
Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity

 , ,
, , 

Abstract
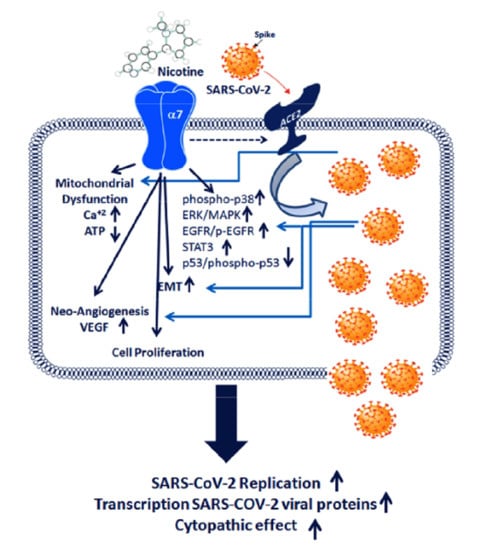
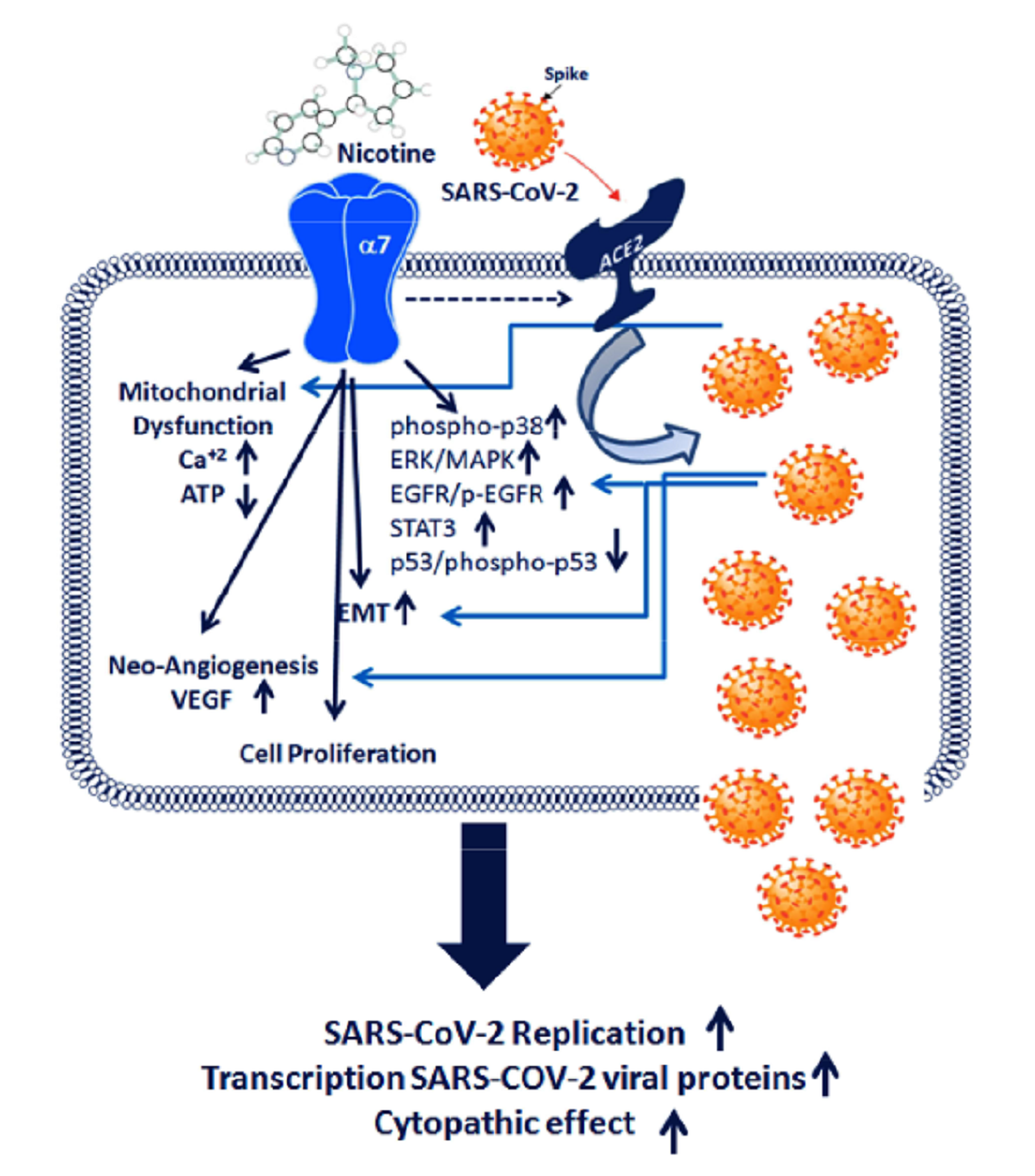
1. Introduction
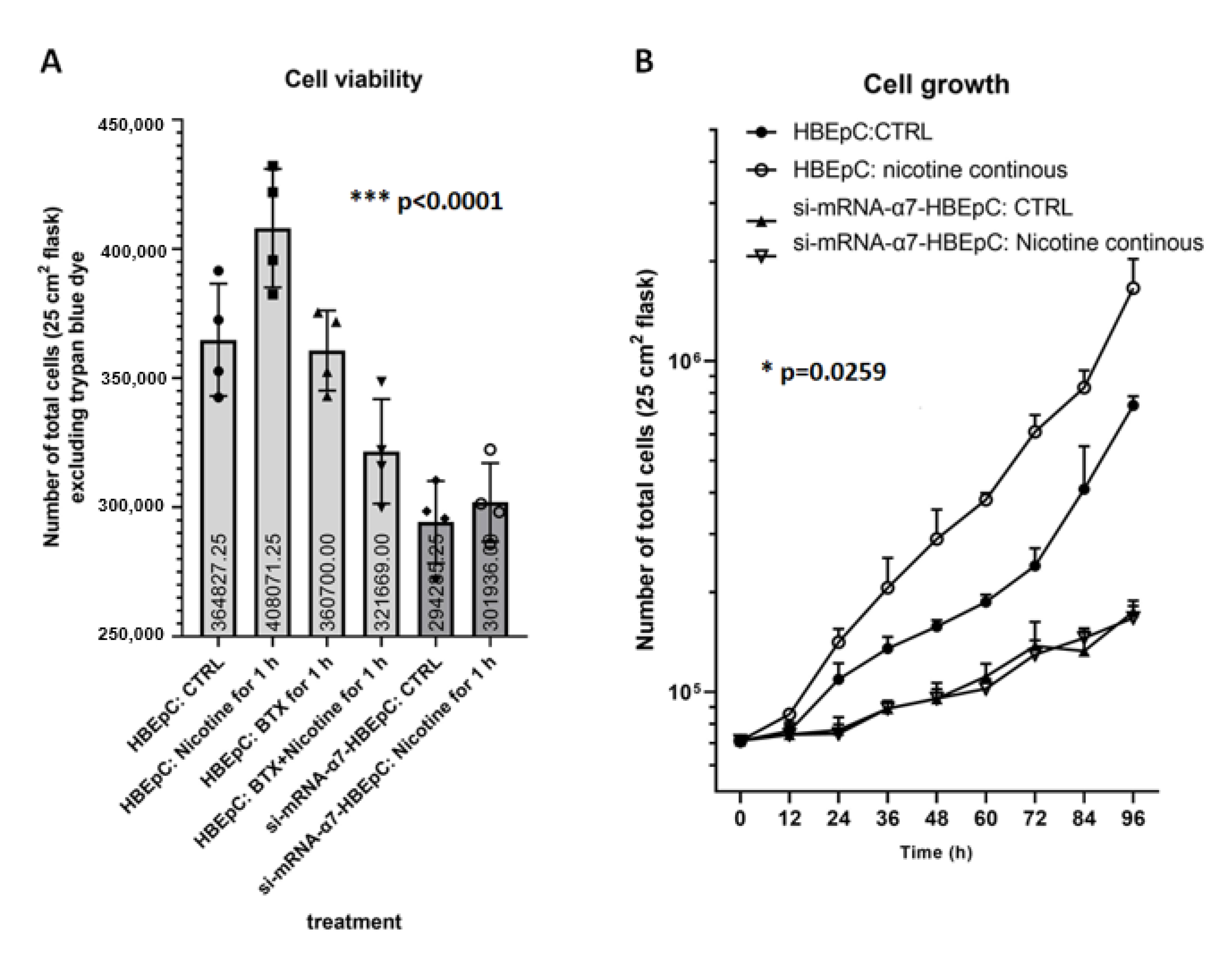
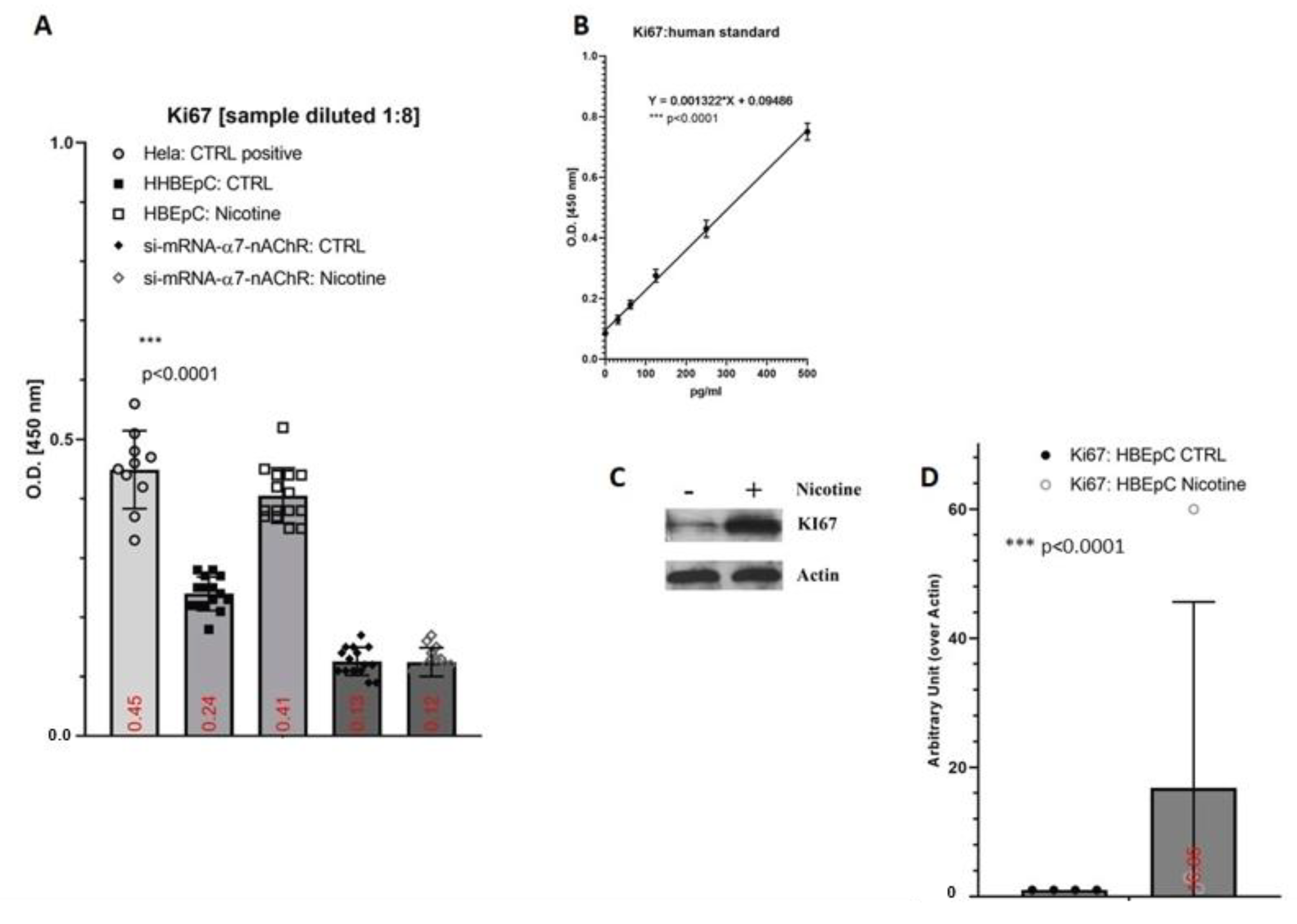
2. Results
3. Discussion
4. Materials and Methods
4.1. Cells and Treatment
4.2. Western Blot Detection
4.3. Evaluation of p53/phosphor-p53, p38/phospo-p38, Calcium, ATP, EGFR/p-EGFR, and VEGF
4.4. Cell Migration
4.5. Cellular Senescence
4.6. Cell Transformation
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| A549 human lung carcinoma cells transfected with human ACE2 | ACE2-A549 |
| Acetylcholine | ACh |
| Acetylcholinesterase | AChE |
| Angiotensin-converting enzyme 2 | ACE2 |
| Butyrylcholinesterase | BuChE |
| Choline acetyltransferase | ChAT |
| Cholinergic Receptor Nicotinic Alpha 7 Subunit gene | CHRNA7 |
| Cholinergic system | CS |
| Cyclic adenosine monophosphate | cAMP |
| Cytomegalovirus | CMV |
| Epithelial cell adhesion molecule | EpCAM |
| Epithelial-mesenchymal transition | EMT |
| Fibronectin | FN |
