
Discovery of Novel Symmetrical 1,4-Dihydropyridines as Inhibitors of Multidrug-Resistant Protein (MRP4) Efflux Pump for Anticancer Therapy

Abstract
1. Introduction
2. Results and Discussion
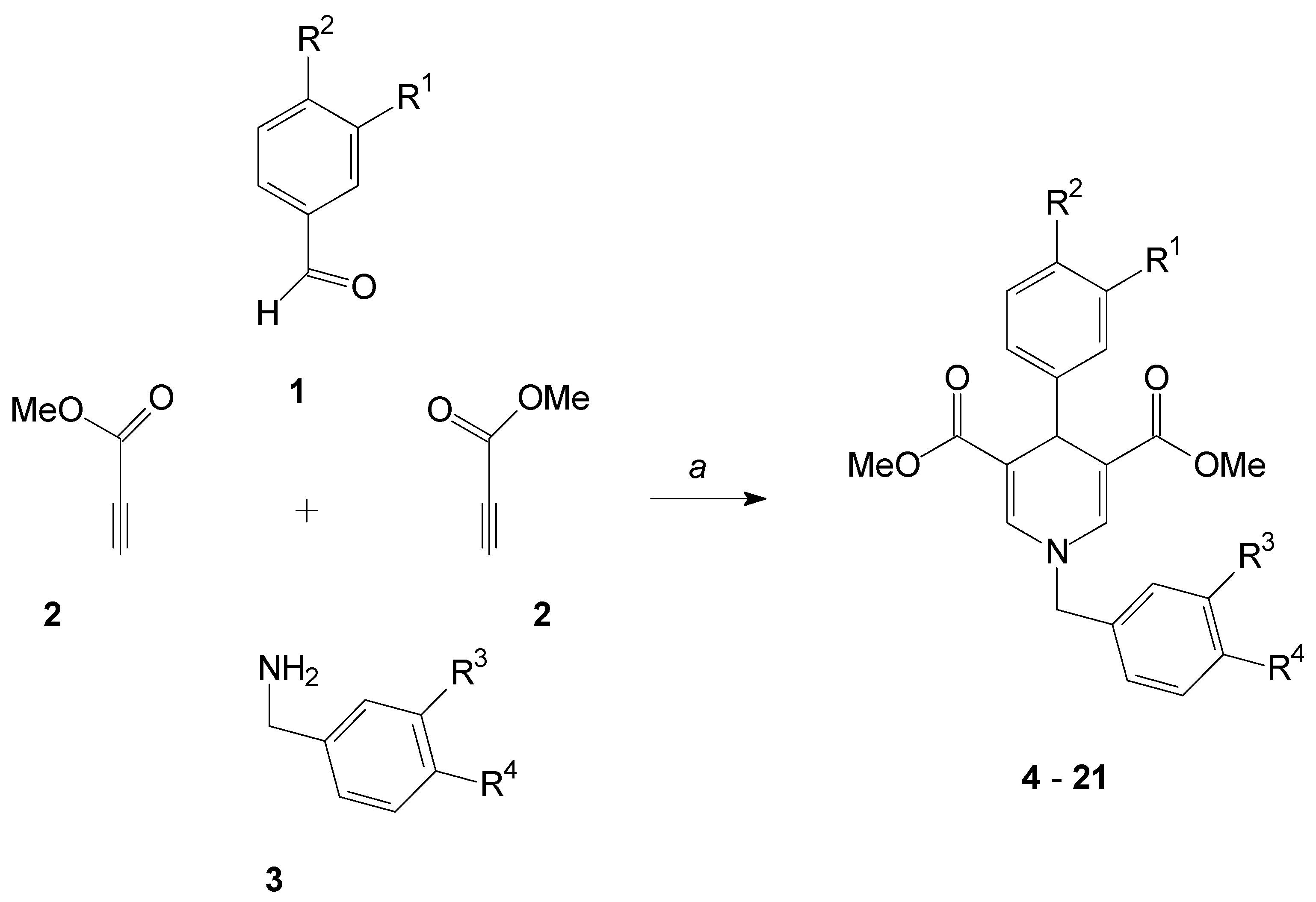
2.1. Formation of the 1,4-Dihydropyridines
2.2. MRP4 Efflux Pump Inhibition with the 1,4-Dihydropyridines
2.3. In Vitro MRP4 Resistance Studies of Drug Reversal
3. Material and Methods
3.1. Chemical Reagents and Instruments
3.2. General Procedure for the Synthesis of Compounds 4–21
3.3. MRP Inhibition Assay
3.4. MRP4 Reversal Assay
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Sample Availability
References
- The International Agency for Research on Cancer. Available online: https://www.wh.int/cancer/PRGlobocanFinal.pdf (accessed on 27 August 2020).
