Antibacterial Activity of Fluorobenzoylthiosemicarbazides and Their Cyclic Analogues with 1,2,4-Triazole Scaffold


 , , ,
, , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
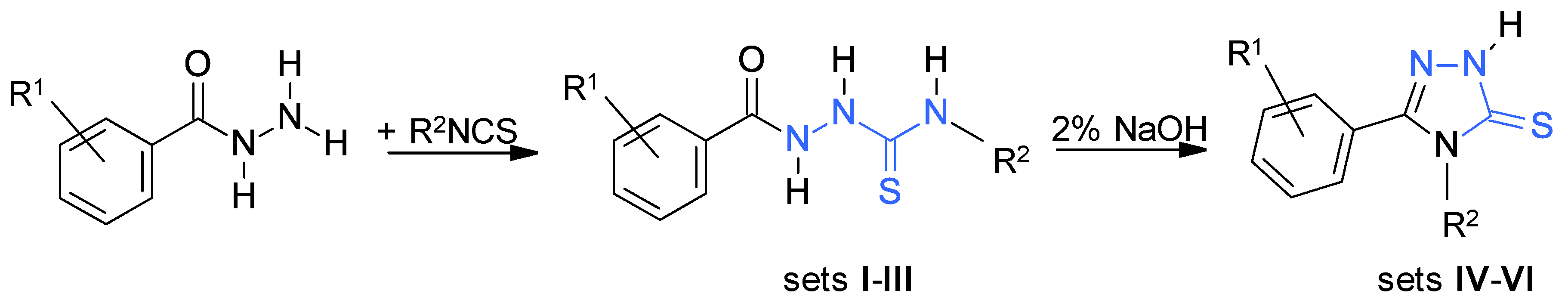
2.1. Synthetic Protocols
2.2. Antibacterial Screening
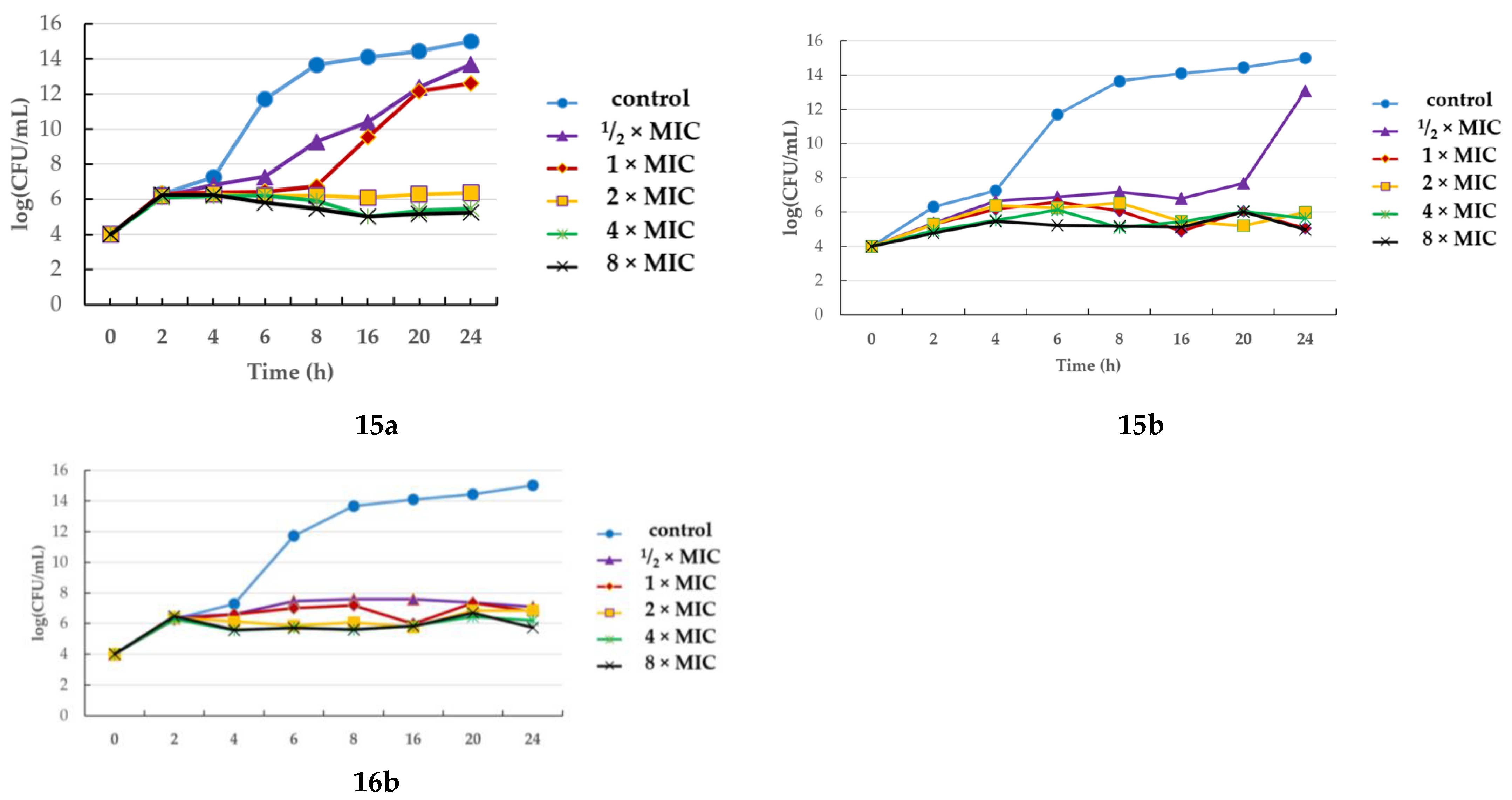
2.3. Assessing Bactericidal/Bacteriostatic Characteristics
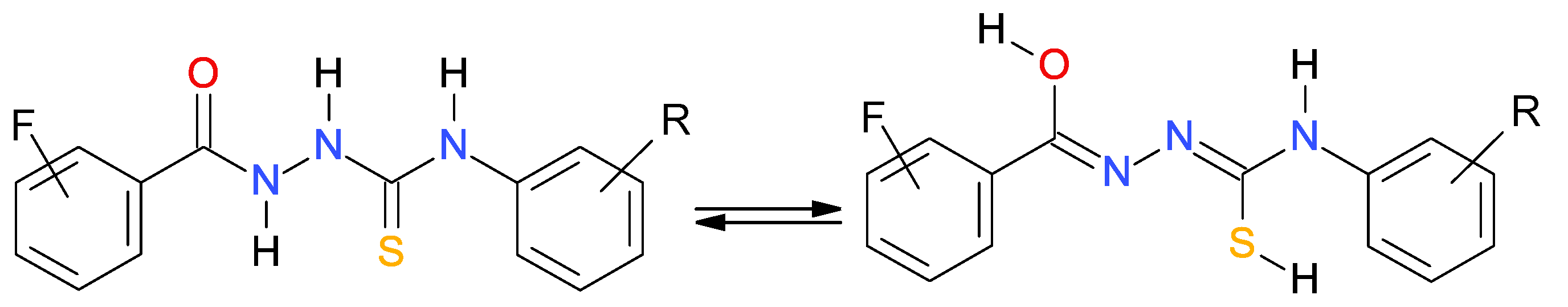
2.4. Thiol−ThioneTautomerismintheThiosemicarbazides
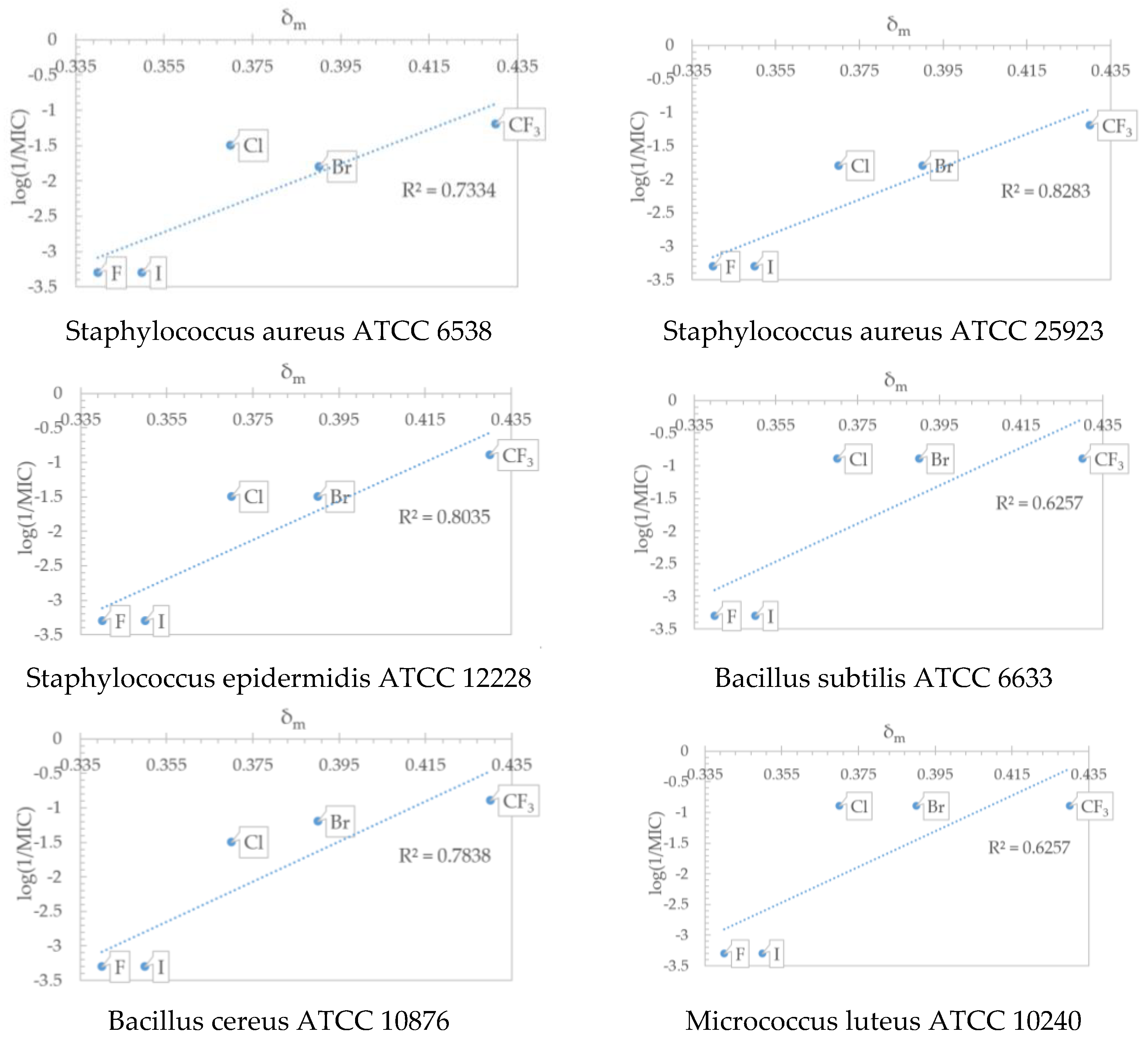
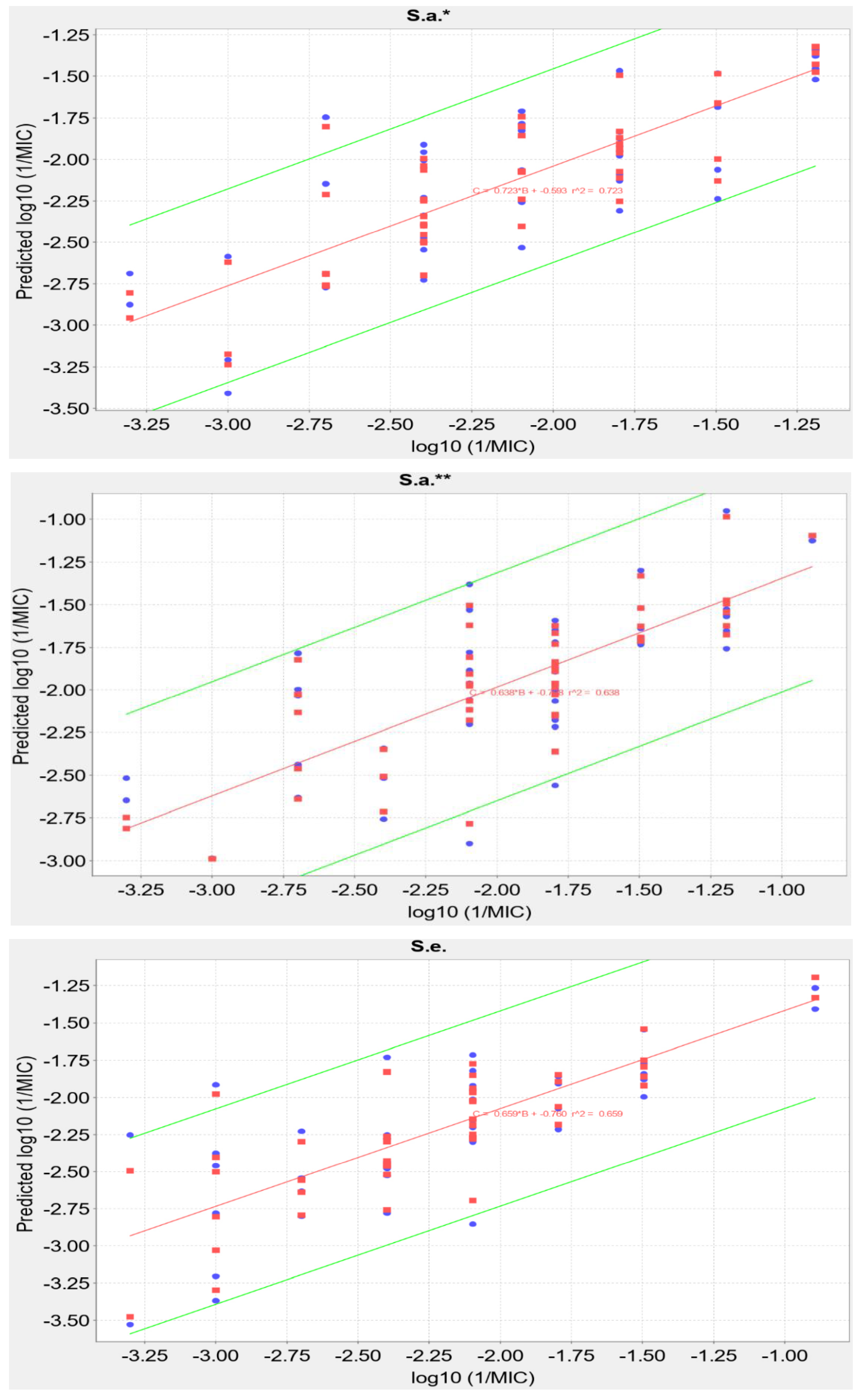
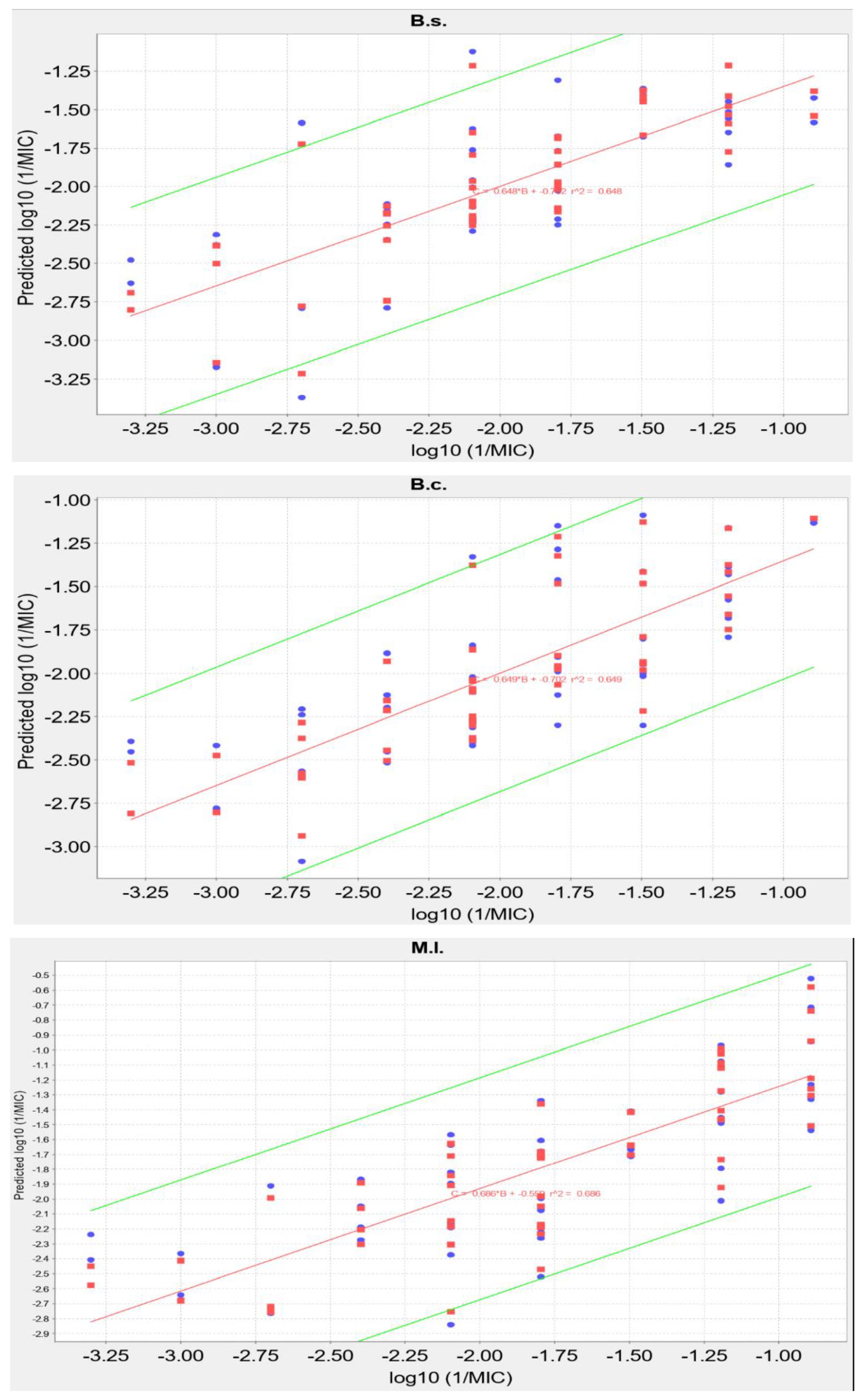
2.5. QSAR Analysis
2.6. Docking Studies
3. Materials and Methods
3.1. Chemistry
3.1.1. General Procedure for Synthesis of the Thiosemicarbazides
3.1.2. General Procedure for Synthesis of the 1,2,4-Triazole-3-thiones
3.2. Antibacterial Screening
Time-Kill Assay
3.3. Docking Studies
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Lu, Y.; Liu, Y.; Xu, Z.; Li, H.; Liu, H.; Zhu, W. Halogen bonding for rational drug design and new drug discovery. Expert Opin. Drug Discov. 2012, 7, 375–383. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. Quantifying intermolecular interactions: Guidelines for the molecular recognition toolbox. Angew. Chemie Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- Richter, F.; Leaver-Fay, A.; Khare, S.D.; Bjelic, S.; Baker, D. De novo enzyme design using Rosetta3. PLoS ONE 2011, 6, e19230. [Google Scholar] [CrossRef] [PubMed]
- Seeman, N.C. Structural DNA nanotechnology: An overview. Methods Mol. Biol. 2005, 303, 143–166. [Google Scholar]
- Hernandes, M.Z.; Cavalcanti, S.M.T.; Moreira, D.R.M.; de Azevedo Junior, W.F.; Leite, A.C.L. Halogen atoms in the modern medicinal chemistry: Hints for the drug design. Curr. Drug Targets 2010, 11, 303–314. [Google Scholar] [CrossRef]
- Shah, P.; Westwell, A.D. The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem. 2007, 22, 527–540. [Google Scholar] [CrossRef]
- Böhm, H.; Banner, D.; Bendels, S.; Kansy, M.; Kuhn, B.; Müller, K.; Obst-Sander, U.; Stahl, M. Fluorine in medicinal chemistry. ChemBioChem 2004, 5, 637–643. [Google Scholar]
- Dugar, S.; Yumibe, N.; Clader, J.W.; Vizziano, M.; Huie, K.; Heek, M.V.; Compton, D.S.; Davis, H.R. Metabolism and structure activity data based drug design: Discovery of (2)SCH 53079 an analog of the potent cholesterol absorption inhibitor(2)SCH 48461. Bioorg. Med. Chem. Lett. 1996, 6, 1271–1274. [Google Scholar] [CrossRef]
- Hutchinson, I.; Chua, M.-S.; Browne, H.L.; Trapani, V.; Bradshaw, T.D.; Westwell, A.D.; Stevens, M.F.G. Antitumor benzothiazoles. 14. Synthesis and in vitro biological properties of fluorinated 2-(4-aminophenyl)benzothiazoles. J. Med. Chem. 2001, 44, 1446–1455. [Google Scholar] [CrossRef]
- Brantley, E.; Trapani, V.; Alley, M.C.; Hose, C.D.; Bradshaw, T.D.; Stevens, M.F.G.; Sausville, E.A.; Stinson, S.F. Fluorinated 2-(4-amino-3-methylphenyl)benzothiazoles induce CYP1A1 expression, become metabolised, and bind to macromolecules in sensitive human cancer cells. Drug Metab. Dispos. 2004, 32, 1392–1401. [Google Scholar] [CrossRef]
- Akama, T.; Ishida, H.; Shida, Y.; Kimura, U.; Gomi, K.; Saito, H.; Fuse, E.; Kobayashi, S.; Yoda, N.; Kasai, M. Design and synthesis of potent antitumor 5,40-diaminoflavone derivatives based on metabolic considerations. J. Med. Chem. 1997, 40, 1894–1900. [Google Scholar] [CrossRef] [PubMed]
