
Desymmetrizing Heteroleptic [Cu(P^P)(N^N)][PF6] Compounds: Effects on Structural and Photophysical Properties, and Solution Dynamic Behavior

 , , ,
, , ,  and
and 
Abstract

1. Introduction
2. Results and Discussion
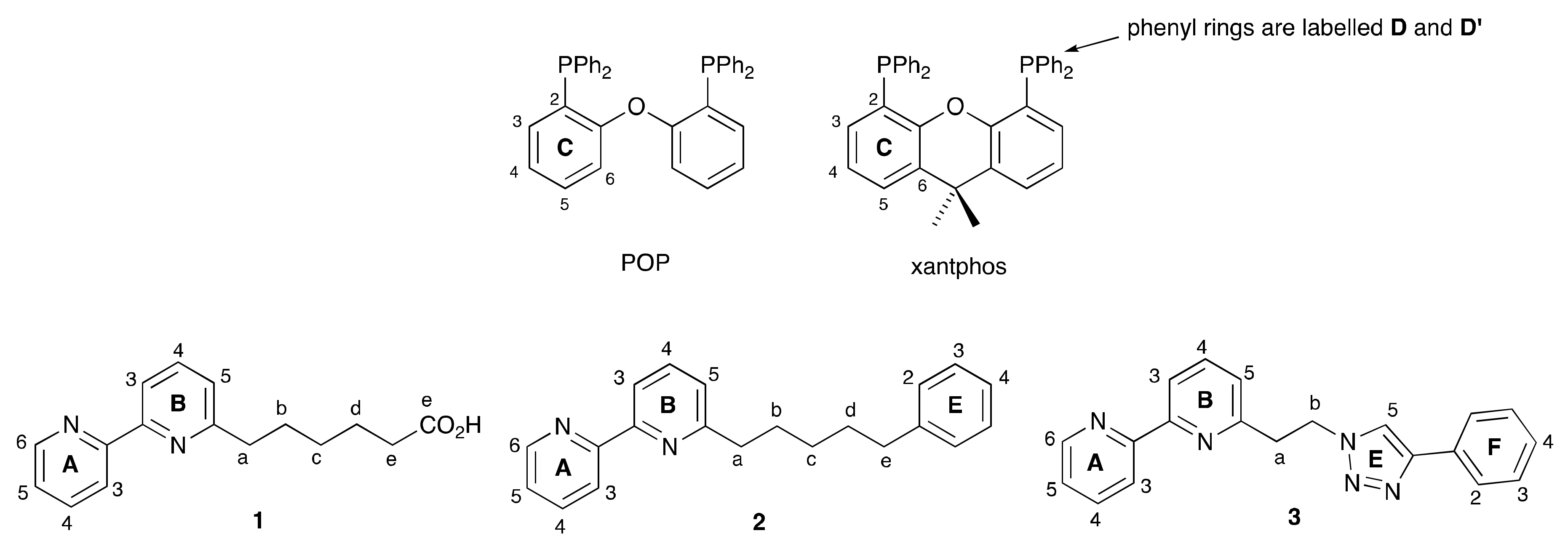
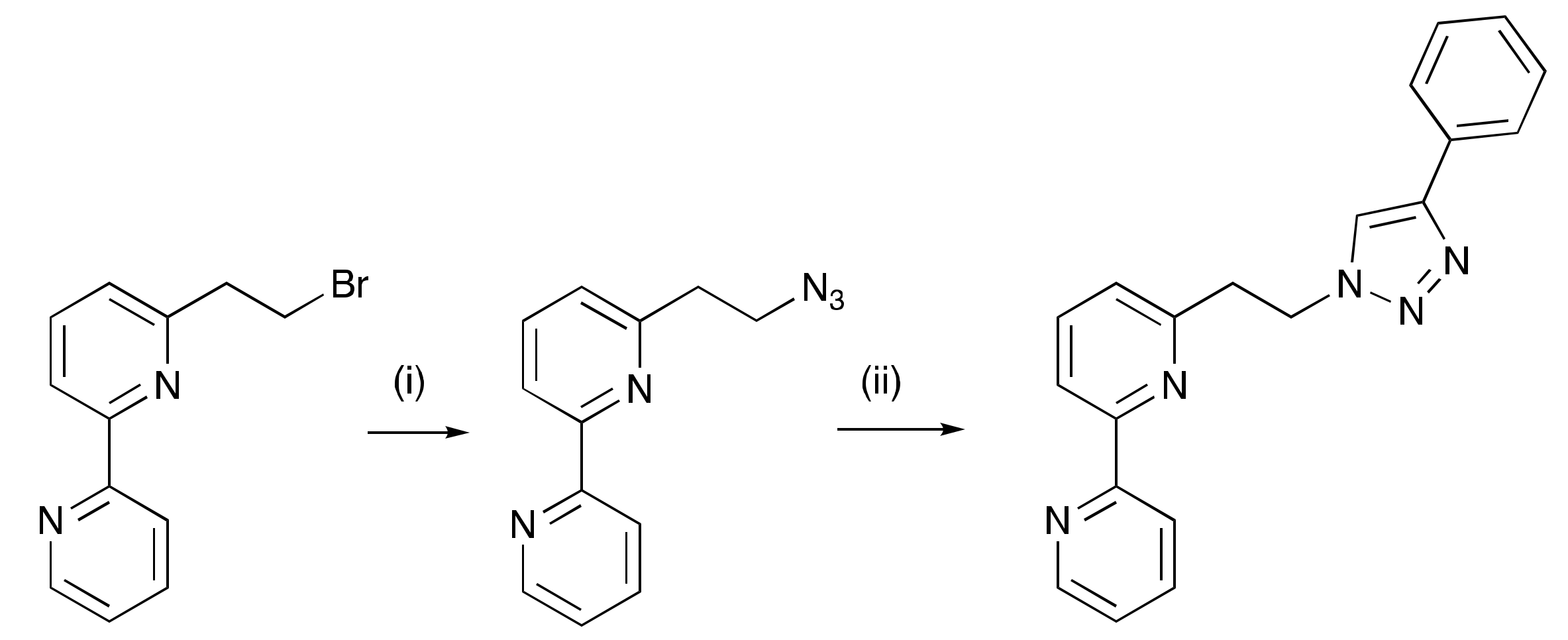
2.1. Ligand Syntheses
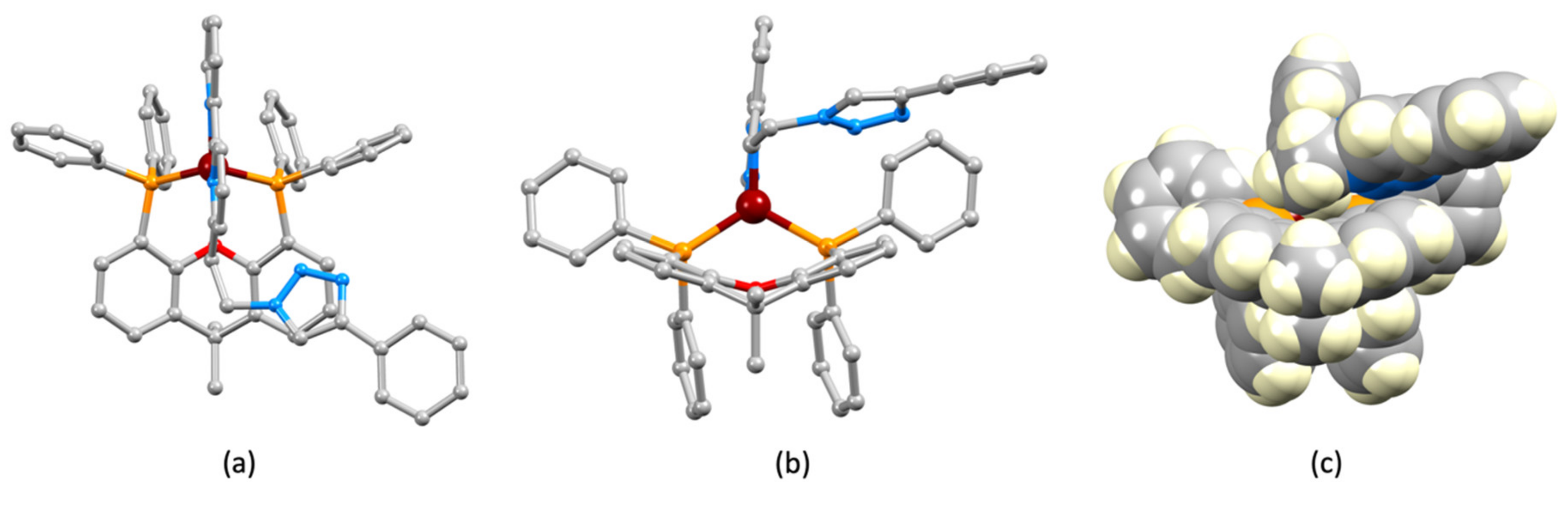
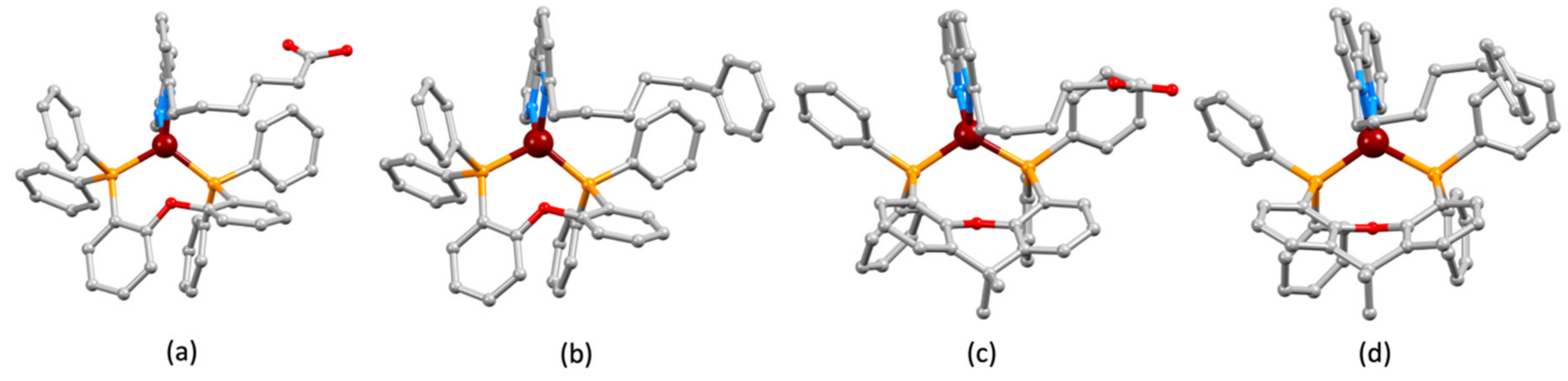
2.2. Synthesis and Characterization of Heteroleptic Copper(I) Complexes
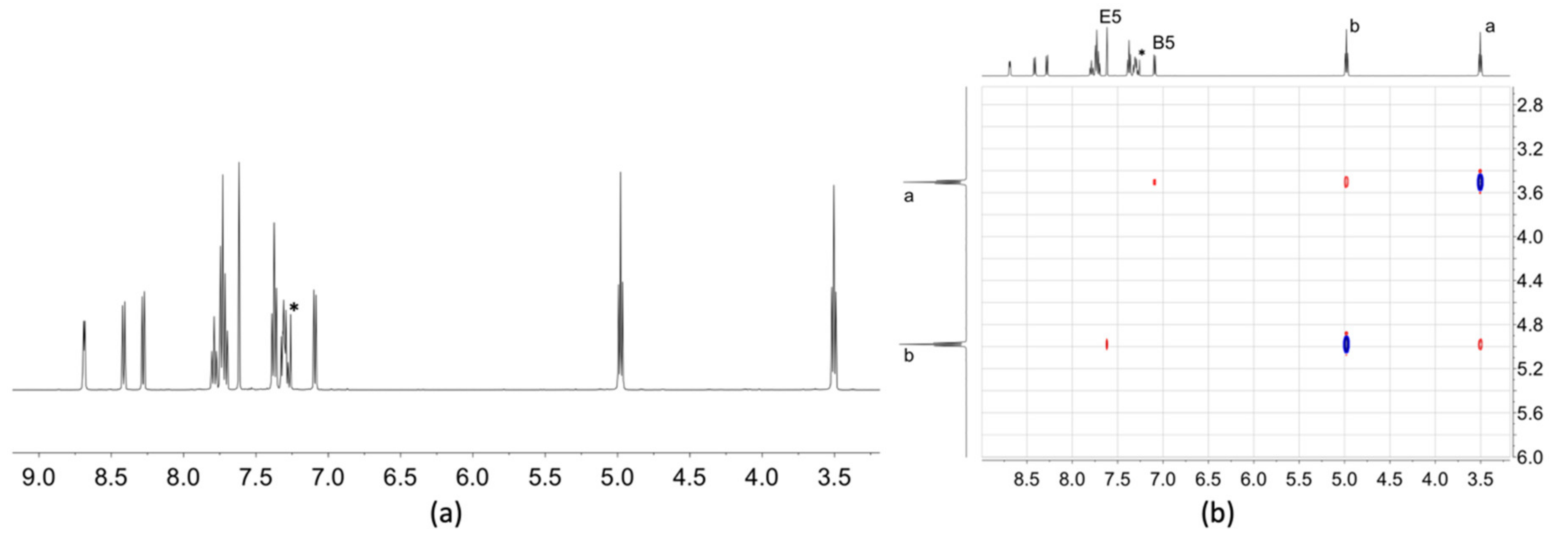
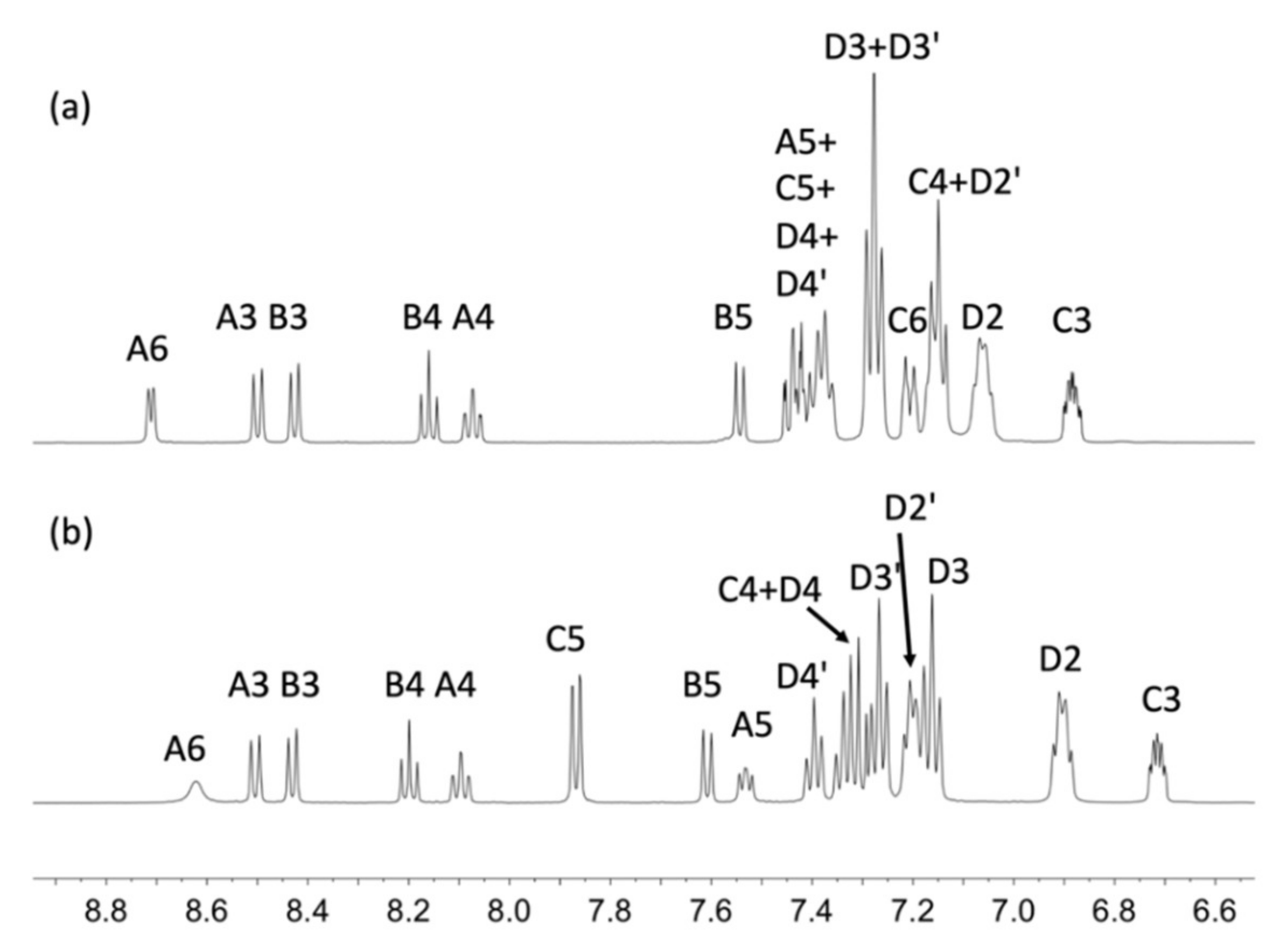
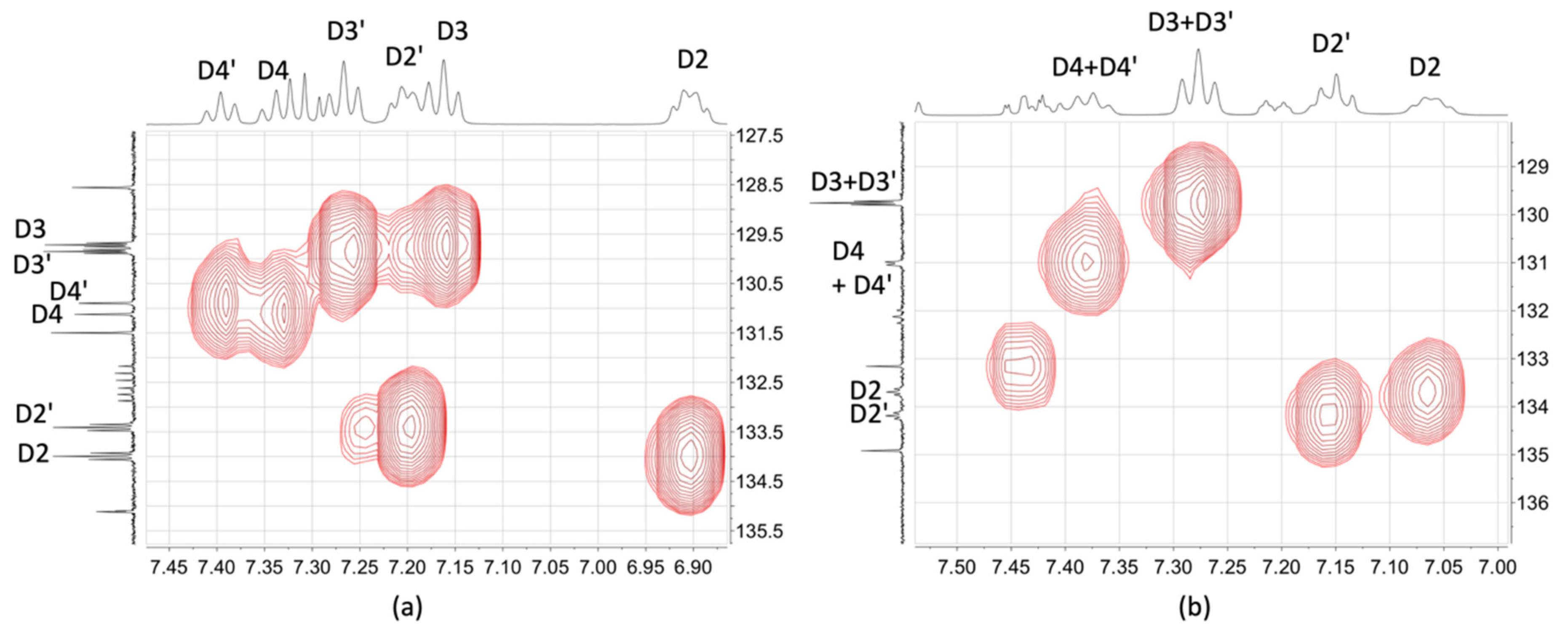
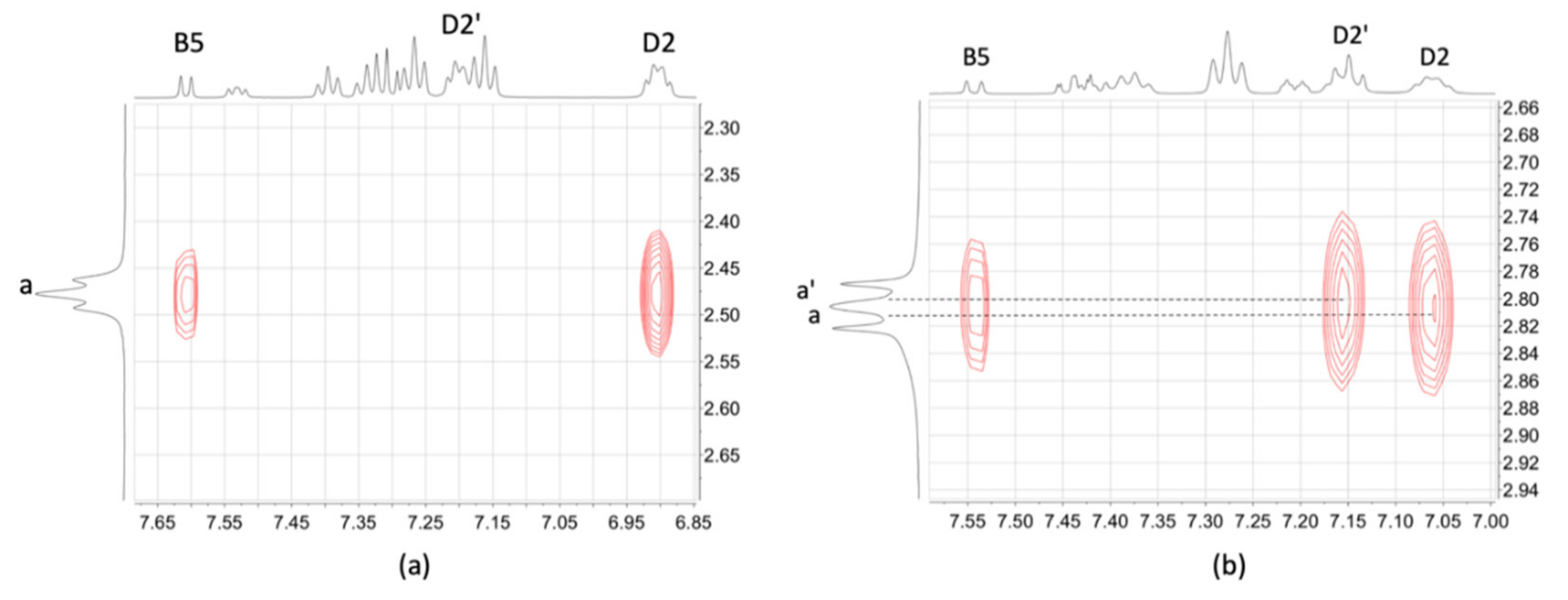
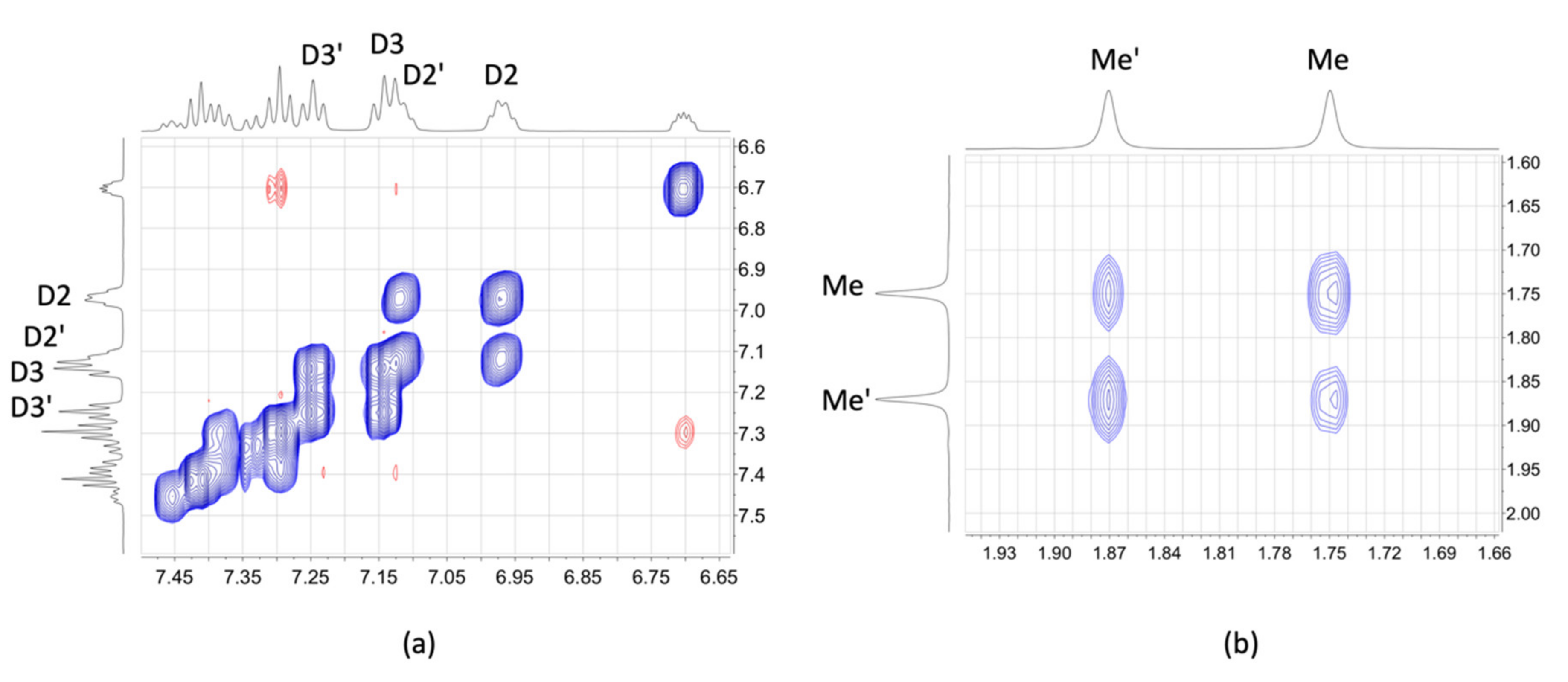
2.3. NMR Spectroscopic Properties and Dynamic Behaviour
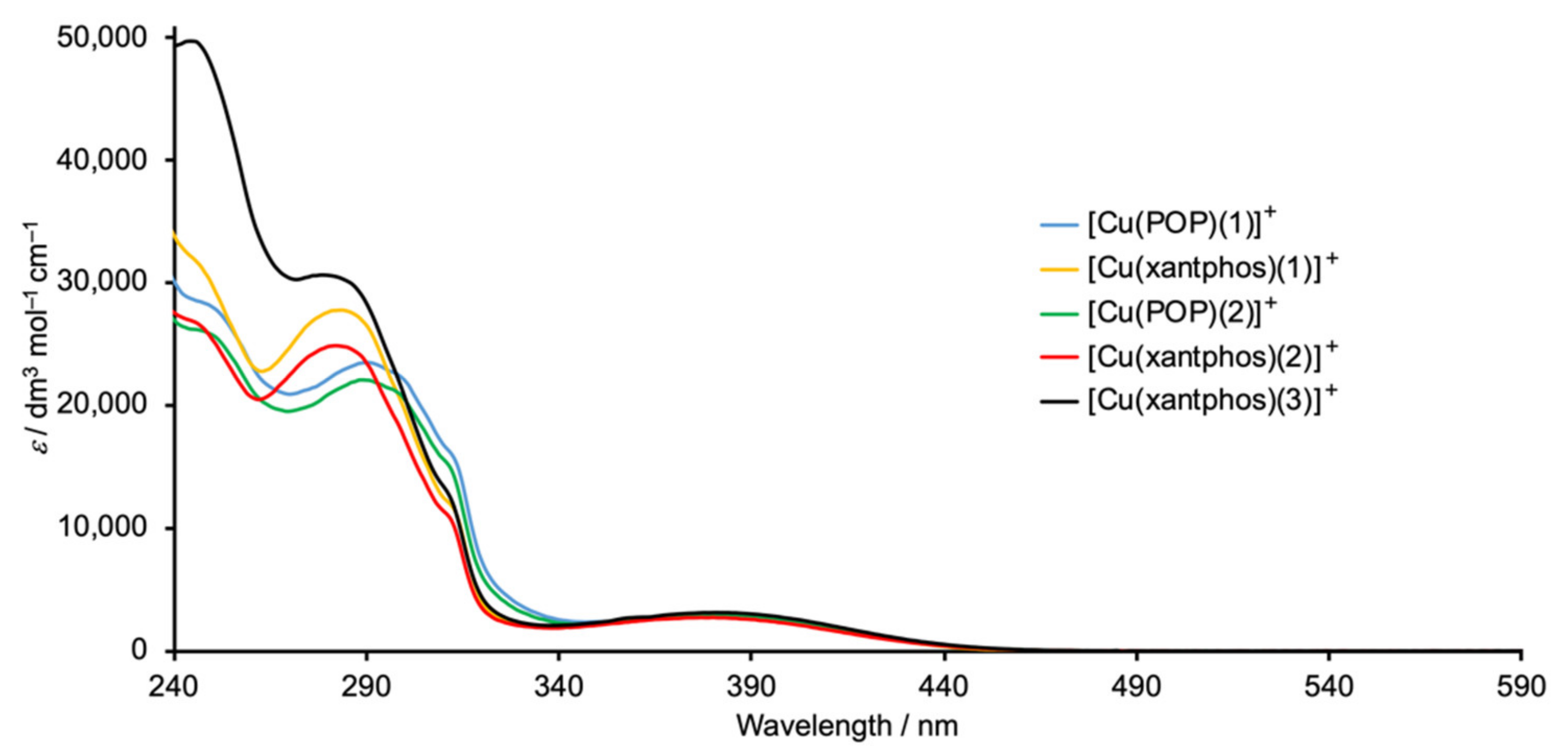
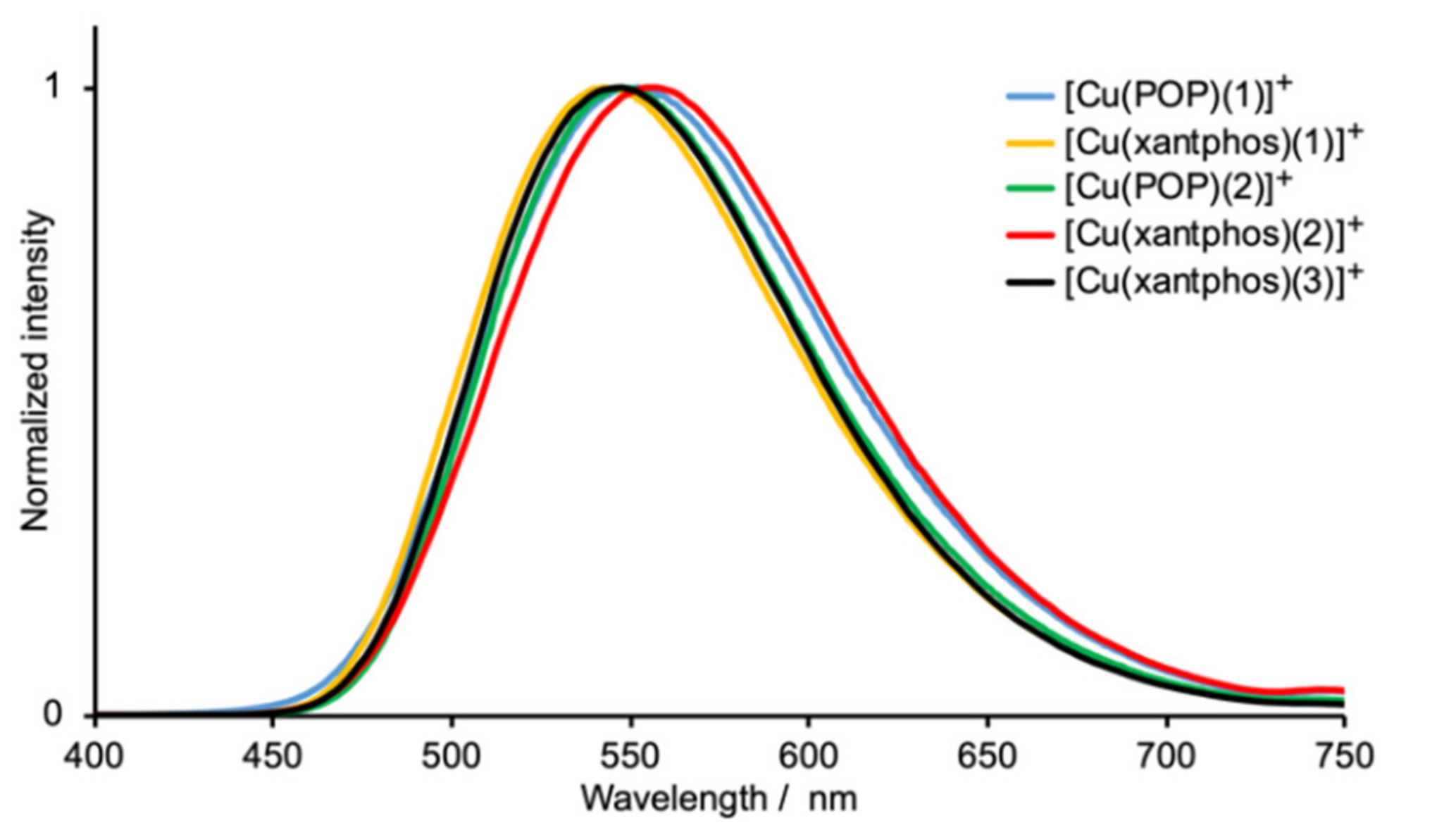
2.4. Electrochemical and Photophysical Properties
3. Materials and Methods
3.1. General
3.2. Compound 2
3.3. Compound 3
3.4. [Cu(POP)(1)][PF6]
3.5. [Cu(xantphos)(1)][PF6]
3.6. [Cu(POP)(2)][PF6]
3.7. [Cu(xantphos)(2)][PF6]
