
Synthesis of Galectin Inhibitors by Regioselective 3′-O-Sulfation of Vanillin Lactosides Obtained under Phase Transfer Catalysis




Abstract
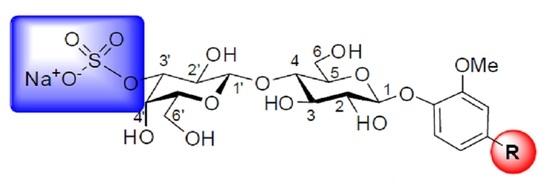
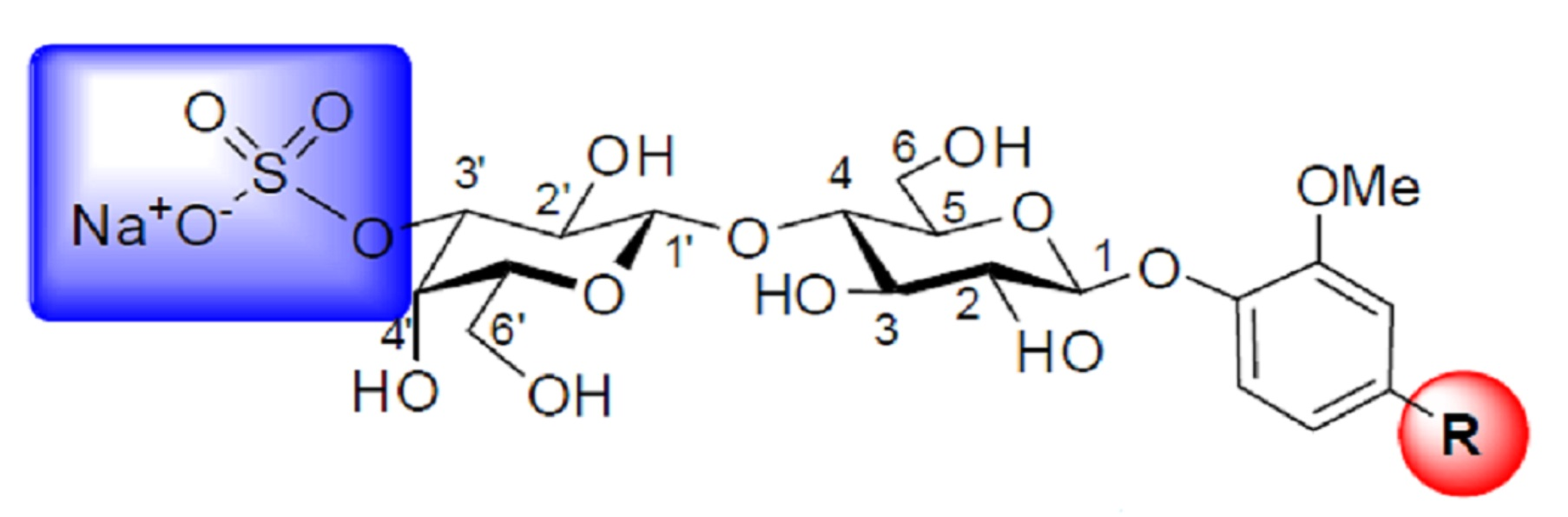{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
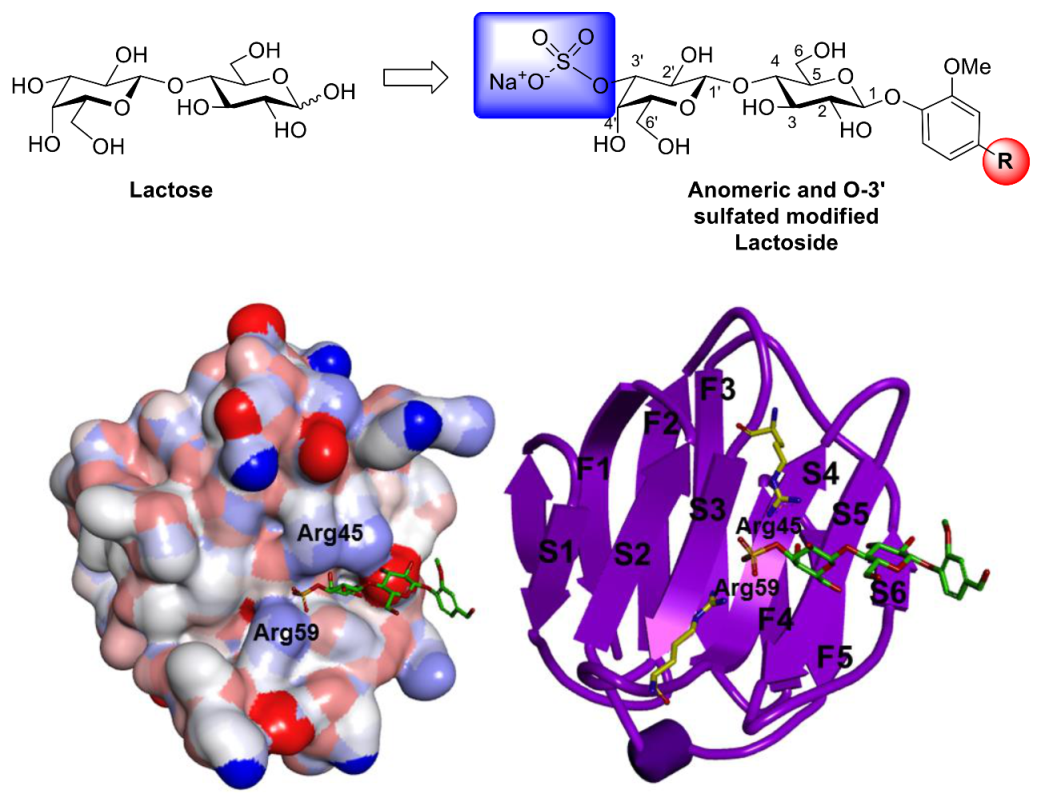
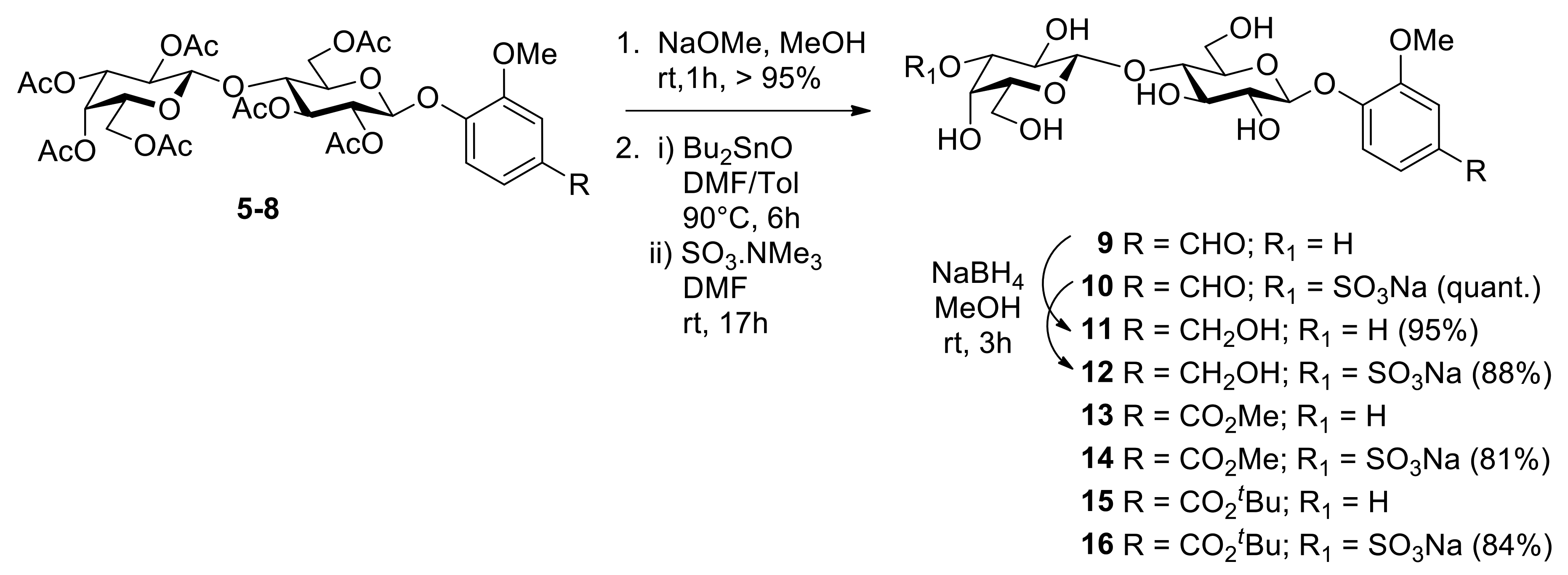
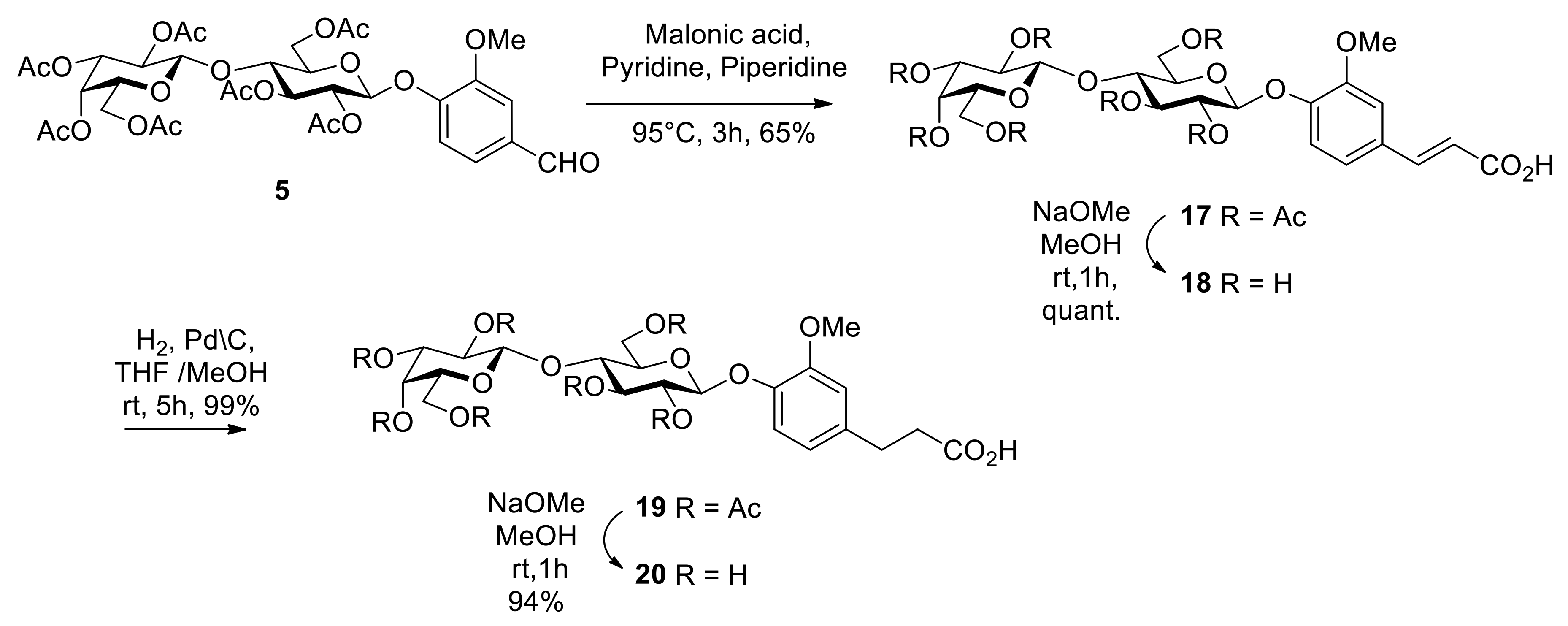
2. Results and Discussion
3. Materials and Methods
3.1. General Synthetic Methods
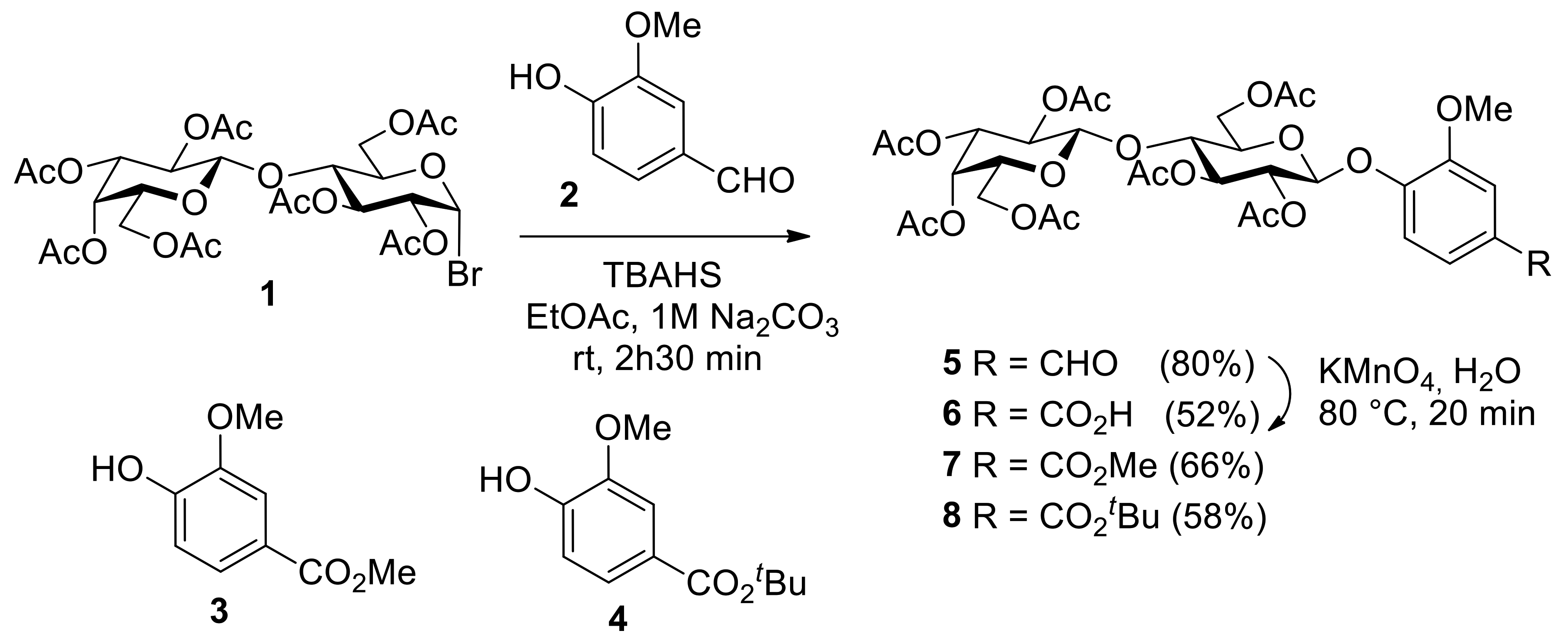
3.2. General Synthetic Procedure A: Phase-Transfer Catalysis (PTC) Reaction
3.3. General Synthetic Procedure B: Zemplén Transesterification Reaction
3.4. General Synthetic Procedure C: Preparation of 3′-O-sulfated Lactosides
3.5. 2,3,4,6-Tetra-O-acetyl-β-d-galactopyranosyl-(1-4)-2,3,6-tri-O-acetyl-α-d-glucopyranosyl bromide (1) (Acetobromolactose)
3.6. 3-Methoxy-4-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-D-lactopyranosyloxy)benzaldehyde (5) (4-formyl-2-methoxyphenyl 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-D-lactopyranoside)
3.7. 3-Methoxy-4-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-d-lactopyranosyloxy)benzoic Acid (6)
3.8. Methyl 3-methoxy-4-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-d-lactopyranosyloxy)benzoate (7)
3.9. Tert-butyl 3-methoxy-4-(2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-d-lactopyranosyloxy)benzoate (8)
3.10. 3-Methoxy-4-(β-d-lactopyranosyloxy)benzaldehyde (9), (4-Formyl-2-methoxyphenyl β-d-lactopyranoside)
3.11. Methyl-3-methoxy-4-(3′-O-sulfo-β-d-lactopyranosyloxy)benzaldehyde, Sodium Salt (10)
3.12. 3-Methoxy-4-(β-d-lactopyranosyloxy)benzylic alcohol (11) (4-hydroxymethyl-2-methoxyphenyl β-d-lactopyranoside)
3.13. Methyl-3-methoxy-4-(3′-O-sulfo-β-d-lactopyranosyloxy)benzylic Alcohol, Sodium Salt (12)
3.14. Methyl 3-methoxy-4-(β-d-lactopyranosyloxy)benzoate (13)
3.15. Methyl 3-methoxy-4-(3′-O-sulfo-β-d-lactopyranosyloxy)benzoate, Sodium Salt (14)
3.16. Tert-butyl 3-methoxy-4-(β-d-lactopyranosyloxy)benzoate (15)