- Mortimer, C.G.; Wells, G.; Crochard, J.-P.; Stone, E.L.; Bradshaw, T.D.; Stevens, M.F.G.; Westwell, A.D. Antitumor benzothiazoles. 26. 2-(3,4-Dimethoxyphenyl)-5-fluorobenzothiazole (GW610, NSC 721648), a simple fluorinated 2-arylbenzothiazole, shows potent and selective inhibitory activity against lung, colon, and breast cancer cell lines. J. Med. Chem. 2006, 49, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Kim, C.-Y.; Chang, J.S.; Doyon, J.B.; Baird, T.T.; Fierke, C.A.; Jain, A.; Christianson, D.W. Contribution of fluorine to protein-ligand affinity in the binding of fluoroaromatic inhibitors to carbonic anhydrase II. J. Am. Chem. Soc. 2000, 122, 12125–12134. [Google Scholar] [CrossRef]
- Maren, T.H.; Conroy, C.W. A new class of carbonic anhydrase inhibitors. J. Biol. Chem. 1993, 268, 26233–26239. [Google Scholar]
- Riley, K.E.; Merz, K.M. Effects of fluorine substitution on edge-to-face interaction of the benzene dimer. J. Phys. Chem. 2005, 109, 17752–17756. [Google Scholar] [CrossRef]
- Hof, F.; Scofield, D.M.; Schweizer, W.B.; Diederich, F. A weak attractive interaction between organic fluorine and an amide group. Ang. Chem. Int. Ed. 2004, 43, 5056–5059. [Google Scholar] [CrossRef]
- Olsen, J.A.; Banner, D.W.; Seiler, P.; Sander, U.O.; D’Arcy, A.; Stihle, M.; Müller, K.; Diederich, F. A fluorine scan of thrombin inhibitors to map the fluorophilicity/fluorophobicity of an enzyme active site: Evidence for C-F···CvO interactions. Ang. Chem. Int. Ed. 2003, 42, 2507–2511. [Google Scholar] [CrossRef]
- Domagala, J.M.; Hanna, L.D.; Heifetz, C.L.; Hutt, M.P.; Mich, T.F.; Sanchez, J.P.; Solomon, M. New structure-activity relationships of the quinolone antibacterials using the target enzyme. The development and application of a DNA gyrase assay. J. Med. Chem. 1986, 29, 394–404. [Google Scholar] [CrossRef]
- Wright, D.H.; Brown, G.H.; Peterson, M.L.; Rotschafer, J.C. Application of fluoroquinolone pharmacodynamics. J. Antimicrob. Chemother. 2000, 46, 669–683. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Ojima, I. Exploration of fluorine chemistry at the multidisciplinary interface of chemistry and biology. J. Org. Chem. 2013, 78, 6358–6383. [Google Scholar] [CrossRef] [PubMed]
- Gillis, E.P.; Eastman, K.J.; Hill, M.D.; Donnelly, D.J.; Meanwell, N.A. Applications of fluorine in medicinal chemistry. J. Med. Chem. 2015, 58, 8315–8359. [Google Scholar] [CrossRef] [PubMed]
- Chudzik-Rzad, B.; Malm, A.; Trotsko, N.; Wujec, M.; Plech, T.; Paneth, A. Synergistic effects of thiosemicarbazides with clinical drugs against S. aureus. Molecules 2020, 25, 2302. [Google Scholar] [CrossRef] [PubMed]
- Ameryckx, A.; Thabault, L.; Pochet, L.; Leimanis, S.; Poupaert, J.H.; Wouters, J.; Joris, B.; Van Bambeke, F.; Frédérick, R. 1-(2-Hydroxybenzoyl)-thiosemicarbazides are promising antimicrobial agents targeting D-alanine-D-alanine ligase in bacterio. Eur. J. Med. Chem. 2018, 159, 324–338. [Google Scholar] [CrossRef] [PubMed]
- Ameryckx, A.; Pochet, L.; Wang, G.; Yildiz, E.; Saadi, B.E.; Wouters, J.; Van Bambeke, F.; Frédérick, R. Pharmacomodulations of the benzoyl-thiosemicarbazide scaffold reveal antimicrobial agents targeting D-alanyl-D-alanine ligase in bacterio. Eur. J. Med. Chem. 2020, 200, 112444. [Google Scholar] [CrossRef]