- Vokinger, K.N.; Hwang, T.J.; Grischott, T.; Reichert, S.; Tibau, A.; Rosemann, T.; Kesselheim, A.S. Prices and clinical benefit of cancer drugs in the USA and Europe: A cost–benefit analysis. Lancet Oncol. 2020, 21, 664–670. [Google Scholar] [CrossRef]
- University of Zürich. High Cost of Cancer Drugs not Always Justified. Science Daily, 30 April 2020. Available online: htpps://www.sciencedaily.com/releases/2020/04/200430191313.htm (accessed on 27 August 2020).
- Meegan, M.J.; O´Boyle, N.M. Special Issue “Anticancer Drugs”. Pharmaceuticals 2019, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.; Ng, J.; Wigmore, T. The side effects of chemotherapeutic agents. Curr. Anaesth. Crit. Care 2008, 19, 70–79. [Google Scholar] [CrossRef]
- Chames, P.; van Regenmortel, M.; Weiss, E.; Baty, D. Therapeutic antibodies: Successes, limitations and hopes for the future. Br. J. Pharmacol. 2009, 157, 220–233. [Google Scholar] [CrossRef]
- Hernandez, I.; Bott, S.W.; Patel, A.S.; Wolf, C.G.; Hospodar, A.R.; Sampathkumar, S.; Shrank, W.H. Pricing of monoclonal antibody therapies: Higher if used for cancer? Am. J. Manag. Care 2018, 24, 109–112. [Google Scholar] [PubMed]
- Barouch-Bentov, R.; Sauer, K. Mechanisms of drug resistance in kinases. Expert Opin. Investig. Drugs 2011, 20, 153–208. [Google Scholar] [CrossRef] [PubMed]
- Gentile, C.; Martorana, A.; Lauria, A.; Bonsignore, R. Kinase Inhibitors in Multitargeted Cancer Therapy. Curr. Med. Chem. 2017, 24, 1671–1686. [Google Scholar] [CrossRef] [PubMed]
- Pottier, C.; Fresnais, M.; Gilon, M.; Jerusalem, G.; Longuespée, R.; Sounni, N.E. Tyrosine Kinase Inhibitors in Cancer: Breakthrough and Challenges of Targeted Therapy. Cancers 2020, 12, 731. [Google Scholar] [CrossRef]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of efflux pumps in multidrug-resistant cancer. Nat. Rev. Cancer 2018, 18, 452–464. [Google Scholar] [CrossRef]
- Gottesman, M.M.; Lavi, O.; Hall, M.D.; Gillet, J.-P. Toward a Better Understanding of the Complexity of Cancer Drug Resistance. Annu. Rev. Pharmacol. Toxicol. 2016, 56, 85–102. [Google Scholar] [CrossRef] [PubMed]
- Sampson, A.; Peterson, B.G.; Tan, K.W.; Iram, S.H. Doxorubicin as a fluorescent reporter identifies novel MRP1 (ABCB1) inhibitors missed by calcein-based high content screening of anticancer agents. Biomed. Pharmacother. 2019, 118, 109289. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhou, X.; Zhou, H.; Jia, J.; Li, L.; Zhai, S.; Yan, B. Reducing Both Pgp Overexpression and Drug Efflux with Anti-Cancer Gold-Paclitaxel Nanoconjugates. PLoS ONE 2016, 11, e0160042. [Google Scholar] [CrossRef] [PubMed]