3.17. Tert-butyl 3-methoxy-4-(3′-O-sulfo-β-d-lactopyranosyloxy)benzoate, Sodium Salt (16)
3.18. 4-E-[(2-Carboxy)ethenyl]-2-methoxyphenyl 2,3,6,2′,3′,4′,6′-hepta-O-acetyl-β-d-lactopyranoside (17)
3.19. 4-E-[(2-Carboxy)ethenyl]-2-methoxyphenyl β-d-lactopyranoside (18)
3.20. 4-(2-Carboxyethyl)-2-methoxyphenyl 2, 3, 6, 2′,3′,4′,6′-hepta-O-acetyl-β-d-lactopyranoside (19)
3.21. 4-(2-Carboxyethyl)-2-methoxyphenyl β-d-lactopyranoside (20)
3.22. Docking Manipulation for Providing Figure 1
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Sample Availability
References
- Solís, D.; Bovin, N.V.; Davis, A.P.; Jiménez-Barbero, J.; Romero, A.; Roy, R.; Smetana Jr, K.; Gabius, H.-J. A guide into glycosciences: How chemistry and biochemistry cooperate to crack the sugar code. Biochim. Biophys. Acta 2015, 1850, 186–235. [Google Scholar]
- Johannes, L.; Jacob, R.; Leffler, H. Galectins at a glance. J. Cell Sci. 2018, 131, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Girotti, M.R.; Salatino, M.; Dalotto-Moreno, T.; Rabinovich, G.A. Sweetening the hallmarks of cancer: Galectins as multifunctional mediators of tumor progression. J. Exp. Med. 2020, 217, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Murphy, P.V.; Gabius, H.-J. Multivalent Carbohydrate-Lectin Interactions: How Synthetic Chemistry Enables Insights into Nanometric Recognition. Molecules 2016, 21, 629. [Google Scholar] [CrossRef] [PubMed]
- Hockl, P.F.; Wolosiuk, A.; Pérez-Sáeza, J.M.; Bordoni, A.V.; Crocia, D.O.; Toum-Terrones, Y.; Soler-Illia, G.J.A.A.; Rabinovich, G.A. Glyco-nano-oncology: Novel therapeutic opportunities by combining small and sweet. Pharmacol. Res. 2016, 109, 45–54. [Google Scholar] [CrossRef]
- Öberga, C.T.; Leffler, H.; Nilsson, U.J. Inhibition of Galectins with Small Molecules. Chimia 2011, 65, 18–23. [Google Scholar] [CrossRef]
- Pieters, R.J. Inhibition and Detection of Galectins. ChemBioChem 2006, 7, 721–728. [Google Scholar] [CrossRef]
- Bertuzzi, S.; Quintana, J.I.; Ardá, A.; Gimeno, A.; Jiménez-Barbero, J. Targeting galectins with glycomimetics. Front. Chem. 2020, 8, 593. [Google Scholar] [CrossRef]