| Hepatitis C virus | HCV |
| Human bronchial epithelial cells | HBEpC |
| Human Coronavirus NL63 | HCoV-NL63 |
| Human immunodeficiency virus | HIV |
| Influenza virus | IAV |
| International Union of Pure and Applied Chemistry | IUPAC |
| Intussusceptive angiogenesis | IA |
| Ly-6/uPAR-related protein 1 | SLURP-1 |
| Measles virus | MV |
| Middle East respiratory syndrome Coronavirus | MERS-CoV |
| Muscarinic receptor | mAChR |
| Nicotinic receptor | nAChR |
| Open Reading Frame 6 | ORF6 |
| Primary Human normal bronchial epithelial | NHBE |
| Renin-Angiotensin System | RAS |
| Respiratory syncytial virus | RSV |
| Rhinovirus | RV |
| Secondary metabolite(s) | SM |
| Senescence-associated β-galactosidase | SA-β-Gal |
| Severe Acute Respiratory Syndrome Corona Virus-2 | SARS-CoV-2 |
| Sprouting angiogenesis | SA |
| Transmembrane protease, serine 2 | TMPRSS2 |
| Vascular endothelial growth factor | VEGF |
| Vesicular acetylcholine transporter | VAChT |
References
- Properties of Nicotine. Available online: http://www.chm.bris.ac.uk/motm/nicotine/E-propriete.html (accessed on 18 December 2020).