- Martinez, L.; Arnaud, O.; Henin, E.; Tao, H.; Chaptal, V.; Doshi, R.; Andrieu, T.; Dussurgey, S.; Tod, M.; di Pietro, A.; et al. Understanding polyspecificity within the substrate-binding cavity of the human multidrug resistance P-glycoprotein. FEBS J. 2014, 281, 673–682. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Chen, Z.-S. Multidrug Resistance Proteins (MRPs) and Cancer Therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The Role of ABC Transporters in Clinical Practice. Oncology 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Binkhathlan, Z. P-glycoprotein Inhibition as a Therapeutic Approach for Overcoming Multidrug Resistance in Cancer: Current Status and Future Perspectives. Curr. Cancer Drug Targets 2013, 13, 326–346. [Google Scholar] [CrossRef]
- Van Leeuwen, F.W.; Buckle, T.; Kersbergen, A.; Rottenberg, S.; Gilhuijs, K.G. Noninvasiv functional imaging of P-glycoprotein-mediated doxorubicin resistance in a mouse model of hereditary breast cancer to predict response and assign P-gp inhibitory sensitivity. Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 406–412. [Google Scholar]
- Pajic, M.; Iyer, J.K.; Kersbergen, A.; van der Burg, E.; Nygren, A.O.; Jonkers, J.; Borst, P.; Rottenberg, S. Moderate Increase in Mdr1a/1b Expression Causes In vivo Resistance to Doxorubicin in a Mouse Model for Hereditary Breast Cancer. Cancer Res. 2009, 69, 6396–6404. [Google Scholar] [CrossRef]
- Dai, C.-L.; Tiwari, A.K.; Wu, C.-P.; Su, X.-D.; Wang, S.-R.; Liu, D.-G.; Ashby, C.R.; Huang, Y.; Robey, R.W.; Liang, Y.-J.; et al. Lapatinib (Tykerb, GW572016) Reverses Multidrug Resistance in Cancer Cells by Inhibiting the Activity of ATP-Binding Cassette Subfamily B Member 1 and G Member 2. Cancer Res. 2008, 68, 7905–7914. [Google Scholar] [CrossRef]
- Mi, Y.-J.; Liang, Y.-J.; Huang, H.-B.; Zhao, H.-Y.; Wu, C.-P.; Wang, F.; Tao, L.-Y.; Zhang, C.-Z.; Dai, C.-L.; Tiwari, A.K.; et al. Apatinib (YN968D1) Reverses Multidrug Resistance by Inhibiting the Efflux Function of Multiple ATP-Binding Cassette Transporters. Cancer Res. 2010, 70, 7981–7991. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.A.; Jedlitschky, G.; Meyer-Schwabedissen, H.; Grube, M.; Köck, K.; Kroemer, H.K. Cellular export of drugs and signaling molecules by the ATP-binding cassette transporters MRP4 (ABCC4) and MRP5 (ABCC5). Drug Metab. Rev. 2005, 37, 253–278. [Google Scholar] [CrossRef] [PubMed]
- Russel, F.G.M.; Koenderink, J.B.; Masereeuw, R. Multidrug resistance protein 4 (MRP4/ABCC4): A versatile efflux transporter for drugs and signalling molecules. Trends Pharmacol. Sci. 2008, 29, 200–207. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.J.; Haber, M.; Porro, A.; Munoz, M.A.; Iraci, N.; Xue, C. ABC multidrug transporters in childhood neuroblastoma: Clinical and biological effects independent of cytotoxic drug efflux. J. Natl. Cancer Inst. 2011, 103, 1236–1251. [Google Scholar] [CrossRef] [PubMed]