- Rauthu, S.R.; Shiao, T.C.; André, S.; Miller, M.C.; Madej, É.; Mayo, K.; Gabius, H.-J.; Roy, R. Defining the potential of aglycone modifications for affinity/selectivity enhancement against medically relevant lectins: Synthesis, activity screening, and HSQC-based NMR analysis. ChemBiochem 2015, 16, 126–139. [Google Scholar] [CrossRef]
- Zetterberg, F.R.; Peterson, K.; Johnsson, R.E.; Brimert, T.; Håkansson, M.; Logan, D.T.; Leffler, H.; Nilsson, U.J. Monosaccharide Derivatives with Low-Nanomolar Lectin Affinity and High Selectivity Based on Combined Fluorine–Amide, Phenyl–Arginine, Sulfur–p, and Halogen Bond Interactions. ChemMedChem 2018, 13, 133–137. [Google Scholar] [CrossRef]
- Peterson, K.; Collins, P.M.; Huang, X.; Kahl-Knutsson, B.; Essén, S.; Zetterberg, F.R.; Oredsson, S.; Leffler, H.; Blanchard, H.; Nilsson, U.J. Aromatic heterocycle galectin-1 interactions for selective single-digit nM affinity ligands. RSC Adv. 2018, 8, 24913–24922. [Google Scholar] [CrossRef]
- Vuong, L.; Kouverianou, E.; Rooney, C.M.; McHugh, B.J.; Howie, S.E.M.; Gregory, C.D.; Forbes, S.J.; Henderson, N.C.; Zetterberg, F.R.; Nilsson, U.J.; et al. An Orally Active Galectin-3 Antagonist Inhibits Lung Adenocarcinoma Growth and Augments Response to PD-L1 Blockade. Cancer Res. 2019, 79, 1480–1492. [Google Scholar] [CrossRef] [PubMed]
- Bratteby, K.; Torkelsson, E.; TampioL’Estrade, E.; Peterson, K.; Shalgunov, V.; Xiong, M.; Leffler, H.; Zetterberg, F.R.; Olsson, T.G.; Gillings, N.; et al. In Vivo Veritas: 18F-Radiolabeled Glycomimetics Allow Insights into the Pharmacological Fate of Galectin-3 Inhibitors. J. Med. Chem. 2020, 63, 747–755. [Google Scholar] [CrossRef] [PubMed]
- Ahmed, H.; Allen, H.J.; Sharma, A.; Matta, K.L. Human Splenic Galaptin: Carbohydrate-Binding Specificity and Characterization of the Combining Site? Biochemistry 1990, 29, 5315–5319. [Google Scholar] [CrossRef]