3.8. [Cu(xantphos)(3)][PF6]
3.9. Crystallography
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Data Availability Statement
Conflicts of Interest
Sample Availability
References
- Ravaro, L.P.; Zanoni, K.P.S.; de Camargo, A.S.S. Luminescent copper(I) complexes as promising materials for the next generation of energy-saving OLED devices. Energy Rep. 2020, 6, 37–45. [Google Scholar] [CrossRef]
- Elie, M.; Gaillard, S.; Renaud, J.L. Light-Emitting Electrochemical Cells: Concepts, Advances and Challenges; Costa, R.D., Ed.; Springer International Publishing: New York City, NY, USA, 2017. [Google Scholar]
- Costa, R.D.; Ortí, E.; Bolink, H.J.; Monti, F.; Accorsi, G.; Armaroli, N. Luminescent ionic transition metal complexes for light-emitting electrochemical cells. Angew. Chem. Int. Ed. 2012, 51, 8178–8211. [Google Scholar] [CrossRef] [PubMed]
- Fresta, E.; Costa, R.D. Beyond traditional light-emitting electrochemical cells—A review of new device designs and emitters. J. Mater. Chem. C 2017, 5, 5643–5675. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Cardinali, F.; Listorti, A. Photochemistry and Photophysics of Coordination Compounds: Copper. Top. Curr. Chem. 2007, 280, 69–115. [Google Scholar] [CrossRef]
- Costa, R.D.; Tordera, D.; Ortí, E.; Bolink, H.J.; Schönle, J.; Graber, S.; Housecroft, C.E.; Constable, E.C.; Zampese, J.A. Copper(I) complexes for sustainable light-emitting electrochemical cells. J. Mater. Chem. C 2011, 21, 16108–16118. [Google Scholar] [CrossRef]
- Yersin, H.; Czerwieniec, R.; Shafikov, M.Z.; Suleymanova, A.F. TADF Material Design: Photophysical Background and Case Studies Focusing on CuI and AgI Complexes. Chem. Phys. Chem. 2017, 18, 3508–3535. [Google Scholar] [CrossRef]
- Czerwieniec, R.; Leitl, M.J.; Homeier, H.H.H.; Yersin, H. Cu(I) complexes—Thermally activated delayed fluorescence. Photophysical approach and material design. Coord. Chem. Rev. 2016, 325, 2–28. [Google Scholar] [CrossRef]