- Al-Mutairi, A.A.; Al-Alshaikh, M.A.; Al-Omary, F.A.M.; Hassan, H.M.; El-Mahdy, A.M.; El-Emam, A.A. Synthesis, antimicrobial, and anti-proliferative activities of novel 4-(adamantan-1-yl)-1-arylidene-3-thiosemicarbazides, 4-arylmethyl N′-(Adamantan-1-yl)piperidine-1-carbothioimidates, and related derivatives. Molecules 2019, 24, 4308. [Google Scholar] [CrossRef]
- Chen, R.; Huo, L.; Jaiswal, Y.; Huang, J.; Zhong, Z.; Zhong, J.; Williams, L.; Xia, X.; Liang, Y.; Yan, Z. Design, synthesis, antimicrobial, and anticancer activities of acridine thiosemicarbazides derivatives. Molecules 2019, 24, 2065. [Google Scholar] [CrossRef]
- El-Sharief, M.A.M.S.; Abbas, S.Y.; El-Bayouki, K.A.M.; El-Gammal, E.W. Synthesis of thiosemicarbazones derived from N-(4-hippuric acid)thiosemicarbazide and different carbonyl compounds as antimicrobial agents. Eur. J. Med. Chem. 2013, 67, 263–268. [Google Scholar] [CrossRef]
- Wang, Y.; Dang, Q.; Liu, C.; Yu, D.; Pu, X.; Wang, Q.; Gao, H.; Zhang, B.; Cha, D. Selective adsorption toward Hg(II) and inhibitory effect on bacterial growth occurring on thiosemicarbazide-functionalized chitosan microsphere surface. ACS Appl. Mater. Interfaces 2018, 10, 40302–40316. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Fang, K.-C.; Sheu, J.-Y.; Hsu, S.-L.; Tzeng, C.-C. Synthesis and antibacterial evaluation of certain quinolone derivatives. J. Med. Chem. 2001, 44, 2374–2377. [Google Scholar] [CrossRef]
- Devar, S.B.; Swamy, B.H.M.; Rao, B.N.; Shivkumar, H.; Shivkumar, B. Synthesis of new tetrazoloquinoline thiocarbohydrazides as potential antimicrobial agents. Indian J. Heterocycl. Chem. 2011, 21, 37–40. [Google Scholar]
- Lalezari, I.; Rezvani, N.; Malekzaseh, F. Synthesis and antimicrobial activity of thiocarbohydrazide-1,5-dicarboxylic acid diesters. J. Pharm. Sci. 1972, 61, 1486–1487. [Google Scholar] [CrossRef] [PubMed]
- Zabin, S.A.; Jejurkar, C.R. Fluorescence, antibacterial and pigmentation studies of some binuclear Schiff base complexes. Asian J. Chem. 1995, 7, 542–550. [Google Scholar]
- Kapron, B.; Luszczki, J.J.; Siwek, A.; Karcz, T.; Nowak, G.; Zagaja, M.; Andres-Mach, M.; Stasilowicz, A.; Cielecka-Piontek, J.; Kocki, J.; et al. Preclinical evaluation of 1,2,4-triazole-based compounds targeting voltage-gated sodium channels (VGSCs) as promising anticonvulsant drug candidates. Bioorg. Chem. 2020, 94, 103355. [Google Scholar] [CrossRef]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 27th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2017. [Google Scholar]
- Molinspiration. Available online: https://www.molinspiration.com/ (accessed on 1 October 2020).
- Hansch, C.; Leo, A.; Taft, R.W. A survey of Hammett substituent constants and resonance and field parameters. Chem. Rev. 1991, 91, 165–195. [Google Scholar] [CrossRef]
- Hammett, L.P. The effect of structure upon the reactions of organic compounds. Benzene derivatives. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Hansch, C.H.; Leo, A. Substituent Constants for Correlation Analysis in Chemistry and Biology; Wiley: New York, NY, USA, 1979. [Google Scholar]
- Taft, R.W., Jr. Linear free energy relationships from rates of esterification and hydrolysis of aliphatic and ortho-substituted benzoate esters. J. Am. Chem. Soc. 1952, 74, 2729–2732. [Google Scholar] [CrossRef]
