 1, 61–69. [Google Scholar] [CrossRef]
- Bartlett, R.J.; Musial, M. Coupled-cluster theory in quantum chemistry. Rev. Mod. Phys. 2007, 79, 291–352. [Google Scholar] [CrossRef]
- Parr, R.G.; Yang, W.T. Density functional approach to the frontier-electron theory of chemical reactivity. J. Am. Chem. Soc. 1984, 106, 4049–4050. [Google Scholar] [CrossRef]
- Morell, C.; Grand, A.; Toro-Labbé, A. New dual descriptor for chemical reactivity. J. Phys. Chem. A 2005, 109, 205–212. [Google Scholar] [CrossRef] [PubMed]
- Gázquez, J.L. Chemical reactivity concepts in density functional theory. In Chemical Reactivity Theory—A Density Functional View; Chattaraj, P.K., Ed.; CRC Press: Boca Ratón, FL, USA, 2009. [Google Scholar]
- Geerlings, P.; De Proft, F.; Langenaeker, W. Conceptual density functional theory. Chem. Rev. 2003, 103, 1793–1873. [Google Scholar] [CrossRef] [PubMed]
- Dennington, R.; Keith, T.; Millam, J. Gauss View; Semichem Inc.: Shawnee, KS, USA, 2009. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecule | Atom | q (N) [a] | q (N + 1) | q (N − 1) | f+ | f− | ∆f |
|---|---|---|---|---|---|---|---|
| CO2 | C | 0.3618 | −0.2960 | 0.5777 | 0.6578 | 0.2159 | 0.4419 |
| O | −0.1808 | −0.3179 | 0.2112 | 0.1371 | 0.3920 | −0.2549 | |
| H2S | H | 0.0616 | −0.0806 | 0.1720 | 0.1421 | 0.1104 | 0.0317 |
| S | −0.1231 | −0.8021 | 0.6560 | 0.6789 | 0.7792 | −0.1002 | |
| cis-HSiSH | H [b] | −0.1150 | −0.1874 | 0.0262 | 0.0724 | 0.1412 | −0.0688 |
| Si | 0.2444 | −0.3987 | 0.7966 | 0.6430 | 0.5523 | 0.0908 | |
| S | −0.1938 | -0.4296 | 0.0482 | 0.2358 | 0.2419 | −0.0061 | |
| H | 0.0644 | 0.0187 | 0.1290 | 0.0457 | 0.0646 | −0.0189 | |
| trans-HSiSH | H [b] | −0.1119 | −0.1841 | 0.0205 | 0.0722 | 0.1324 | −0.0602 |
| Si | 0.2394 | −0.4024 | 0.7784 | 0.6418 | 0.5390 | 0.1029 | |
| S | −0.1880 | −0.4252 | 0.0827 | 0.2373 | 0.2707 | −0.0334 | |
| H | 0.0605 | 0.0149 | 0.1184 | 0.0456 | 0.0580 | −0.0124 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liu, X. Hydrogenation of CO2 Promoted by Silicon-Activated H2S: Origin and Implications. Molecules 2021, 26, 50. https://doi.org/10.3390/molecules26010050
Liu X. Hydrogenation of CO2 Promoted by Silicon-Activated H2S: Origin and Implications. Molecules. 2021; 26(1):50. https://doi.org/10.3390/molecules26010050
Chicago/Turabian StyleLiu, Xing. 2021. "Hydrogenation of CO2 Promoted by Silicon-Activated H2S: Origin and Implications" Molecules 26, no. 1: 50. https://doi.org/10.3390/molecules26010050
APA StyleLiu, X. (2021). Hydrogenation of CO2 Promoted by Silicon-Activated H2S: Origin and Implications. Molecules, 26(1), 50. https://doi.org/10.3390/molecules26010050