- Leitl, M.J.; Krylova, V.A.; Djurovich, P.I.; Thompson, M.E.; Yersin, H. Phosphorescence versus Thermally Activated Delayed Fluorescence. Controlling Singlet–Triplet Splitting in Brightly Emitting and Sublimable Cu(I) Compounds. J. Am. Chem. Soc. 2014, 136, 16032–16038. [Google Scholar] [CrossRef]
- Buckner, M.T.; McMillin, D.R. Photoluminescence from copper(I) complexes with low-lying metal-to-ligand charge transfer excited states. J. Chem. Soc. Chem. Commun. 1978, 759–761. [Google Scholar] [CrossRef]
- Rader, R.A.; McMillin, D.R.; Buckner, M.T.; Matthews, T.G.; Casadonte, D.J.; Lengel, R.K.; Whittaker, S.B.; Darmon, L.M.; Lytle, F.E. Photostudies of 2,2′-bipyridine bis(triphenylphosphine)copper(1+), 1,10-phenanthroline bis(triphenylphosphine)copper(1+), and 2,9-dimethyl-1,10-phenanthroline bis(triphenylphosphine)copper(1+) in solution and in rigid, low-temperature glasses. Simultaneous multiple emissions from intraligand and charge-transfer states. J. Am. Chem. Soc. 1981, 103, 5906–5912. [Google Scholar] [CrossRef]