- Sahores, A.; Carozzo, A.; May, M.; Gómez, N.; di Siervi, N.; Serro, M.D.S.; Yaneff, A.; Rodríguez-González, A.; Abba, M.; Shayo, C.; et al. Multidrug transporter MRP4/ABCC4 as a key determinant of pancreatic cancer aggressiveness. Sci. Rep. 2020, 10, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Colavita, J.P.M.; Todaro, J.S.; de Sousa, M.; May, M.; Gómez, N.; Yaneff, A.; di Siervi, N.; Aguirre, M.V.; Guijas, C.; Ferrini, L.; et al. Multidrug resistance protein 4 (MRP4/ABCC4) is overexpressed in clear cell renal cell carcinoma (ccRCC) and is essential to regulate cell proliferation. Int. J. Biol. Macromol. 2020, 161, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Gao, J.; Cheung, L.; Bongers, A.; Somers, K.; Clifton, M.; Ramsay, E.E.; Russell, A.J.; Valli, E.; Gifford, A.J.; et al. ABCC4/MRP4 contributes to the aggressiveness of Myc-associated epithelial ovarian cancer. Int. J. Cancer 2020, 147, 2225–2238. [Google Scholar] [CrossRef]
- Wijaya, J.; Vo, B.T.; Liu, J.; Xu, B.; Wu, G.; Wang, Y.; Peng, J.; Zhang, J.; Janke, L.J.; Orr, B.A.; et al. An ABC Transporter Drives Medulloblastoma Pathogenesis by Regulating Sonic Hedgehog Signaling. Cancer Res. 2020, 80, 1524–1537. [Google Scholar] [CrossRef]
- Murray, J.; Valli, E.; Yu, D.M.; Truong, A.M.; Gifford, A.J.; Eden, G.L.; Gamble, L.D.; Hanssen, K.M.; Flemming, C.L.; Tan, A.; et al. Suppression of the ATP-binding cassette transporter ABCC4 impairs neuroblastoma tumour growth and sensitises to irinotecan in vivo. Eur. J. Cancer 2017, 83, 132–141. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, L.; Li, X.; Wang, X.; Li, L.; Fu, X.; Sun, Z.; Li, Z.; Nan, F.; Chang, Y.; et al. ATP-binding cassette sub-family C member 4 (ABCC4) is overexpressed in human NK/T-cell lymphoma and regulates chemotherapy sensitivity: Potential as a functional therapeutic target. Leuk. Res. 2015, 39, 1448–1454. [Google Scholar] [CrossRef] [PubMed]
- Lin, F.; He, Z.; Hu, B.; Tang, L.; Zheng, S.; Sun, Y.; Sheng, Z.; Yao, Y. The overexpression of MRP4 is related to multidrug resistance in osteosarcoma cells. J. Cancer Res. Ther. 2015, 11, 18. [Google Scholar] [CrossRef] [PubMed]
- Copsel, S.; Garcia, C.; Diez, F.; Vermeulem, M.; Baldi, A.; Bianciotti, L.G.; Russel, F.G.; Shayo, C.; Davio, C. Multidrug Resistance Protein 4 (MRP4/ABCC4) Regulates cAMP Cellular Levels and Controls Human Leukemia Cell Proliferation and Differentiation. J. Biol. Chem. 2011, 286, 6979–6988. [Google Scholar] [CrossRef] [PubMed]