- Yildiz, D. Nicotine, its metabolism and an overview of its biological effects. Toxicon 2004, 43, 619–632. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Nastrucci, C.; Alzetta, G.; Szalai, C. Tobacco habit: Historical, cultural, neurobiological, and genetic features of people’s relationship with an addictive drug. Perspect. Biol. Med. 2011, 54, 557–577. [Google Scholar] [CrossRef] [PubMed]
- Steppuhn, A.; Gase, K.; Krock, B.; Halitschke, R.; Baldwin, I.T. Nicotine’s defensive function in nature. PLoS Biol. 2004, 2, E217. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Pandit, S.S.; Steppuhn, A.; Baldwin, I.T. Natural history-driven, plant-mediated RNAi-based study reveals CYP6B46’s role in a nicotine-mediated antipredator herbivore defense. Proc. Natl. Acad. Sci. USA 2014, 111, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Moghbel, N.; Ryu, B.; Ratsch, A.; Steadman, K.J. Nicotine alkaloid levels, and nicotine to nornicotine conversion, in Australian Nicotiana species used as chewing tobacco. Heliyon 2017, 3, e00469. [Google Scholar] [CrossRef] [PubMed]
- Mayer, B. How much nicotine kills a human? Tracing back the generally accepted lethal dose to dubious self-experiments in the nineteenth century. Arch. Toxicol. 2014, 88, 5–7. [Google Scholar] [CrossRef]
- Alkam, T.; Nabeshima, T. Molecular mechanisms for nicotine intoxication. Neurochem. Int. 2019, 125, 117–126. [Google Scholar] [CrossRef]
- Matta, S.G.; Balfour, D.J.; Benowitz, N.L.; Boyd, R.T.; Buccafusco, J.J.; Caggiula, A.R.; Craig, C.R.; Collins, A.C.; Damaj, M.I.; Donny, E.C.; et al. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology 2007, 190, 269–319. [Google Scholar] [CrossRef]
- Changeux, J.P. The nicotinic acetylcholine receptor: A typical ‘allosteric machine’. Philos. Trans. R. Soc. Lond B Biol. Sci. 2018, 373. [Google Scholar] [CrossRef]
- Zoli, M.; Pucci, S.; Vilella, A.; Gotti, C. Neuronal and Extraneuronal Nicotinic Acetylcholine Receptors. Curr. Neuropharmacol. 2018, 16, 338–349. [Google Scholar] [CrossRef]
- Cardinale, A.; Nastrucci, C.; Cesario, A.; Russo, P. Nicotine: Specific role in angiogenesis, proliferation and apoptosis. Crit. Rev. Toxicol. 2012, 42, 68–89. [Google Scholar] [CrossRef] [PubMed]
- Dwoskin, L.P.; Smith, A.M.; Wooters, T.E.; Zhang, Z.; Crooks, P.A.; Bardo, M.T. Nicotinic receptor-based therapeutics and candidates for smoking cessation. Biochem. Pharmacol. 2009, 78, 732–743. [Google Scholar] [CrossRef] [PubMed]
- Daly, J.W. Nicotinic agonists, antagonists, and modulators from natural sources. Cell Mol. Neurobiol. 2005, 25, 513–552. [Google Scholar] [CrossRef] [PubMed]
- Xing, H.; Andrud, K.W.; Soti, F.; Rouchaud, A.; Jahn, S.C.; Lu, Z.; Cho, Y.H.; Habibi, S.; Corsino, P.; Slavov, S.; et al. A Methyl Scan of the Pyrrolidinium Ring of Nicotine Reveals Significant Differences in its Interactions with alpha7 and alpha4beta2 Nicotinic Acetylcholine Receptors. Mol. Pharmacol. 2020, 98, 168–180. [Google Scholar] [CrossRef] [PubMed]
- Picciotto, M.R.; Kenny, P.J. Mechanisms of Nicotine Addiction. Cold Spring Harb. Perspect. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Paleari, L.; Grozio, A.; Cesario, A.; Russo, P. The cholinergic system and cancer. Semin. Cancer Biol. 2008, 18, 211–217. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, B.J.; Williams, M.; Plummer, H.K.; Schuller, H.M. Activation of voltage-operated Ca2+-channels in human small cell lung carcinoma by the tobacco-specific nitrosamine 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Int. J. Oncol. 2000, 16, 513–521. [Google Scholar] [CrossRef]
- Schuller, H.M. Is cancer triggered by altered signalling of nicotinic acetylcholine receptors? Nat. Rev. Cancer 2009, 9, 195–205. [Google Scholar] [CrossRef]
- Hurst, R.; Rollema, H.; Bertrand, D. Nicotinic acetylcholine receptors: From basic science to therapeutics. Pharmacol. Ther. 2013, 137, 22–54. [Google Scholar] [CrossRef]
- Grando, S.A.; Kawashima, K.; Wessler, I. A historic perspective on the current progress in elucidation of the biologic significance of non-neuronal acetylcholine. Int. Immunopharmacol. 2020, 81, 106289. [Google Scholar] [CrossRef]
- Cox, M.A.; Bassi, C.; Saunders, M.E.; Nechanitzky, R.; Morgado-Palacin, I.; Zheng, C.; Mak, T.W. Beyond neurotransmission: Acetylcholine in immunity and inflammation. J. Intern. Med. 2020, 287, 120–133. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.K.; Kirkpatrick, C.J. Activation of Muscarinic Receptors by Non-neuronal Acetylcholine. In Muscarinic Receptors; Fryer, A.D., Christopoulos, A., Nathanson, N.M., Eds.; Springer: Berlin/Heidelberg, Germany, 2012; pp. 469–491. [Google Scholar]
- Wessler, I.; Kilbinger, H.; Bittinger, F.; Kirkpatrick, C.J. The biological role of non-neuronal acetylcholine in plants and humans. Jpn. J. Pharmacol. 2001, 85, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.K.; Kirkpatrick, C.J. Non-neuronal acetylcholine involved in reproduction in mammals and honeybees. J. Neurochem. 2017, 142, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Wessler, I.; Kilbinger, H.; Bittinger, F.; Unger, R.; Kirkpatrick, C.J. The non-neuronal cholinergic system in humans: Expression, function and pathophysiology. Life Sci. 2003, 72, 2055–2061. [Google Scholar] [CrossRef]