- Weber, M.D.; Viciano-Chumillas, M.; Armentano, D.; Cano, J.; Costa, R.D. σ-Hammett parameter: A strategy to enhance both photo- and electro-luminescence features of heteroleptic copper(I) complexes. Dalton Trans. 2017, 46, 6312–6323. [Google Scholar] [CrossRef] [PubMed]
- Alkan-Zambada, M.; Constable, E.C.; Housecroft, C.E. The Role of Percent Volume buried in the Characterization of Copper(I) Complexes for Lighting Purposes. Molecules 2020, 25, 2647. [Google Scholar] [CrossRef] [PubMed]
- Keller, S.; Alkan-Zambada, M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Extended π-Systems in Diimine Ligands in [Cu(P^P)(N^N)][PF6] Complexes: From 2,2′-Bipyridine to 2-(Pyridin-2-yl)quinoline. Crystals 2020, 10, 255. [Google Scholar] [CrossRef]
- Leoni, E.; Mohanraj, J.; Holler, M.; Mohankumar, M.; Nierengarten, I.; Monti, F.; Sournia-Saquet, A.; Delavaux-Nicot, B.; Nierengarten, J.-F.; Armaroli, N. Heteroleptic Copper(I) Complexes Prepared from Phenanthroline and Bis-Phosphine Ligands: Rationalization of the Photophysical and Electrochemical Properties. Inorg. Chem. 2018, 57, 15537–15549. [Google Scholar] [CrossRef] [PubMed]