- Oevermann, L.; Scheitz, J.; Starke, K.; Köck, K.; Kiefer, T.; Dölken, G.; Nießen, J.; Greinacher, A.; Siegmund, W.; Zygmunt, M.; et al. Hematopoietic stem cell differentiation affects expression and function of MRP4 (ABCC4), a transport protein for signaling molecules and drugs. Int. J. Cancer 2009, 124, 2303–2311. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Köck, K.; Ritter, C.A.; Chen, Z.-S.; Grube, M.; Jedlitschky, G.; Illmer, T.; Ayres, M.; Beck, J.F.; Siegmund, W.; et al. Expression of ABCC-Type Nucleotide Exporters in Blasts of Adult Acute Myeloid Leukemia: Relation to Long-term Survival. Clin. Cancer Res. 2009, 15, 1762–1769. [Google Scholar] [CrossRef] [PubMed]
- Cheung, L.; Yu, D.M.; Neiron, Z.; Failes, T.W.; Arndt, G.M.; Fletcher, J.I. Identification of new MRP4 inhibitors from a library of FDA approved drugs using a high-throughput bioluminescence screen. Biochem. Pharmacol. 2015, 93, 380–388. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.-H.; Zhou, L.; Wang, S.-S.; Jing, X.; Guo, H.-L.; Sun, F.; Zhang, Y.; Chen, F.; Xu, J.; Ji, X. Methotrexate Disposition in Pediatric Patients with Acute Lymphoblastic Leukemia: What Have We Learnt from the Genetic Variants of Drug Transporters. Curr. Pharm. Des. 2019, 25, 627–634. [Google Scholar] [CrossRef] [PubMed]
- Drenberg, C.D.; Hu, S.; Li, L.; Buelow, D.R.; Orwick, S.J.; Gibson, A.A.; Schuetz, J.D.; Sparreboom, A.; Baker, S.D. ABCC4 Is a Determinant of Cytarabine-Induced Cytotoxicity and Myelosuppression. Clin. Transl. Sci. 2016, 9, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Yuan, B.; Yoshino, Y.; Fukushima, H.; Markova, S.; Takagi, N.; Toyoda, H.; Kroetz, D.L. Multidrug resistance-associated protein 4 is a determinant of arsenite resistance. Oncol. Rep. 2015, 35, 147–154. [Google Scholar] [CrossRef]
- Tsukamoto, M.; Yamashita, M.; Nishi, T.; Nakagawa, H. A Human ABC Transporter ABCC4 Gene SNP (rs11568658, 559 G > T, G187W) Reduces ABCC4-Dependent Drug Resistance. Cells 2019, 8, 39. [Google Scholar] [CrossRef]
- Chen, Q.; Meng, F.; Wang, L.; Mao, Y.; Zhou, H.; Hua, D.; Zhang, H.; Wang, W. A polymorphism in ABCC4 is related to efficacy of 5-FU/capecitabine-based chemotherapy in colorectal cancer patients. Sci. Rep. 2017, 7, 1–7. [Google Scholar] [CrossRef]
- Choi, J.-S.; Coi, I.; Choi, D.-H. Effects of Nifedipine on the Pharmakokinetics of Repaglidine in Rats: Possible Role of CYP3A4 and P-glycoprotein Inhibition by Nifedipine. Pharmacol. Rep. 2013, 65, 1411–1430. [Google Scholar] [CrossRef]
- Nasr-Esfahani, M.; Montazerozohori, M.; Raeatikia, R. An efficient Hantzsch synthesis of 1,4-dihydropyridines using p-toluensulfonic acid under solvent-free condition. Maejo Int. J. Sci. Technol. 2014, 8, 32–40. [Google Scholar]
- Xie, M.; Rich, T.C.; Scheitrum, C.; Conti, M.; Richter, W. Inactivation of Multidrug Resistance Proteins Disrupts Both Cellular Extrusion and Intracellular Degradation of cAMP. Mol. Pharmacol. 2011, 80, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Yuan, X.; Xiao, Z.; Jin, H.; Zhang, L.; Liu, Z. Discovery of novel multidrug resistance protein 4 (MRP4) inhibitors as active agents reducing ressistance to anticancer drug 6-Mercaptopurine (6-MP) by structure and ligand-based virtual screening. PLoS ONE 2018, 0205175. [Google Scholar]