- Aluigi, M.G.; Diaspro, A.; Ramoino, P.; Russo, P.; Falugi, C. The sea urchin, Paracentrotus lividus, as a model to investigate the onset of molecules immunologically related to the alpha-7 subunit of nicotinic receptors during embryonic and larval development. Curr. Drug Targets 2012, 13, 587–593. [Google Scholar] [CrossRef]
- Wang, H.; Yu, M.; Ochani, M.; Amella, C.A.; Tanovic, M.; Susarla, S.; Li, J.H.; Wang, H.; Yang, H.; Ulloa, L.; et al. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature 2003, 421, 384–388. [Google Scholar] [CrossRef]
- Paleari, L.; Cesario, A.; Fini, M.; Russo, P. alpha7-Nicotinic receptor antagonists at the beginning of a clinical era for NSCLC and Mesothelioma? Drug Discov. Today 2009, 14, 822–836. [Google Scholar] [CrossRef]
- Maouche, K.; Medjber, K.; Zahm, J.M.; Delavoie, F.; Terryn, C.; Coraux, C.; Pons, S.; Cloez-Tayarani, I.; Maskos, U.; Birembaut, P.; et al. Contribution of alpha7 nicotinic receptor to airway epithelium dysfunction under nicotine exposure. Proc. Natl. Acad. Sci. USA 2013, 110, 4099–4104. [Google Scholar] [CrossRef]
- Krais, A.M.; Hautefeuille, A.H.; Cros, M.P.; Krutovskikh, V.; Tournier, J.M.; Birembaut, P.; Thepot, A.; Paliwal, A.; Herceg, Z.; Boffetta, P.; et al. CHRNA5 as negative regulator of nicotine signaling in normal and cancer bronchial cells: Effects on motility, migration and p63 expression. Carcinogenesis 2011, 32, 1388–1395. [Google Scholar] [CrossRef]
- Sartelet, H.; Maouche, K.; Totobenazara, J.L.; Petit, J.; Burlet, H.; Monteau, M.; Tournier, J.M.; Birembaut, P. Expression of nicotinic receptors in normal and tumoral pulmonary neuroendocrine cells (PNEC). Pathol. Res. Pract. 2008, 204, 891–898. [Google Scholar] [CrossRef]
- Lam, D.C.L.; Luo, S.Y.; Fu, K.H.; Lui, M.M.S.; Chan, K.H.; Wistuba, I.I.; Gao, B.; Tsao, S.W.; Ip, M.S.M.; Minna, J.D. Nicotinic acetylcholine receptor expression in human airway correlates with lung function. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 310, L232–L239. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Zhou, X.; Kolosov, V.P.; Perelman, J.M. The expression and pharmacological characterization of nicotinic acetylcholine receptor subunits in HBE16 airway epithelial cells. Cell Biochem. Biophys. 2012, 62, 421–431. [Google Scholar] [CrossRef] [PubMed]
- Russo, P.; Bonassi, S.; Giacconi, R.; Malavolta, M.; Tomino, C.; Maggi, F. COVID-19 and smoking: Is nicotine the hidden link? Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.M.; Yang, C.X.; Sin, D.D. COVID-19 and nicotine as a mediator of ACE-2. Eur. Respir. J. 2020, 55. [Google Scholar] [CrossRef] [PubMed]
- Olds, J.L.; Kabbani, N. Is nicotine exposure linked to cardiopulmonary vulnerability to COVID-19 in the general population? FEBS J. 2020. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef]
- Costa, L.B.; Perez, L.G.; Palmeira, V.A.; Macedo, E.C.T.; Ribeiro, V.T.; Lanza, K.; Simoes, E.S.A.C. Insights on SARS-CoV-2 Molecular Interactions with the Renin-Angiotensin System. Front. Cell Dev. Biol. 2020, 8, 559841. [Google Scholar] [CrossRef]
- van de Veerdonk, F.; Netea, M.G.; van Deuren, M.; van der Meer, J.W.M.; de Mast, Q.; Bruggemann, R.J.; van der Hoeven, H. Kinins and Cytokines in COVID-19: A Comprehensive Pathophysiological Approach. 2020. 2020040023. Available online: 10.20944/preprints202004.0023.v1. (accessed on 18 December 2020).
- Hou, Y.J.; Okuda, K.; Edwards, C.E.; Martinez, D.R.; Asakura, T.; Dinnon, K.H., 3rd; Kato, T.; Lee, R.E.; Yount, B.L.; Mascenik, T.M.; et al. SARS-CoV-2 Reverse Genetics Reveals a Variable Infection Gradient in the Respiratory Tract. Cell 2020, 182, 429–446e14. [Google Scholar] [CrossRef]
- Milne, S.; Yang, C.X.; Timens, W.; Bosse, Y.; Sin, D.D. SARS-CoV-2 receptor ACE2 gene expression and RAAS inhibitors. Lancet Respir. Med. 2020, 8, e50–e51. [Google Scholar] [CrossRef]
- Lukassen, S.; Chua, R.L.; Trefzer, T.; Kahn, N.C.; Schneider, M.A.; Muley, T.; Winter, H.; Meister, M.; Veith, C.; Boots, A.W.; et al. SARS-CoV-2 receptor ACE2 and TMPRSS2 are primarily expressed in bronchial transient secretory cells. EMBO J. 2020, 39, e105114. [Google Scholar] [CrossRef] [PubMed]
- Cai, G.; Bosse, Y.; Xiao, F.; Kheradmand, F.; Amos, C.I. Tobacco Smoking Increases the Lung Gene Expression of ACE2, the Receptor of SARS-CoV-2. Am. J. Respir. Crit. Care Med. 2020, 201, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Leung, J.M.; Niikura, M.; Yang, C.W.T.; Sin, D.D. COVID-19 and COPD. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Russo, P. Expert view smoking, nicotine, and COVID-19 myths and facts. Available online: https://www.sciencemediacentre.org/expert-reaction-to-questions-about-smoking-and-covid-19/ (accessed on 18 December 2020).