- Taft, R.W., Jr. Linear steric energy relationships. J. Am. Chem. Soc. 1953, 75, 4538–4539. [Google Scholar] [CrossRef]
- Taft, R.W., Jr. Separation of polar, steric, and resonance effects in reactivity. In Steric Effects in Organic Chemistry; Newman, M.S., Ed.; Wiley: New York, NY, USA, 1969; pp. 556–675. [Google Scholar]
- Charton, M. Nature of the ortho effect. II. Composition of the Taft steric parameters. J. Am. Chem. Soc. 1969, 91, 615–618. [Google Scholar] [CrossRef]
- Charton, M. Steric effects. I. Esterification and acid-catalyzed hydrolysis of esters. J. Am. Chem. Soc. 1975, 97, 1552–1556. [Google Scholar] [CrossRef]
- Charton, M. Steric effects. II. Base-catalyzed ester hydrolysis. J. Am. Chem. Soc. 1975, 97, 3691–3693. [Google Scholar] [CrossRef]
- Charton, M. Steric effects. III. Bimolecular nucleophilic substitution. J. Am. Chem. Soc. 1975, 97, 3694–3697. [Google Scholar] [CrossRef]
- Fujita, T.; Iwasa, J.; Hansch, C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]
- Röhrig, U.F.; Majjigapu, S.R.; Grosdidier, A.; Bron, S.; Stroobant, V.; Pilotte, L.; Colau, D.; Vogel, P.; Van den Eynde, B.J.; Zoete, V.; et al. Rational design of 4-aryl-1,2,3-triazoles for indoleamine 2,3-dioxygenase 1. Inhibition J. Med. Chem. 2012, 55, 5270–5290. [Google Scholar] [CrossRef]
- Paneth, A.; Stączek, P.; Plech, T.; Strzelczyk, A.; Dzitko, K.; Wujec, M.; Kuśmierz, E.; Kosikowska, U.; Grzegorczyk, A.; Paneth, P. Biological evaluation and molecular modelling study of thiosemicarbazide derivatives as bacterial type IIA topoisomerases inhibitors. J. Enzym. Inhib. Med. Chem. 2016, 31, 14–22. [Google Scholar] [CrossRef]
- Siwek, A.; Stączek, P.; Wujec, M.; Stefańska, J.; Kosikowska, U.; Malm, A.; Jankowski, S.; Paneth, P. Biological and docking studies of topoisomerase IV inhibition by thiosemicarbazides. J. Mol. Model. 2011, 17, 2297–2303. [Google Scholar] [CrossRef]
- Siwek, A.; Stączek, P.; Stefańska, J. Synthesis and structure-activity relationship studies of 4-arylthiosemicarbazides as topoisomerase IV inhibitors with Gram-positive antibacterial activity. Search for molecular basis of antibacterial activity of thiosemicarbazides. Eur. J. Med. Chem. 2011, 46, 5717–5726. [Google Scholar] [CrossRef]
- Finberg, R.W.; Moellering, R.C.; Tally, F.P.; Craig, W.A.; Pankey, G.A.; Dellinger, E.P.; West, M.A.; Joshi, M.; Linden, P.K.; Rolston, K.V.; et al. The importance of bactericidal drugs: Future directions in infectious disease. Clin. Infect. Dis. 2004, 39, 1314–1320. [Google Scholar] [CrossRef]
- French, G.L. Bactericidal agents in the treatment of MRSA infections—The potential role of daptomycin. J. Antimicrob. Chemother. 2006, 58, 1107–1117. [Google Scholar] [CrossRef]
- Kohanski, M.A.; Dwyer, D.J.; Collins, J.J. How antibiotics kill bacteria: From targets to networks. Nat. Rev. Microbiol. 2010, 8, 423–435. [Google Scholar] [CrossRef]
- Hall, I.H.; Chen, S.; Barnes, B.J.; West, D.X. The hypolipidemic activity of heterocyclic thiosemicarbazones, thioureas and their metal complexes in Sprague dawley male rats. Met-Based Drugs 1999, 6, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Abu-Melha, S. Pyridyl thiosemicarbazide: Synthesis, crystal structure, DFT/B3LYP, molecular docking studies and its biological investigations. Chem. Cent. J. 2018, 12, 101. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [PubMed]