- Mohankumar, M.; Holler, M.; Meichsner, E.; Nierengarten, J.-F.; Niess, F.; Sauvage, J.-P.; Delavaux-Nicot, B.; Leoni, E.; Monti, F.; Malicka, J.M.; et al. Heteroleptic Copper(I) Pseudorotaxanes Incorporating Macrocyclic Phenanthroline Ligands of Different Sizes. J. Am. Chem. Soc. 2018, 140, 2336–2347. [Google Scholar] [CrossRef]
- Keller, S.; Constable, E.C.; Housecroft, C.E.; Neuburger, M.; Prescimone, A.; Longo, G.; Pertegás, A.; Sessolo, M.; Bolink, H.J. [Cu(bpy)(P^P)]+ containing light-emitting electrochemical cells: Improving performance through simple substitution. Dalton Trans. 2014, 43, 16593–16596. [Google Scholar] [CrossRef]
- Keller, S.; Pertegás, A.; Longo, G.; Martínez, L.; Cerdá, J.; Junquera-Hernández, J.M.; Prescimone, A.; Constable, E.C.; Housecroft, C.E.; Ortí, E.; et al. Shine bright or live long: Substituent effects in [Cu(N^N)(P^P)]+-based light-emitting electrochemical cells where N^N is a 6-substituted 2,2′-bipyridine. J. Mater. Chem. C 2016, 4, 3857–3871. [Google Scholar] [CrossRef]