- Lupacchini, L.T.C.; Russo, P. COVID-19 infection, and Nicotine. Systematic revision of the literature. Tabaccologia 2020, 3, 8. [Google Scholar]
- Farsalinos, K.; Barbouni, A.; Niaura, R. Systematic review of the prevalence of current smoking among hospitalized COVID-19 patients in China: Could nicotine be a therapeutic option? Intern. Emerg. Med. 2020, 15, 845–852. [Google Scholar] [CrossRef]
- Giannouchos, T.V.; Sussman, R.A.; Mier, J.M.; Poulas, K.; Farsalinos, K. Characteristics and risk factors for COVID-19 diagnosis and adverse outcomes in Mexico: An analysis of 89,756 laboratory-confirmed COVID-19 cases. Eur. Respir. J. 2020. [Google Scholar] [CrossRef]
- Israel, A.; Feldhamer, I.; Lahad, A.; Levin-Zamir, D.; Lavie, G. Smoking and the risk of COVID-19 in a large observational population study. medRxiv 2020. [Google Scholar] [CrossRef]
- Miyara, M.; Tubach, F.; Martinez, V.; Morelot-Panzini, C.; Pernet, J.; Haroche, J.; Lebbah, S.; Morawiec, E.; Gorochov, G.; Caumes, E.; et al. Low rate of daily smokers in patients with symptomatic COVID-19. medRxiv 2020. [Google Scholar] [CrossRef]
- Zureik, M.; Baricault, B.; Vabre, C.; Semenzato, L.; Drouin, J.; cuenot, F.; penso, L.; Herlemont, P.; Sbidian, E.; Weill, A.; et al. Nicotine-replacement therapy, as a surrogate of smoking, and the risk of hospitalization with Covid-19 and all-cause mortality: A nationwide, observational cohort study in France. medRxiv 2020. [Google Scholar] [CrossRef]
- Tattan-Birch, H.; Perski, O.; Jackson, S.; Shahab, L.; West, R.; Brown, J. COVID-19, smoking, vaping and quitting: A representative population survey in England. Addiction 2020. [Google Scholar] [CrossRef]
- Vardavas, C.I.; Nikitara, K. COVID-19 and smoking: A systematic review of the evidence. Tob. Induc. Dis. 2020, 18, 20. [Google Scholar] [CrossRef] [PubMed]
- Berlin, I.; Thomas, D.; Le Faou, A.L.; Cornuz, J. COVID-19 and Smoking. Nicotine Tob. Res. 2020, 22, 1650–1652. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Meng, M.; Kumar, R.; Wu, Y.; Huang, J.; Lian, N.; Deng, Y.; Lin, S. The impact of COPD and smoking history on the severity of COVID-19: A systemic review and meta-analysis. J. Med. Virol. 2020, 92, 1915–1921. [Google Scholar] [CrossRef] [PubMed]
- Van Zyl-Smit, R.N.; Richards, G.; Leone, F.T. Tobacco smoking and COVID-19 infection. Lancet Respir. Med. 2020, 8, 664–665. [Google Scholar] [CrossRef]
- Alqahtani, J.S.; Oyelade, T.; Aldhahir, A.M.; Alghamdi, S.M.; Almehmadi, M.; Alqahtani, A.S.; Quaderi, S.; Mandal, S.; Hurst, J.R. Prevalence, Severity and Mortality associated with COPD and Smoking in patients with COVID-19: A Rapid Systematic Review and Meta-Analysis. PLoS ONE 2020, 15, e0233147. [Google Scholar] [CrossRef]
- Tsigaris, P.; Teixeira da Silva, J.A. Smoking Prevalence and COVID-19 in Europe. Nicotine Tob. Res. 2020, 22, 1646–1649. [Google Scholar] [CrossRef]
- Cattaruzza, M.S.; Zaga, V.; Gallus, S.; D’Argenio, P.; Gorini, G. Tobacco smoking and COVID-19 pandemic: Old and new issues. A summary of the evidence from the scientific literature. Acta Biomed. 2020, 91, 106–112. [Google Scholar] [CrossRef]
- Cattaruzza, M.S.; Gorini, G.; Bosetti, C.; Boffi, R.; Lugo, A.; Veronese, C.; Carreras, G.; Santucci, C.; Stival, C.; Pacifici, R.; et al. Covid-19 and the role of smoking: The protocol of the multicentric prospective study COSMO-IT (COvid19 and SMOking in ITaly). Acta Bio-Medica: Atenei Parm. 2020, 91, e2020062. [Google Scholar] [CrossRef]
- Gaiha, S.M.; Cheng, J.; Halpern-Felsher, B. Association Between Youth Smoking, Electronic Cigarette Use, and COVID-19. J. Adolesc Health 2020, 67, 519–523. [Google Scholar] [CrossRef]
- Wang, Q.; Sundar, I.K.; Li, D.; Lucas, J.H.; Muthumalage, T.; McDonough, S.R.; Rahman, I. E-cigarette-induced pulmonary inflammation and dysregulated repair are mediated by nAChR alpha7 receptor: Role of nAChR alpha7 in SARS-CoV-2 Covid-19 ACE2 receptor regulation. Respir. Res. 2020, 21, 154. [Google Scholar] [CrossRef]
- Changeux, J.P.; Amoura, Z.; Rey, F.A.; Miyara, M. A nicotinic hypothesis for Covid-19 with preventive and therapeutic implications. C R Biol. 2020, 343, 33–39. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Farsalinos, K.; Eliopoulos, E.; Leonidas, D.D.; Papadopoulos, G.E.; Tzartos, S.; Poulas, K. Nicotinic Cholinergic System and COVID-19: In Silico Identification of an Interaction between SARS-CoV-2 and Nicotinic Receptors with Potential Therapeutic Targeting Implications. Int. J. Mol. Sci. 2020, 21, 5807. [Google Scholar] [CrossRef]
- Oliveira, A.S.F.; Ibarra, A.A.; Bermudez, I.; Casalino, L.; Gaieb, Z.; Shoemark, D.K.; Gallagher, T.; Sessions, R.B.; Amaro, R.E.; Mulholland, A.J. Simulations support the interaction of the SARS-CoV-2 spike protein with nicotinic acetylcholine receptors and suggest subtype specificity. BioRxiv 2020. [Google Scholar] [CrossRef]
- Hasanagic, S.; Serdarevic, F. Potential role of memantine in the prevention and treatment of COVID-19: Its antagonism of nicotinic acetylcholine receptors and beyond. Eur. Respir. J. 2020, 56. [Google Scholar] [CrossRef] [PubMed]
- Capo-Velez, C.M.; Delgado-Velez, M.; Baez-Pagan, C.A.; Lasalde-Dominicci, J.A. Nicotinic Acetylcholine Receptors in HIV: Possible Roles During HAND and Inflammation. Cell Mol. Neurobiol. 2018, 38, 1335–1348. [Google Scholar] [CrossRef] [PubMed]
- Sifat, A.E.; Nozohouri, S.; Villalba, H.; Vaidya, B.; Abbruscato, T.J. The Role of Smoking and Nicotine in the Transmission and Pathogenesis of COVID-19. J. Pharmacol. Exp. Ther. 2020, 375, 498–509. [Google Scholar] [CrossRef]
- Li, W.; Sui, J.; Huang, I.C.; Kuhn, J.H.; Radoshitzky, S.R.; Marasco, W.A.; Choe, H.; Farzan, M. The S proteins of human coronavirus NL63 and severe acute respiratory syndrome coronavirus bind overlapping regions of ACE2. Virology 2007, 367, 367–374. [Google Scholar] [CrossRef]
- Li, G.; He, X.; Zhang, L.; Ran, Q.; Wang, J.; Xiong, A.; Wu, D.; Chen, F.; Sun, J.; Chang, C. Assessing ACE2 expression patterns in lung tissues in the pathogenesis of COVID-19. J. Autoimmun. 2020, 112, 102463. [Google Scholar] [CrossRef]