- Roy, R.; Cao, Y.; Kaltner, H.; Kottari, N.; Shiao, T.C.; Belkhadem, K.; André, S.; Manning, J.C.; Murphy, P.V.; Gabius, H.-J. Teaming up synthetic chemistry and histochemistry for activity screening in galectin-directed inhibitor design. Histochem. Cell Biol. 2017, 147, 285–301. [Google Scholar] [CrossRef]
- Sirois, S.; Giguère, D.; Roy, R. A first QSAR model for galectin-3 glycomimetic inhibitors based on 3D docked structures. Med. Chem. 2006, 2, 481–489. [Google Scholar] [CrossRef]
- Giguère, D.; Bonin, M.-A.; Cloutier, P.; Patnam, R.; St-Pierre, C.; Sato, S.; Roy, R. Syntheis of Stable and Selective Inhibitors of Human Galectin-1 and -3. Bioorg. Med. Chem. 2008, 16, 7811–7823. [Google Scholar]
- Giguère, D.; André, S.; Bonin, M.-A.; Bellefleur, M.A.; Provencal, A.; Cloutier, P.; Roy, R.; Gabius, H.-J. Inhibitory potential of chemical substitutions at bioinspired sites of β-D-galactopyranoside on glycoprotein/cell surface binding of two classes of medically relevant lectins. Bioorg. Med. Chem. 2011, 19, 3280–3287. [Google Scholar]
- Giguère, D.; Patnam, R.; Bellefleur, M.A.; St-Pierre, C.; Sato, S.; Roy, R. Carbohydrate triazoles and isoxazoles as inhibitors of galectins-1 and -3. Chem. Commun. 2006, 2379–2381. [Google Scholar] [CrossRef]
- André, S.; Giguère, D.; Dam, T.K.; Brewer, C.F.; Gabius, H.-J.; Roy, R. O/S-Glycosides with aglyconic extensions at the anomeric position: Synthesis and novel HTS screening for inhibitory activity on medically relevant galactoside-specific lectins in assays of increasing biorelevance. New J. Chem. 2010, 34, 2229–2240. [Google Scholar] [CrossRef]
- Ruiz, F.M.; Gilles, U.; Ludwig, A.-K.; Sehad, C.; Shiao, T.C.; Caballero, G.G.; Kaltner, H.; Lindner, I.; Roy, R.; Reusch, D.; et al. Chicken GRIFIN: Structural characterization in crystals and in solution. Biochimie 2018, 146, 127–138. [Google Scholar] [CrossRef] [PubMed]
- Dahlqvist, A.; Leffler, H.; Nilsson, U.J. C1-Galactopyranosyl Heterocycle Structure Guides Selectivity: Triazoles Prefer Galectin-1 and Oxazoles Prefer Galectin-3. ACS Omega 2019, 4, 7047–7053. [Google Scholar] [CrossRef]