- Alkan-Zambada, M.; Keller, S.; Martínez-Sarti, L.; Prescimone, A.; Junquera-Hernández, J.M.; Constable, E.C.; Bolink, H.J.; Sessolo, M.; Ortí, E.; Housecroft, C.E. [Cu(P^P)(N^N)][PF6] compounds with bis(phosphane) and 6-alkoxy, 6-alkylthio, 6-phenyloxy and 6-phenylthio-substituted 2,2′-bipyridine ligands for light-emitting electrochemical cells. J. Mater. Chem. C 2018, 6, 8460–8471. [Google Scholar] [CrossRef]
- Liu, B.Y.; Ganzel, P.; Traylor, T.G. Synthesis and structural characterization of a novel binuclear copper complex: Bis[μ-O,O’-6(2,2′-bipyridyl)hexalato]dicopper(II) bis(perchlorate). Inorg. Chim. Acta 1997, 254, 407–410. [Google Scholar] [CrossRef]
- Bevilacqua, V.; King, M.; Chaumontet, M.; Nothisen, M.; Gabillet, S.; Buisson, D.; Puente, C.; Wagner, A.; Taran, F. Copper-chelating Azides for Efficient Click Conjugation Reactions in Complex Media. Angew. Chem. Int. Ed. 2014, 53, 5872–5876. [Google Scholar] [CrossRef]
- Spartan’18; v. 1.4.4; Wavefunction, Inc.: Irvine, CA, USA, 2019.