- Bouhaddou, M.; Memon, D.; Meyer, B.; White, K.M.; Rezelj, V.V.; Correa Marrero, M.; Polacco, B.J.; Melnyk, J.E.; Ulferts, S.; Kaake, R.M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712e19. [Google Scholar] [CrossRef]
- Ma, Q.; Pan, W.; Li, R.; Liu, B.; Li, C.; Xie, Y.; Wang, Z.; Zhao, J.; Jiang, H.; Huang, J.; et al. Liu Shen capsule shows antiviral and anti-inflammatory abilities against novel coronavirus SARS-CoV-2 via suppression of NF-kappaB signaling pathway. Pharmacol. Res. 2020, 158, 104850. [Google Scholar] [CrossRef] [PubMed]
- Kopecky-Bromberg, S.A.; Martinez-Sobrido, L.; Palese, P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J. Virol. 2006, 80, 785–793. [Google Scholar] [CrossRef] [PubMed]
- Catassi, A.; Servent, D.; Paleari, L.; Cesario, A.; Russo, P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: Implications on lung carcinogenesis. Mutat. Res. 2008, 659, 221–231. [Google Scholar] [CrossRef] [PubMed]
- Bojkova, D.; Klann, K.; Koch, B.; Widera, M.; Krause, D.; Ciesek, S.; Cinatl, J.; Munch, C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. [Google Scholar] [CrossRef] [PubMed]
- Pruijssers, A.J.; Denison, M.R. Nucleoside analogues for the treatment of coronavirus infections. Curr. Opin. Virol. 2019, 35, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Thoms, M.; Buschauer, R.; Ameismeier, M.; Koepke, L.; Denk, T.; Hirschenberger, M.; Kratzat, H.; Hayn, M.; Mackens-Kiani, T.; Cheng, J.; et al. Structural basis for translational shutdown and immune evasion by the Nsp1 protein of SARS-CoV-2. Science 2020, 369, 1249–1255. [Google Scholar] [CrossRef]
- Stewart, C.A.; Gay, C.M.; Ramkumar, K.; Cargill, K.R.; Cardnell, R.J.; Nilsson, M.B.; Heeke, S.; Park, E.M.; Kundu, S.T.; Diao, L.; et al. SARS-CoV-2 infection induces EMT-like molecular changes, including ZEB1-mediated repression of the viral receptor ACE2, in lung cancer models. BioRxiv 2020. [Google Scholar] [CrossRef]
- Carlisle, D.L.; Liu, X.; Hopkins, T.M.; Swick, M.C.; Dhir, R.; Siegfried, J.M. Nicotine activates cell-signaling pathways through muscle-type and neuronal nicotinic acetylcholine receptors in non-small cell lung cancer cells. Pulm. Pharmacol. Ther. 2007, 20, 629–641. [Google Scholar] [CrossRef]
- Schaal, C.; Chellappan, S.P. Nicotine-mediated cell proliferation and tumor progression in smoking-related cancers. Mol. Cancer Res. 2014, 12, 14–23. [Google Scholar] [CrossRef]
- Sacar Demirci, M.D.; Adan, A. Computational analysis of microRNA-mediated interactions in SARS-CoV-2 infection. Peer J. 2020, 8, e9369. [Google Scholar] [CrossRef]
- Turk, C.; Turk, S.; Temirci, E.S.; Malkan, U.Y.; Haznedaroglu, I.C. In vitro analysis of the renin-angiotensin system and inflammatory gene transcripts in human bronchial epithelial cells after infection with severe acute respiratory syndrome coronavirus. J. Renin. Angiotensin. Aldosterone Syst. 2020, 21. [Google Scholar] [CrossRef] [PubMed]
- Puliyappadamba, V.T.; Cheriyan, V.T.; Thulasidasan, A.K.; Bava, S.V.; Vinod, B.S.; Prabhu, P.R.; Varghese, R.; Bevin, A.; Venugopal, S.; Anto, R.J. Nicotine-induced survival signaling in lung cancer cells is dependent on their p53 status while its down-regulation by curcumin is independent. Mol. Cancer 2010, 9, 220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Ganapathy, S.; Avraham, H.; Nishioka, T.; Chen, C. Nicotine exposure potentiates lung tumorigenesis by perturbing cellular surveillance. Br. J. Cancer 2020, 122, 904–911. [Google Scholar] [CrossRef] [PubMed]
- Ramaiah, M.J. mTOR inhibition and p53 activation, microRNAs: The possible therapy against pandemic COVID-19. Gene Rep. 2020, 20, 100765. [Google Scholar] [CrossRef]
- Aloni-Grinstein, R.; Charni-Natan, M.; Solomon, H.; Rotter, V. p53 and the Viral Connection: Back into the Future (double dagger). Cancers 2018, 10, 178. [Google Scholar] [CrossRef]
- Zhang, J.; Kamdar, O.; Le, W.; Rosen, G.D.; Upadhyay, D. Nicotine induces resistance to chemotherapy by modulating mitochondrial signaling in lung cancer. Am. J. Respir. Cell Mol. Biol 2009, 40, 135–146. [Google Scholar] [CrossRef]
- Manevski, M.; Muthumalage, T.; Devadoss, D.; Sundar, I.K.; Wang, Q.; Singh, K.P.; Unwalla, H.J.; Chand, H.S.; Rahman, I. Cellular stress responses and dysfunctional Mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol. 2020, 33, 101443. [Google Scholar] [CrossRef]
- Malinska, D.; Wieckowski, M.R.; Michalska, B.; Drabik, K.; Prill, M.; Patalas-Krawczyk, P.; Walczak, J.; Szymanski, J.; Mathis, C.; Van der Toorn, M.; et al. Mitochondria as a possible target for nicotine action. J. Bioenerg. Biomembr. 2019, 51, 259–276. [Google Scholar] [CrossRef]
- Singh, K.; Chen, Y.C.; Judy, J.T.; Seifuddin, F.; Tunc, I.; Pirooznia, M. Network Analysis and Transcriptome Profiling Identify Autophagic and Mitochondrial Dysfunctions in SARS-CoV-2 Infection. BioRxiv 2020. [Google Scholar] [CrossRef]
- Wang, Y.; He, J.; Jiang, H.; Zhang, Q.; Yang, H.; Xu, X.; Zhang, C.; Xu, C.; Wang, J.; Lu, W. Nicotine enhances storeoperated calcium entry by upregulating HIF1alpha and SOCC components in nonsmall cell lung cancer cells. Oncol. Rep. 2018, 40, 2097–2104. [Google Scholar]
- Zia, S.; Ndoye, A.; Nguyen, V.T.; Grando, S.A. Nicotine enhances expression of the alpha 3, alpha 4, alpha 5, and alpha 7 nicotinic receptors modulating calcium metabolism and regulating adhesion and motility of respiratory epithelial cells. Res. Commun. Mol. Pathol. Pharmacol. 1997, 97, 243–262. [Google Scholar] [PubMed]
- Straus, M.R.; Tang, T.; Lai, A.L.; Flegel, A.; Bidon, M.; Freed, J.H.; Daniel, S.; Whittaker, G.R. Ca(2+) Ions Promote Fusion of Middle East Respiratory Syndrome Coronavirus with Host Cells and Increase Infectivity. J. Virol. 2020, 94. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.L.; Millet, J.K.; Daniel, S.; Freed, J.H.; Whittaker, G.R. The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium-Dependent Manner. J. Mol. Biol. 2017, 429, 3875–3892. [Google Scholar] [CrossRef] [PubMed]