- Giguère, D.; Sato, S.; St-Pierre, C.; Sirois, S.; Roy, R. Aryl O- and S-galactosides and lactosides as specific inhibitors of human galectins-1 and -3: Role of electrostatic potential at O-3. Bioorg. Med. Chem. Lett. 2006, 16, 1668–1672. [Google Scholar] [CrossRef] [PubMed]
- Allen, H.J.; Ahmed, H.; Matta, L. Binding of synthetic sulfated ligands by human splenic galectin 1, a β-galactoside-binding lectin. Glycoconjugate J. 1998, 15, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Ideo, H.; Seko, A.; Yamashita, K. Galectin-4 Binds to Sulfated Glycosphingolipids and Carcinoembryonic Antigen in Patches on the Cell Surface of Human Colon Adenocarcinoma Cells. J. Biol. Chem. 2005, 280, 4730–4737. [Google Scholar] [CrossRef] [PubMed]
- Bum-Erdene, K.; Leffler, H.; Nilsson, U.J.; Blanchard, H. Structural characterisation of human galectin-4 N-terminal carbohydrate recognition domain in complex with glycerol, lactose, 3′-sulfo-lactose, and 2′-fucosyllactose. Sci. Rep. 2016, 6, 20289. [Google Scholar] [CrossRef] [PubMed]
- Ideo, H.; Matsuzaka, T.; Nonaka, T.; Seko, A.; Yamashita, K. Galectin-8-N-domain Recognition Mechanism for Sialylated and Sulfated Glycans. J. Biol. Chem. 2011, 286, 11346–11355. [Google Scholar] [CrossRef]
- André, S.; Cejas Ortega, P.J.; Alamino Perez, M.; Roy, R.; Gabius, H.-J. Lactose-containing Starburst Dendrimers: Influence of Dendrimer Generation and Binding-Site Orientation of Receptors (Plant/animal Lectins and Immunoglobulins) on Binding Properties. Glycobiol. 1999, 9, 1253–1261. [Google Scholar] [CrossRef]
- Ahmad, N.; Gabius, H.-J.; André, S.; Kaltner, H.; Sabesan, S.; Roy, R.; Liu, B.; Macaluso, F.; Brewer, C.F. Galectin-3 precipitates as a pentamer with synthetic multivalent carbohydrates and forms heterogeneous cross-linked complexes. J. Biol. Chem. 2004, 279, 10841–10847. [Google Scholar] [CrossRef]
- Abbassi, L.; Chabre, Y.M.; Kottari, N.; Arnold, A.A.; André, S.; Josserand, J.; Gabius, H.-J.; Roy, R. Multifaceted Glycodendrimers With Programmable Bioactivity Through Convergent, Divergent, and Accelerated Approaches Using Polyfunctional Cyclotriphosphazenes. Polym.Chem. 2015, 6, 7666–7683. [Google Scholar] [CrossRef]