- Keller, S.; Brunner, F.; Junquera-Hernández, J.M.; Pertegás, A.; La-Placa, M.-G.; Prescimone, A.; Constable, E.C.; Bolink, H.J.; Ortí, E.; Housecroft, C.E. CF3 Substitution of [Cu(P^P)(bpy)][PF6] complexes: Effects on Photophysical Properties and Light-emitting Electrochemical Cell Performance. ChemPlusChem 2018, 83, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Rieke, R.D. 2-Pyridyl and 3-pyridylzinc bromides: Direct preparation and coupling reaction. Tetrahedron 2010, 66, 3135–3146. [Google Scholar] [CrossRef]
- Ziessel, R.; Toupet, L.; Chardon-Noblat, S.; Deronzier, A.; Matt, D. Co-ordinative properties of a hybrid phosphine–bipyridine ligand. J. Chem. Soc. Dalton Trans. 1997, 3777–3784. [Google Scholar] [CrossRef]
- Kubas, G.J.; Monzyk, B.; Crumbliss, A.L. Tetrakis(acetonitrile)copper(I) hexafluorophosphate. Inorg. Synth. 1979, 19, 90–92. [Google Scholar] [CrossRef]
- APEX2, version 2, User Manual, M86-E01078; Bruker Analytical X-ray Systems, Inc.: Madison, WI, USA, 2016.
- Betteridge, P.W.; Carruthers, J.R.; Cooper, R.I.; Prout, K.; Watkin, D.J. CRYSTALS version 12: Software for guided crystal structure analysis. J. Appl. Cryst. 2003, 36, 1487. [Google Scholar] [CrossRef]
- Macrae, C.F.; Sovago, I.; Cottrell, S.J.; Galek, P.T.A.; McCabe, P.; Pidcock, E.; Platings, M.; Shields, G.P.; Stevens, J.S.; Towler, M.; et al. Mercury 4.0: From visualization to analysis, design and prediction. J. Appl. Cryst. 2020, 53, 226–235. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Crystallogr. 2015, 71, 9–18. [Google Scholar] [CrossRef]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Bond | Bond Length/Å | Angle | Bond Angle/o |
|---|---|---|---|
| Cu1–P1 | 2.2673(7) | P1–Cu1–P2 | 114.33(3) |
| Cu1–P2 | 2.2591(7) | P1–Cu1–N1 | 113.50(7) |
| Cu1–N1 | 2.063(2) | P2–Cu1–N1 | 111.67(7) |
| Cu1–N2 | 2.066(2) | P1–Cu1–N2 | 113.80(7) |
| O1–C20 | 1.384(3) | P2–Cu1–N2 | 118.78(7) |
| O1–C22 | 1.388(3) | N1–Cu1–N2 | 80.18(10) |
| N3–N4 | 1.332(5) | C20–O1–C22 | 114.96(19) |
| N4–N5 | 1.316(5) | N3–N4–N5 | 108.8(3) |
| Compound | Epca/V | E1/2/V | Epc − Epa/mV |
|---|---|---|---|
| [Cu(POP)(1)][PF6] | +0.85 | −2.07 | 80 |
| [Cu(xantphos)(1)][PF6] | +0.93 | −2.06 | 90 |
| [Cu(POP)(2)][PF6] | +0.92 | −2.10 | 120 |
| [Cu(xantphos)(2)][PF6] | +0.94 | −2.13 | 130 |
| [Cu(xantphos)(3)][PF6] | +0.81 | −2.02 | 90 |
| Compound | λmax/nm (εmax/dm3 mol−1 cm−1) | |
|---|---|---|
| π*←π | MLCT | |
| [Cu(POP)(1)][PF6] | 247 sh (28,500), 292 (23,400), 299 sh (22,300), 312 sh (14,800) | 381 (2900) |
| [Cu(xantphos)(1)][PF6] | 247 sh (31,400), 285 (27,700), 315 sh (11,500) | 383 (2900) |
| [Cu(POP)(2)][PF6] | 250 sh (25,600), 292 (21,900), 300 sh (20,500), 312 sh (14,800) | 384 (2850) |
| [Cu(xantphos)(2)][PF6] | 248 sh (26,200), 285 (24,800), 312 sh (10,700) | 382 (2700) |
| [Cu(xantphos)(3)][PF6] | 247 (49,400), 282 (30,600), 312 sh (12,400) | 383 (3100) |
| Compound | λexc /nm | λmaxem/ nm | PLQY/% | τ/μs a | τ(1)/μs (A1) | τ(2)/μs (A2) |
|---|---|---|---|---|---|---|
| [Cu(POP)(1)][PF6] | 365 | 552 | 17 | 5.4 | 5.94 (0.82) | 2.30 (0.14) |
| [Cu(xantphos)(1)][PF6] | 365 | 542 | 28 | 7.2 | 8.29 (0.77) | 2.16 (0.88) |
| [Cu(POP)(2)][PF6] | 365 | 555 | 13 | 5.1 | 5.47 (0.85) | 1.88 (0.09) |
| [Cu(xantphos)(2)][PF6] | 365 | 546 | 26 | 8.7 | 9.64 (0.83) | 2.26 (0.12) |
| [Cu(xantphos)(3)][PF6] | 365 | 548 | 25 | 7.1 | 8.64 (0.71) | 2.24 (0.22) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Meyer, M.; Brunner, F.; Prescimone, A.; Constable, E.C.; Housecroft, C.E. Desymmetrizing Heteroleptic [Cu(P^P)(N^N)][PF6] Compounds: Effects on Structural and Photophysical Properties, and Solution Dynamic Behavior. Molecules 2021, 26, 125. https://doi.org/10.3390/molecules26010125
Meyer M, Brunner F, Prescimone A, Constable EC, Housecroft CE. Desymmetrizing Heteroleptic [Cu(P^P)(N^N)][PF6] Compounds: Effects on Structural and Photophysical Properties, and Solution Dynamic Behavior. Molecules. 2021; 26(1):125. https://doi.org/10.3390/molecules26010125
Chicago/Turabian StyleMeyer, Marco, Fabian Brunner, Alessandro Prescimone, Edwin C. Constable, and Catherine E. Housecroft. 2021. "Desymmetrizing Heteroleptic [Cu(P^P)(N^N)][PF6] Compounds: Effects on Structural and Photophysical Properties, and Solution Dynamic Behavior" Molecules 26, no. 1: 125. https://doi.org/10.3390/molecules26010125
APA StyleMeyer, M., Brunner, F., Prescimone, A., Constable, E. C., & Housecroft, C. E. (2021). Desymmetrizing Heteroleptic [Cu(P^P)(N^N)][PF6] Compounds: Effects on Structural and Photophysical Properties, and Solution Dynamic Behavior. Molecules, 26(1), 125. https://doi.org/10.3390/molecules26010125