- Weigend, F.; Ahlrichs, R. Balanced basis sets of split valence, triple zeta valence and quadruple zeta valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B. Gaussian, Version 16; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Siwek, A.; Wujec, M.; Wawrzycka-Gorczyca, I.; Dobosz, M.; Paneth, P. Thiol-thione tautomeric forms recognition on the example of 4-[3-(2-methyl-furan-3-yl)-5-thioxo-1,2,4-triazolin-4-yl]acetic acid. Heteroatom Chem. 2008, 19, 337–344. [Google Scholar] [CrossRef]
- Liu, S.; Chang, J.S.; Herberg, J.T.; Horng, M.-M.; Tomich, P.K.; Lin, A.H.; Marotti, K.R. Allosteric inhibition of Staphylococcus aureus D-alanine:D-alanine ligase revealed by crystallographic studies. Proc. Nat. Acad. Sci. USA 2006, 103, 15178–15183. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Y.; Lin, Q.; Wei, T. Compound N-Fluorobenzamido-N’-Phenylthiocarbamide as Well as Preparation and Application Thereof. Patent No CN 101514176, 21 November 2012. [Google Scholar]
- Sarac, K. Synthesis, spectroscopic properties, quantum chemical calculations, and biological activities of 2-{[5-(2-fluorophenyl)-4-(4-methylphenyl)-4H-1,2,4-triazol-3-yl]sulfanyl}-1-[3-methyl-3-(2,4,6-trimethylphenyl)-cyclobutyl]ethan-1-one. Russ. J. Org. Chem. 2020, 56, 119–128. [Google Scholar] [CrossRef]
- Joshi, K.C.; Mehta, D.S. Synthesis of some 3-(fluorinated aryl)-4-alkyl-/aryl-5-mercapto-1,2,4-triazoles and related compounds as possible CNS [central nervous system] depressants. J. Indian Chem. Soc. 1974, 51, 613–615. [Google Scholar]
- Carter, D.I.; Cheeseright, T.J.; Vinter, J.G. Preparation of N-Thiadiazolyl Acetamides for Treating and Preventing Diseases. Patent No WO 2008107677, 30 October 2008. [Google Scholar]
- Aouad, M. Synthesis, characterization and antimicrobial evaluation of some new Schiff, Mannich and acetylenic Mannich bases incorporating a 1,2,4-triazole nucleus. Molecules 2014, 19, 18897–18910. [Google Scholar] [CrossRef]
- Gulerman, N.; Rollas, S.; Kiraz, M.; Ekinci, A.C.; Vidin, A. Evaluation of antimycobacterial and anticonvulsant activities of new 1-(4-fluorobenzoyl)-4-substituted-thiosemicarbazide and 5-(4-fluorophenyl)-4-substituted-2,4-dihydro-3H-1,2,4-triazole-3-thione derivatives. Farmaco 1997, 52, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Paneth, A.; Trotsko, N.; Popiolek, L.; Grzegorczyk, A.; Krzanowski, T.; Janowska, S.; Malm, A.; Wujec, M. Synthesis and antibacterial evaluation of mannich bases derived from 1,2,4-triazole. Chem. Biodivers. 2019, 16, e1900377. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Tong, J.; Li, B.; Shi, Y.; Cao, Y. (Fluorophenyl)Triazole Derivatives as Fungicides and Their Preparation, Agrochemical Compositions and Use in the Treatment of Plant Fungal Infection. Patent No. CN 102766102, 3 December 2014. [Google Scholar]
- CLSI. Performance Standards for Antimicrobial Susceptibility Testing, 30th ed.; CLSI Document M100; Clinical Laboratory Standards Institute: Wayne, PA, USA, 2020. [Google Scholar]
- Mun, S.-H.; Kang, O.-H.; Joung, D.-K.; Kim, S.-B.; Choi, J.-G.; Shin, D.-W.; Kwon, D.-Y. In vitro anti-MRSA activity of carvone with gentamicin. Exp. Ther. Med. 2014, 7, 891–896. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| R1 | R2 | cLogP | S. a.* | S. a.** | S. e. | B. s. | B. c. | M. l. | |
|---|---|---|---|---|---|---|---|---|---|