- Deeley, R.G.; Westlake, C.; Cole, S.P. Transmembrane Transport of Endo- and Xenobiotics by Mammalian ATP-Binding Cassette Multidrug Resistance Proteins. Physiol. Rev. 2006, 86, 849–899. [Google Scholar] [CrossRef] [PubMed]
- Eva, A.; Robbins, K.C.; Andersen, P.R.; Srinivasan, A.; Tronick, S.R.; Reddy, E.P.; Ellmore, N.W.; Galen, A.T.; Lautenberg, J.A.; Papas, T.S.; et al. Cellular genes analogous to retroviral onco genes are transcribed in human tumor cells. Nature 1982, 295, 116–119. [Google Scholar] [CrossRef] [PubMed]
- Rius, M.; Nies, A.T.; Hummel-Eisenbeiss, J.; Jedlitschky, G.; Keppler, D. Cotransport of reduced glutathione with bile salts by MRP4 (ABCC4) localized to the basolateral hepatocyte membrane. Hepatology 2003, 38, 374–384. [Google Scholar] [CrossRef]


{kind=link}
| Compound | R1 | R2 | R3 | R4 | FAR |
|---|---|---|---|---|---|
| Value [a] | |||||
| 4 | CF3 | H | CF3 | H | 1.28 |
| 5 | CF3 | H | OMe | H | 1.21 |
| 6 | CF3 | H | H | OMe | 1.11 |
| 7 | CF3 | H | OMe | OMe | 1.10 |
| 8 | H | CF3 | OMe | H | 1.28 |
| 9 | H | CF3 | H | OMe | 1.11 |
| 10 | H | CF3 | OMe | OMe | 1.26 |
| 11 | F | F | OMe | H | 1.43 |
| 12 | F | F | H | OMe | 1.40 |
| 13 | F | F | OMe | OMe | 1.23 |
| 14 | F | H | OMe | OMe | 1.21 |
| 15 | H | F | OMe | OMe | 1.12 |
| 16 | CF3 | OMe | CF3 | H | 1.19 |
| 17 | CF3 | OMe | OMe | H | 1.55 |
| 18 | CF3 | OMe | H | OMe | 0.95 |
| 19 | CF3 | OMe | OMe | OMe | 1.22 |
| 20 | OMe | OMe | CF3 | H | 0.98 |
| 21 | OBn | OBn | CF3 | H | 1.30 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Döring, H.; Kreutzer, D.; Ritter, C.; Hilgeroth, A. Discovery of Novel Symmetrical 1,4-Dihydropyridines as Inhibitors of Multidrug-Resistant Protein (MRP4) Efflux Pump for Anticancer Therapy. Molecules 2021, 26, 18. https://doi.org/10.3390/molecules26010018
Döring H, Kreutzer D, Ritter C, Hilgeroth A. Discovery of Novel Symmetrical 1,4-Dihydropyridines as Inhibitors of Multidrug-Resistant Protein (MRP4) Efflux Pump for Anticancer Therapy. Molecules. 2021; 26(1):18. https://doi.org/10.3390/molecules26010018
Chicago/Turabian StyleDöring, Henry, David Kreutzer, Christoph Ritter, and Andreas Hilgeroth. 2021. "Discovery of Novel Symmetrical 1,4-Dihydropyridines as Inhibitors of Multidrug-Resistant Protein (MRP4) Efflux Pump for Anticancer Therapy" Molecules 26, no. 1: 18. https://doi.org/10.3390/molecules26010018
APA StyleDöring, H., Kreutzer, D., Ritter, C., & Hilgeroth, A. (2021). Discovery of Novel Symmetrical 1,4-Dihydropyridines as Inhibitors of Multidrug-Resistant Protein (MRP4) Efflux Pump for Anticancer Therapy. Molecules, 26(1), 18. https://doi.org/10.3390/molecules26010018