- Chabre, Y.M.; Roy, R. Design and Creativity in Multivalent Neoglycoconjugate Synthesis. Adv. Carbohydr. Chem. Biochem. 2010, 63, 165–393. [Google Scholar] [PubMed]
- André, S.; Pieters, R.J.; Vrasidas, I.; Kaltner, H.; Kuwabara, I.; Liu, F.-T.; Liskamp, R.M.J.; Gabius, H.-J. Wedgelike Glycodendrimers as Inhibitors of Binding of Mammalian Galectins to Glycoproteins, Lactose Maxiclusters, and Cell Surface Glycoconjugates. ChemBioChem 2001, 2, 822–830. [Google Scholar] [CrossRef]
- Ennist, J.H.; Termuehlen, H.R.; Bernhard, S.P.; Fricke, M.S.; Cloninger, M.J. Chemoenzymatic Synthesis of Galectin Binding Glycopolymers. Bioconjugate Chem. 2018, 29, 4030–4039. [Google Scholar] [CrossRef] [PubMed]
- Cousin, J.M.; Cloninger, M.J. Glycodendrimers: Tools to explore multivalent galectin-1 interactions. Beilstein J. Org. Chem. 2015, 11, 739–747. [Google Scholar] [CrossRef]
- Tavares, M.R.; Bláhová, M.; Sedláková, L.; Elling, L.; Pelantová, H.; Konefał, R.; Etrych, T.; Křen, V.; Bojarová, P.; Chytil, P. High-Affinity N-(2-Hydroxypropyl)methacrylamide Copolymers with Tailored N-Acetyllactosamine Presentation Discriminate between Galectins. Biomacromolecules 2020, 21, 641–652. [Google Scholar] [CrossRef]
- Filipová, M.; Bojarová, P.; Tavares, M.R.; Bumba, L.; Elling, L.; Chytil, P.; Gunár, K.; Křen, V.; Etrych, T.; Janoušková, O. Glycopolymers for Efficient Inhibition of Galectin-3: In Vitro Proof of Efficacy Using Suppression of T Lymphocyte Apoptosis and Tumor Cell Migration. Biomacromolecules 2020, 21, 3122–3133. [Google Scholar]
- Percec, V.; Leowanawat, P.; Sun, H.-J.; Kulikov, O.; Nusbaum, C.D.; Tran, T.M.; Bertin, A.; Wilson, D.A.; Peterca, M.; Zhang, S.; et al. Modular Synthesis of Amphiphilic Janus Glycodendrimers and their Self-Assembly into Glycodendrimersomes and Other Complex Architectures with Bioactivity to Biomedically Relevant Lectins. J. Am. Chem. Soc. 2013, 135, 9055–9077. [Google Scholar] [CrossRef]