| 3a | 2F | Ph | 2.26 | 500 [1.73] | 500 [1.73] | 500 [1.73] | 250 [0.87] | 250 [0.87] | 250 [0.87] |
| 4a | 2F | 1-naph | 3.42 | 250 [0.74] | 125 [0.37] | 250 [0.74] | 125 [0.37] | 125 [0.37] | 125 [0.37] |
| 8a | 2F | para-FPh | 2.42 | 250 [0.81] | 250 [0.81] | 250 [0.81] | 125 [0.41] | 125 [0.41] | 62.5 [0.20] |
| 9a | 2F | meta-ClPh | 2.91 | 31.25 [0.10] | 62.5 [0.19] | 31.25 [0.10] | 15.63 [0.05] | 31.25 [0.10] | 7.82 [0.02] |
| 10a | 2F | para-ClPh | 2.94 | 250 [0.77] | 500 [1.54] | 250 [0.77] | 500 [1.54] | 31.25 [0.10] | 7.82 [0.02] |
| 11a | 2F | meta-BrPh | 3.04 | 62.5 [0.17] | 62.5 [0.17] | 31.25 [0.09] | 15.63 [0.04] | 15.63 [0.04] | 7.82 [0.02] |
| 15a | 2F | meta-CF3Ph | 3.13 | 15.63 [0.04] | 15.63 [0.04] | 7.82 [0.02] | 7.82 [0.02] | 7.82 [0.02] | 7.82 [0.02] |
| 16a | 2F | para-CF3Ph | 3.15 | 500 [1.40] | 500 [1.40] | 125 [0.35] | 7.82 [0.02] | 15.63 [0.04] | 7.82 [0.02] |
| 3b | 3F | Ph | 2.28 | 250 [0.87] | 250 [0.87] | 1000 [3.46] | 250 [0.87] | 250 [0.87] | 125 [0.44] |
| 4b | 3F | 1-naph | 3.44 | 250 [0.74] | 125 [0.37] | 250 [0.74] | 125 [0.37] | 125 [0.37] | 125 [0.37] |
| 5b | 3F | meta-tol | 2.71 | n.a. | 125 [0.41] | n.a. | n.a. | n.a. | 250 [0.83] |
| 6b | 3F | para-tol | 2.73 | 1000 [3.30] | 1000 [3.30] | 1000 [3.30] | 1000 [3.30] | 1000 [3.30] | 1000 [3.30] |
| 7b | 3F | meta-FPh | 2.42 | 62.5 [0.20] | 62.5 [0.20] | 125 [0.41] | 62.5 [0.20] | 62.5 [0.20] | 31.25 [0.10] |
| 8b | 3F | para-FPh | 2.44 | 250 [0.81] | 125 [0.41] | 500 [1.62] | 125 [0.41] | 250 [0.81] | 250 [0.81] |
| 9b | 3F | meta-ClPh | 2.94 | 15.63 [0.05] | 15.63 [0.05] | 31.25 [0.10] | 31.25 [0.10] | 15.63 [0.05] | 7.82 [0.02] |
| 10b | 3F | para-ClPh | 2.96 | 15.63 [0.05] | 7.82 [0.02] | 7.82 [0.02] | 15.63 [0.05] | 62.5 [0.19] | 7.82 [0.02] |
| 11b | 3F | meta-BrPh | 3.07 | 31.25 [0.09] | 15.63 [0.04] | 62.5 [0.17] | 15.63 [0.04] | 15.63 [0.04] | 15.63 [0.04] |
| 12b | 3F | para-BrPh | 3.09 | 62.5 [0.17] | 31.25 [0.09] | 1000 [2.72] | 125 [0.34] | 62.5 [0.17] | 15.63 [0.04] |
| 13b | 3F | meta-IPh | 3.34 | 31.25 [0.08] | 15.63 [0.04] | 62.5 [0.15] | 15.63 [0.04] | 15.63 [0.04] | 15.63 [0.04] |
| 15b | 3F | meta-CF3Ph | 3.15 | 15.63 [0.04] | 31.25 [0.09] | 31.25 [0.09] | 31.25 [0.09] | 31.25 [0.09] | 15.63 [0.04] |
| 16b | 3F | para-CF3Ph | 3.18 | 15.63 [0.04] | 15.63 [0.04] | 31.25 [0.09] | 31.25 [0.09] | 15.63 [0.04] | 15.63 [0.04] |
| 8c | 4F | para-FPh | 2.47 | 250 [0.81] | 62.5 [0.20] | 250 [0.81] | 125 [0.41] | 125 [0.41] | 62.5 [0.20] |
| 9c | 4F | meta-ClPh | 2.96 | 250 [0.77] | n.a. | n.a. | 62.5 [0.19] | 62.5 [0.19] | 15.63 [0.05] |
| 10c | 4F | para-ClPh | 2.98 | 62.5 [0.19] | 31.25 [0.10] | 125 [0.39] | 62.5 [0.19] | 31.25 [0.10] | 15.63 [0.05] |
| 11c | 4F | meta-BrPh | 3.09 | 62.5 [0.17] | 125 [0.34] | 125 [0.34] | 31.25 [0.09] | 31.25 [0.09] | 15.63 [0.04] |
| 12c | 4F | para-BrPh | 3.12 | 62.5 [0.17] | 31.25 [0.09] | 62.5 [0.17] | 31.25 [0.09] | 31.25 [0.09] | 15.63 [0.04] |
| 13c | 4F | meta-IPh | 3.36 | 31.25 [0.08] | 15.63 [0.04] | 125 [0.30] | 62.5 [0.15] | 31.25 [0.08] | 15.63 [0.04] |
| 14c | 4F | para-IPh | 3.39 | 125 [0.30] | 62.5 [0.15] | 125 [0.30] | 62.5 [0.15] | 62.5 [0.15] | 15.63 [0.04] |
| 15c | 4F | meta-CF3Ph | 3.18 | 125 [0.35] | 125 [0.35] | 125 [0.35] | 125 [0.35] | 125 [0.35] | 62.5 [0.18] |