- Jayaseelan, V.P.; Paramasivam, A. Repurposing calcium channel blockers as antiviral drugs. J. Cell Commun. Signal 2020, 14, 467–468. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, P.; Rizwani, W.; Pillai, S.; Kinkade, R.; Kovacs, M.; Rastogi, S.; Banerjee, S.; Carless, M.; Kim, E.; Coppola, D.; et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int. J. Cancer 2009, 124, 36–45. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Zou, Y.; Zhao, Z.; Li, B.; Ran, P. Nicotine-induced epithelial-mesenchymal transition via Wnt/beta-catenin signaling in human airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2013, 304, L199–L209. [Google Scholar] [CrossRef] [PubMed]
- Roman, J.; Ritzenthaler, J.D.; Gil-Acosta, A.; Rivera, H.N.; Roser-Page, S. Nicotine and fibronectin expression in lung fibroblasts: Implications for tobacco-related lung tissue remodeling. FASEB J. 2004, 18, 1436–1438. [Google Scholar] [CrossRef]
- Zheng, Y.; Ritzenthaler, J.D.; Roman, J.; Han, S. Nicotine stimulates human lung cancer cell growth by inducing fibronectin expression. Am. J. Respir. Cell Mol. Biol. 2007, 37, 681–690. [Google Scholar] [CrossRef]
- Xu, J.; Xu, X.; Jiang, L.; Dua, K.; Hansbro, P.M.; Liu, G. SARS-CoV-2 induces transcriptional signatures in human lung epithelial cells that promote lung fibrosis. Respir. Res. 2020, 21, 182. [Google Scholar] [CrossRef]
- Zhang, C.; Yu, P.; Zhu, L.; Zhao, Q.; Lu, X.; Bo, S. Blockade of alpha7 nicotinic acetylcholine receptors inhibit nicotine-induced tumor growth and vimentin expression in non-small cell lung cancer through MEK/ERK signaling way. Oncol. Rep. 2017, 38, 3309–3318. [Google Scholar]
- Yu, Y.T.; Chien, S.C.; Chen, I.Y.; Lai, C.T.; Tsay, Y.G.; Chang, S.C.; Chang, M.F. Surface vimentin is critical for the cell entry of SARS-CoV. J. Biomed. Sci. 2016, 23, 14. [Google Scholar] [CrossRef] [PubMed]
- Fararjeh, A.S.; Tu, S.H.; Chen, L.C.; Cheng, T.C.; Liu, Y.R.; Chang, H.L.; Chang, H.W.; Huang, C.C.; Wang, H.R.; Hwang-Verslues, W.W.; et al. Long-term exposure to extremely low-dose of nicotine and 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) induce non-malignant breast epithelial cell transformation through activation of the a9-nicotinic acetylcholine receptor-mediated signaling pathway. Environ. Toxicol. 2019, 34, 73–82. [Google Scholar] [PubMed]
- Ackermann, M.; Verleden, S.E.; Kuehnel, M.; Haverich, A.; Welte, T.; Laenger, F.; Vanstapel, A.; Werlein, C.; Stark, H.; Tzankov, A.; et al. Pulmonary Vascular Endothelialitis, Thrombosis, and Angiogenesis in Covid-19. N. Engl. J. Med. 2020, 383, 120–128. [Google Scholar] [CrossRef] [PubMed]
- Vrancken, K.; Vervaeke, P.; Balzarini, J.; Liekens, S. Viruses as key regulators of angiogenesis. Rev. Med. Virol. 2011, 21, 181–200. [Google Scholar] [CrossRef]
- Chernyavsky, A.I.; Shchepotin, I.B.; Grando, S.A. Mechanisms of growth-promoting and tumor-protecting effects of epithelial nicotinic acetylcholine receptors. Int. Immunopharmacol. 2015, 29, 36–44. [Google Scholar] [CrossRef]
- Zacharias, W.J.; Frank, D.B.; Zepp, J.A.; Morley, M.P.; Alkhaleel, F.A.; Kong, J.; Zhou, S.; Cantu, E.; Morrisey, E.E. Regeneration of the lung alveolus by an evolutionarily conserved epithelial progenitor. Nature 2018, 555, 251–255. [Google Scholar] [CrossRef]
- Hayflick, L. The Limited in Vitro Lifetime of Human Diploid Cell Strains. Exp. Cell Res. 1965, 37, 614–636. [Google Scholar] [CrossRef]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Kagan, J.C. Signaling organelles of the innate immune system. Cell 2012, 151, 1168–1178. [Google Scholar] [CrossRef]
- Novoa, R.R.; Calderita, G.; Arranz, R.; Fontana, J.; Granzow, H.; Risco, C. Virus factories: Associations of cell organelles for viral replication and morphogenesis. Biol. Cell 2005, 97, 147–172. [Google Scholar] [CrossRef] [PubMed]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef] [PubMed]
- Duarte, S.; Viedma-Poyatos, A.; Navarro-Carrasco, E.; Martinez, A.E.; Pajares, M.A.; Perez-Sala, D. Vimentin filaments interact with the actin cortex in mitosis allowing normal cell division. Nat. Commun. 2019, 10, 4200. [Google Scholar] [CrossRef] [PubMed]
- Cheng, F.; Eriksson, J.E. Intermediate Filaments and the Regulation of Cell Motility during Regeneration and Wound Healing. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef]
- Gan, Z.; Ding, L.; Burckhardt, C.J.; Lowery, J.; Zaritsky, A.; Sitterley, K.; Mota, A.; Costigliola, N.; Starker, C.G.; Voytas, D.F.; et al. Vimentin Intermediate Filaments Template Microtubule Networks to Enhance Persistence in Cell Polarity and Directed Migration. Cell Syst. 2016, 3, 252–263. [Google Scholar] [CrossRef]
- Snyder, L.D.; Mosher, C.; Holtze, C.H.; Lancaster, L.H.; Flaherty, K.R.; Noth, I.; Neely, M.L.; Hellkamp, A.S.; Bender, S.; Conoscenti, C.S.; et al. Time to diagnosis of idiopathic pulmonary fibrosis in the IPF-PRO Registry. BMJ Open Respir. Res. 2020, 7, e000567. [Google Scholar] [CrossRef]
- Ramos, I.; Stamatakis, K.; Oeste, C.L.; Perez-Sala, D. Vimentin as a Multifaceted Player and Potential Therapeutic Target in Viral Infections. Int. J. Mol. Sci. 2020, 21, 4675. [Google Scholar] [CrossRef]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Cruz, G.N.F.; Christoff, A.P.; de Oliveira, L.F.V. Equivolumetric protocol generates library sizes proportional to total microbial load in nextgeneration sequencing. BioRxiv 2020. [Google Scholar] [CrossRef]
- Webb, B.J.; Peltan, I.D.; Jensen, P.; Hoda, D.; Hunter, B.; Silver, A.; Starr, N.; Buckel, W.; Grisel, N.; Hummel, E.; et al. Clinical criteria for COVID-19-associated hyperinflammatory syndrome: A cohort study. Lancet Rheumatol. 2020, 2, e754–e763. [Google Scholar] [CrossRef]
- Polidoro, R.B.; Hagan, R.S.; de Santis Santiago, R.; Schmidt, N.W. Overview: Systemic Inflammatory Response Derived from Lung Injury Caused by SARS-CoV-2 Infection Explains Severe Outcomes in COVID-19. Front. Immunol. 2020, 11, 1626. [Google Scholar] [CrossRef] [PubMed]
- Cimpean, A.M.; Raica, M. Historical Overview of In Vivo and In Vitro Angiogenesis Assays. Methods Mol. Biol. 2021, 2206, 1–13. [Google Scholar] [PubMed]
- De Spiegelaere, W.; Casteleyn, C.; Van den Broeck, W.; Plendl, J.; Bahramsoltani, M.; Simoens, P.; Djonov, V.; Cornillie, P. Intussusceptive angiogenesis: A biologically relevant form of angiogenesis. J. Vasc. Res. 2012, 49, 390–404. [Google Scholar] [CrossRef] [PubMed]