- Zhang, S.; Moussodia, R.-O.; Sun, H.-J.; Leowanawat, P.; Muncan, A.; Nusbaum, C.D.; Chelling, K.M.; Heiney, P.A.; Klein, M.L.; André, S.; et al. Mimics of Biological Membranes with Programmable Glycan Ligands Self-Assembled from Amphiphilic Janus Glycodendrimers. Angew. Chem Int. Ed. 2014, 53, 10899–10903. [Google Scholar] [CrossRef]
- Zhang, S.; Moussodia, R.-O.; Murzeau, C.; Sun, H.-J.; Klein, M.L.; Vértesy, S.; André, S.; Roy, R.; Gabius, H.-J.; Percec, V. Dissecting molecular aspects of cell interactions using glycodendrimersomes with programmable glycan presentation and engineered human lectins. Angew. Chem. Int. Ed. 2015, 54, 4036–4040. [Google Scholar] [CrossRef]
- Freichel, T.; Laaf, D.; Hoffmann, M.; Konietzny, P.B.; Heine, V.; Wawrzinek, R.; Rademacher, C.; Snyder, N.L.; Elling, L.; Hartmann, L. Effects of linker and liposome anchoring on lactose-functionalized glycomacromolecules as multivalent ligands for binding galectin-3. RSC Adv. 2019, 9, 23484–23497. [Google Scholar] [CrossRef]
- Zhang, H.; Laaf, D.; Elling, L.; Pieters, R.J. Thiodigalactoside−Bovine Serum Albumin Conjugates as High-Potency Inhibitors of Galectin-3: An Outstanding Example of Multivalent Presentation of Small Molecule Inhibitors. Bioconjugate Chem. 2018, 29, 1266–1275. [Google Scholar] [CrossRef] [PubMed]
- Deseo, M.A.; Elkins, A.; Rochfort, S.; Kitchen, B. Antioxidant activity and polyphenol composition of sugarcane molasses extract. Food Chem. 2020, 314, 126180. [Google Scholar] [CrossRef] [PubMed]
- Roy, R. Phase transfer catalysis in carbohydrate chemistry. In Phase Transfer Catalysis; Sasson, Y., Neumann, R., Eds.; Chapman and Hall: Heidelberg, Germany, 1997; pp. 244–275. [Google Scholar]
- Roy, R.; Tropper, F.D.; Cao, S.; Kim, J.M. Anomeric Group Transformations Under PTC. ACS Symp. Ser. 1997, 659, 163–180. [Google Scholar]
- Wang, L.-X.; Pavlova, N.V.; Yang, M.; Li, S.-C.; Li, Y.-T.; Lee, Y.C. Synthesis of aryl 3’-sulfo-β-lactosides as fluorogenic and chromogenic substrates for ceramide glycanases. Carbohydr. Res. 1998, 306, 341–348. [Google Scholar] [CrossRef]