| 16c | 4F | para-CF3Ph | 3.20 | 125 [0.35] | 62.5 [0.18] | 125 [0.35] | 62.5 [0.18] | 62.5 [0.18] | 31.25 [0.09] |
| Cef. | 0.98 [0.002] | 0.49 [0.001] | 0.24 [0.0006] | 15.63 [0.04] | 31.25 [0.07] | 0.98 [0.002] |
| Strain | 15a | 15b | 16b | Vancomycin |
|---|---|---|---|---|
| MSSA-1 | 15.63 [0.04] | 31.25 [0.09] | 7.82 [0.02] | 0.39 [0.0003] |
| MSSA-2 | 15.63 [0.04] | 31.25 [0.09] | 15.63 [0.04] | 0.78 [0.0005] |
| MSSA-3 | 7.82 [0.02] | 15.63 [0.04] | 7.82 [0.02] | 0.39 [0.0003] |
| MSSA-4 | 15.63 [0.04] | 31.25 [0.09] | 15.63 [0.04] | 0.78 [0.0005] |
| MSSA-5 | 7.82 [0.02] | 15.63 [0.04] | 15.63 [0.04] | 0.78 [0.0005] |
| MSSA-6 | 7.82 [0.02] | 15.63 [0.04] | 15.63 [0.04] | 0.78 [0.0005] |
| MSSA-7 | 7.82 [0.02] | 15.63 [0.04] | 7.82 [0.02] | 0.78 [0.0005] |
| MSSA-8 | 7.82 [0.02] | 15.63 [0.04] | 15.63 [0.04] | 1.56 [0.001] |
| MRSA-11 | 15.63 [0.04] | 31.25 [0.09] | 7.82 [0.02] | 0.78 [0.0005] |
| MRSA-12 | 7.82 [0.02] | 31.25 [0.09] | 3.92 [0.01] | 0.39 [0.0003] |
| MRSA-13 | 7.82 [0.02] | 31.25 [0.09] | 7.82 [0.02] | 0.78 [0.0005] |
| MRSA-14 | 15.63 [0.04] | 15.63 [0.04] | 7.82 [0.02] | 0.78 [0.0005] |
| MRSA-15 | 15.63 [0.04] | 31.25 [0.09] | 7.82 [0.02] | 0.78 [0.0005] |
| MRSA-16 | 7.82 [0.02] | 15.63 [0.04] | 7.82 [0.02] | 0.78 [0.0005] |
| MRSA-17 | 15.63 [0.04] | 15.63 [0.04] | 3.92 [0.01] | 0.78 [0.0005] |
| MRSA-18 | 15.63 [0.04] | 15.63 [0.04] | 7.82 [0.02] | 0.78 [0.0005] |
| R1 | R2 | cLogP | S. a.* | S. a.** | S. e. | B. s. | B. c. | M. l. | |
|---|---|---|---|---|---|---|---|---|---|
| 1at | 2F | Pr | 2.56 | 1000 [4.21] | 125 [0.53] | 1000 [4.21] | 1000 [4.21] | 500 [2.11] | 500 [2.11] |
| 8at | 2F | para-FPh | 3.12 | 500 [1.73] | 250 [0.86] | n.a. | 250 [0.86] | 500 [1.73] | 125 [0.43] |
| 11at | 2F | meta-BrPh | 3.95 | 500 [1.43] | 125 [0.36] | 125 [0.36] | 125 [0.36] | 500 [1.43] | 500 [1.43] |
| 12at | 2F | para-BrPh | 3.76 | 1000 [2.86] | 500 [1.43] | 1000 [2.86] | 1000 [2.86] | 1000 [2.86] | 1000 [2.86] |
| 13at | 2F | meta-IPh | 4.22 | 125 [0.31] | 62.5 [0.16] | 125 [0.31] | 15.63 [0.04] | 125 [0.31] | 125 [0.31] |
| 14at | 2F | para-IPh | 4.04 | 62.5 [0.16] | 125 [0.31] | 62.5 [0.16] | 62.5 [0.16] | 250 [0.63] | 62.5 [0.16] |
| 15at | 2F | meta-CF3Ph | 4.03 | 62.5 [0.18] | 62.5 [0.18] | 125 [0.37] | 62.5 [0.18] | 31.25 [0.09] | 62.5 [0.18] |
| 16at | 2F | para-CF3Ph | 3.85 | 250 [0.74] | 62.5 [0.18] | 250 [0.74] | 250 [0.74] | 125 [0.37] | 62.5 [0.18] |
| 9bt | 3F | meta-ClPh | 3.84 | 125 [0.41] | 62.5 [0.20] | 250 [0.82] | 250 [0.82] | 125 [0.41] | 62.5 [0.20] |
| 10bt | 3F | para-ClPh | 3.65 | 250 [0.82] | 31.25 [0.10] | 500 [1.64] | 125 [0.41] | 250 [0.82] | 31.25 [0.10] |
| 11bt | 3F | meta-BrPh | 3.97 | 62.5 [0.18] | 62.5 [0.18] | 125 [0.36] | 125 [0.36] | 62.5 [0.18] | 125 [0.36] |
| 16bt | 3F | para-CF3Ph | 3.87 | 250 [0.74] | 125 [0.37] | 500 [1.47] | 250 [0.74] | 250 [0.74] | 125 [0.37] |
| 8ct | 4F | para-FPh | 3.16 | 500 [1.73] | 500 [1.73] | 1000 [3.46] | 500 [1.73] | 500 [1.73] | 500 [1.73] |
| 10ct | 4F | para-ClPh | 3.68 | 125 [0.41] | 31.25 [0.10] | 125 [0.41] | 125 [0.41] | 125 [0.41] | 125 [0.41] |
| 12ct | 4F | para-BrPh | 3.81 | 125 [0.36] | 62.5 [0.18] | 125 [0.36] | 62.5 [0.18] | 125 [0.36] | 62.5 [0.18] |
| 13ct | 4F | meta-IPh | 4.27 | 125 [0.31] | 62.5 [0.16] | 125 [0.31] | 62.5 [0.16] | 62.5 [0.16] | 62.5 [0.16] |
| 15ct | 4F | meta-CF3Ph | 4.08 | 250 | 125 [0.37] | 250 [0.74] | 125 [0.37] | 125 [0.37] | 62.5 [0.18] |