- Grindley, T.B. Applications of tin-containing intermediates to carbohydrate chemistry. Adv. Carbohydr. Chem. Biochem. 1998, 53, 17–142. [Google Scholar] [PubMed]
- David, S.; Hanessian, S. Regioselective manipulation of hydroxyl groups via organotin derivatives. Tetrahedron 1985, 41, 643–663. [Google Scholar] [CrossRef]
- de Andrade, S.F.; Figueiredo, R.C.; de Souza Filho, J.D.; Alves, R.J. An Efficient Synthesis of D-Galactose-Based Multivalent Neoglycoconjugates. J. Braz. Chem. Soc. 2012, 6, 1062–1069. [Google Scholar] [CrossRef]
- Shiao, T.C.; Giguère, D.; Galanos, N.; Roy, R. Efficient Synthesis of Hepta-O-acetyl-β-lactosyl azide via Phase Transfer Catalysis. In Carbohydrate Chemistry: Proven Synthetic Methods; Kovac, P., Ed.; CRC Press: Boca Raton, FL, USA; Taylor & Francis Group: Abingdon, UK, 2014; Volume 2, pp. 253–256. [Google Scholar]
- Mousavifar, L.; Touaibia, M.; Roy, R. Development of Mannopyranoside Therapeutics Against Adherent-invasive E. coli Infections. Acc. Chem. Res. 2018, 51, 2937–2948. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belkhadem, K.; Cao, Y.; Roy, R. Synthesis of Galectin Inhibitors by Regioselective 3′-O-Sulfation of Vanillin Lactosides Obtained under Phase Transfer Catalysis. Molecules 2021, 26, 115. https://doi.org/10.3390/molecules26010115
Belkhadem K, Cao Y, Roy R. Synthesis of Galectin Inhibitors by Regioselective 3′-O-Sulfation of Vanillin Lactosides Obtained under Phase Transfer Catalysis. Molecules. 2021; 26(1):115. https://doi.org/10.3390/molecules26010115
Chicago/Turabian StyleBelkhadem, Karima, Yihong Cao, and René Roy. 2021. "Synthesis of Galectin Inhibitors by Regioselective 3′-O-Sulfation of Vanillin Lactosides Obtained under Phase Transfer Catalysis" Molecules 26, no. 1: 115. https://doi.org/10.3390/molecules26010115
APA StyleBelkhadem, K., Cao, Y., & Roy, R. (2021). Synthesis of Galectin Inhibitors by Regioselective 3′-O-Sulfation of Vanillin Lactosides Obtained under Phase Transfer Catalysis. Molecules, 26(1), 115. https://doi.org/10.3390/molecules26010115