| 16ct | 4F | para-CF3Ph | 3.90 | n.a. | n.a. | n.a. | n.a. | 125 [0.37] | 62.5 [0.18] |
| Cef. | 0.98 [0.002] | 0.49 [0.001] | 0.24 [0.0006] | 15.63 [0.04] | 31.25 [0.07] | 0.98 [0.002] |
| Species | R2 | CV r2 | Equation |
|---|---|---|---|
| S. a.* | 0.7229 | 0.6346 | Log10(1/MIC) = 0.4570 × Chlorine count + 5851.7193 × highest electrophilic susceptibility on O − 275.6544 × ring count all aromatic/MW − 1600.7357 × highest nucleophilic susceptibility/MW − 0.7411 × ln(highest nucleophilic susceptibility on H) − 3.6088 |
| S. a.** | 0.6376 | 0.5042 | Log10(1/MIC) = −0.8910 × Bromine count + 68.6203 × highest electrophilic susceptibility on C − 10.8611 × highest radical susceptibility on C + 3544.5345 × highest electrophilic susceptibility on O + 0.0205 × hydrophobicity weighted positive area − 6.2195 |
| S. e. | 0.6589 | 0.5465 | Log10(1/MIC) = 0.6767 × Chlorine count + 23.1042 × highest radical susceptibility on C + 0.0282 × atomic charge weighted positive area-atomic charge weighted negative area − 5541.1048 × highest nucleophilic susceptibility on C/MW − 1.7821 × ln(Csp2 bonded to 2 C) + 4.8737 |
| B. s. | 0.6477 | 0.5163 | Log10(1/MIC) = 0.3005 × Chlorine count − 40.9730 × all count/MW − 5.3291 × highest nucleophilic susceptibility2 + 1.3923 × ln(rotatable bond count) + 2.26699e-03 × 1.0/highest nucleophilic susceptibility on H − 0.0468 |
| B. c. | 0.6493 | 0.5250 | Log10(1/MIC) = 8728.8101 × highest electrophilic susceptibility on O − 16.0298 × highest radical susceptibility on C2 − 16.8597 × 1.0/log P + 1.64514e-03 × 1.0/highest nucleophilic susceptibility on H − 2.7091 × sqrt(ring count all aromatic) + 4.5796 |
| M. l. | 0.6858 | 0.5990 | Log10(1/MIC) = 0.7892 × Chlorine count + 5623.4960 × highest electrophilic susceptibility on O − 344.9316 × ring count all aromatic/MW − 1613.2362 × highest nucleophilic susceptibility/MW + 1.84660e-03 × 1.0/highest radical susceptibility on H + 0.3114 |
| 9a | 11a | 15a | 16a | 9b | 10b | 11b | 13b | 15b | 16b | PR | NI |
|---|---|---|---|---|---|---|---|---|---|---|---|
| −25.9 | −23.4 | −28.4 | −25.4 | −27.9 | −28.3 | −28.5 | −26.0 | −27.9 | −28.2 | −27.7 | −14.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kosikowska, U.; Wujec, M.; Trotsko, N.; Płonka, W.; Paneth, P.; Paneth, A. Antibacterial Activity of Fluorobenzoylthiosemicarbazides and Their Cyclic Analogues with 1,2,4-Triazole Scaffold. Molecules 2021, 26, 170. https://doi.org/10.3390/molecules26010170
Kosikowska U, Wujec M, Trotsko N, Płonka W, Paneth P, Paneth A. Antibacterial Activity of Fluorobenzoylthiosemicarbazides and Their Cyclic Analogues with 1,2,4-Triazole Scaffold. Molecules. 2021; 26(1):170. https://doi.org/10.3390/molecules26010170
Chicago/Turabian StyleKosikowska, Urszula, Monika Wujec, Nazar Trotsko, Wojciech Płonka, Piotr Paneth, and Agata Paneth. 2021. "Antibacterial Activity of Fluorobenzoylthiosemicarbazides and Their Cyclic Analogues with 1,2,4-Triazole Scaffold" Molecules 26, no. 1: 170. https://doi.org/10.3390/molecules26010170
APA StyleKosikowska, U., Wujec, M., Trotsko, N., Płonka, W., Paneth, P., & Paneth, A. (2021). Antibacterial Activity of Fluorobenzoylthiosemicarbazides and Their Cyclic Analogues with 1,2,4-Triazole Scaffold. Molecules, 26(1), 170. https://doi.org/10.3390/molecules26010170