- Bychkov, M.; Shenkarev, Z.; Shulepko, M.; Shlepova, O.; Kirpichnikov, M.; Lyukmanova, E. Water-soluble variant of human Lynx1 induces cell cycle arrest and apoptosis in lung cancer cells via modulation of alpha7 nicotinic acetylcholine receptors. PLoS ONE 2019, 14, e0217339. [Google Scholar] [CrossRef]
- Shulepko, M.A.; Bychkov, M.L.; Shlepova, O.V.; Shenkarev, Z.O.; Kirpichnikov, M.P.; Lyukmanova, E.N. Human secreted protein SLURP-1 abolishes nicotine-induced proliferation, PTEN down-regulation and alpha7-nAChR expression up-regulation in lung cancer cells. Int. Immunopharmacol. 2020, 82, 106303. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, T.; Kubli, S.P.; Yoshinaga, S.K.; Pfeffer, K.; Mak, T.W. An aberrant STAT pathway is central to COVID-19. Cell Death Differ. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, P.; Breuer, M.; Hirst, N. COVID-19 breakthroughs: Separating fact from fiction. FEBS J. 2020. [Google Scholar] [CrossRef]
- Farsalinos, K.; Niaura, R.; Le Houezec, J.; Barbouni, A.; Tsatsakis, A.; Kouretas, D.; Vantarakis, A.; Poulas, K. Editorial: Nicotine and SARS-CoV-2: COVID-19 may be a disease of the nicotinic cholinergic system. Toxicol. Rep. 2020, 7, 658–663. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Name | Nicotine |
|---|---|
| IUPAC Name | 3-(1-methyl-2-pyrrolidinyl)pyridine |
| Chemical Specification | Bicyclic molecule characterized by a pyridine cycle and a pyrrolidine cycle existing in natures only in the S shape (i.e., levogyre) [1] |
| Chemical Characteristics | Chemical formula: C10H14N2 Molecular Weight: 162.234. |
| Physical Characteristics | Light yellow liquid that turns dark-brown after exposure to light/air with fishy odor when warm. Boiling point: 274.58 °C at 760 Tor [2] |
| Synthesized as SM by plants | Familia: Solanaceae, Genus: Nicotiana, Species: Nicotiana tabacum |
| History | 1492 Cristoforo Colombo discovery of the plant Nicotiana. Nicotine owes is name to Jean Nicot (1530-1604) who introduced the use of tobacco to the French court (Caterina de Medici) in the sixteenth century, thus helping the spread of tobacco into all Europe [3] 1828 the German chemists Wilhelm Heinrich Posselt and Karl Ludwig Reimann were the first to isolate nicotine from tobacco. 1843 Louis Melsens described the chemical formula. 1893 Adolf Pinner and Richard Wolffenstein described the structure. 1904 Amé Pictet and Arnold Rotschy, synthesized nicotine [3] |
| Production, Use and Natural Resistance | Plants produce SM, metabolites not essential for plant reproduction, as direct defenses against pathogens (i.e., insects) and herbivores. Among SM, nicotine is one of the best-studied drug representing one of the first insecticides utilized to control pests in agriculture [4]. Although nicotine is very toxic, some insects develop resistance, indeed the tobacco hornworm Manduca sexta may survive to nicotine concentrations that are considered toxic for non-adapted herbivores [4]. Although the precise mechanisms for Manduca sexta’s nicotine resistance are not completely understood, an efficient excretion and metabolism seem to be involved. Manduca sexta uses nicotine to protect herself by the attack of its native major nocturnal predator wolf spider Camptocosa parallela [5] |
| Concentration in Tobacco leaves | The percentage of nicotine on dry weight of tobacco is between 0.3 to 8.3% depending on plant variety, as for Nicotiana gossei and Nicotiana velutina, respectively, and 6.7% in Virginia variety, and cultivation [6] |
| Lethal Doses | In animals the lethal dose may be from 3 mg/kg in mice to 50 mg/kg in rats. There is no consensus on the human lethal dose of nicotine and 60 mg of nicotine given orally (resulting in a plasma concentration of ~180 ng/mL) is often suggested as the lethal dose in the literature. However, it has been reported that adults may survive to dosages much higher than this (up to 500 mg) [7] |
| Metabolism | Nicotine is metabolized in the liver, principally to cotinine that in turn is metabolized to trans-3′-hydroxycotinine excreted via renal [8] |
| Nicotine concentrations into cigarette | The amount of nicotine in one tobacco cigarette is approximately 1–2% = 1–2 g/100. Considering the human body weight average equal to 68 kg one cigarette may deliver approximately 10–30 μg/Kg, resulting in a peak plasma level of 10–50 ng/mL. A concentration equal to 50 ng/mL can be converted to molarity dividing by nicotine MW (i.e., 162) (50 ng/mL divided by 162) = 0.309 = 3.1 × 10−7 M [9]. This concentration is 3.6 times lesser than the lethal dose. 60 mg dose means a 0.8 mg/kg in humans equivalent approximately to the amount found in five cigarettes. |
| Effects induced by Nicotine in Human Airway Epithelial Cells [References] | Effects Induced by SARS-CoV-2 [References] | Effects Induced by SARS-CoV or MERS-CoV [References] | Effects Induced by Non-Tumorigenic Virus Infection [References] | |
|---|---|---|---|---|
| α7-nAChR Down-Stream Pathways | ||||
| α7-nAChR | Increase in HBEpC and A549 cells [35,47] | Silico experiments show the possibility that SARS-CoV-2 may interact with nAChR [67,68]. Nemantine, α7-nAChR antagonist, decreases ACE2 and reduces oxidative stress and inflammation. Nemantine may reduce SARS-CoV-2 virulence [69]. | HIV-gp120 induces and regulates mucus formation on airway epithelial cells through a CXCR4-α7-nAChR-GABAAR dependent pathway. Nicotine enhances production of HIV of in vitro-infected alveolar macrophages from healthy cigarette smokers. Nicotine-treated microglia show increased HIV-1 expression in a concentration dependent manner In neuronal cells the α7-nAChR is upregulated after gp120 exposure (Reviewed in [70]). | |
| ACE2 | Increase in HBEpC and A549 cells [35,47] Correlation between α7-nAChR and ACE2 [36] and reviewed in [71] | Bioinformatics modeling and in vitro experiments indicate that SARS-CoV-2 utilizes ACE2 as a receptor to gain entry into human cells [39]. | It is the entry receptor for SARS-CoV and HCoV-NL63 [72]. The expression of ACE2 is increased 24 h after SARS-CoV infection and remains at a high level after 48 h [73]. | |
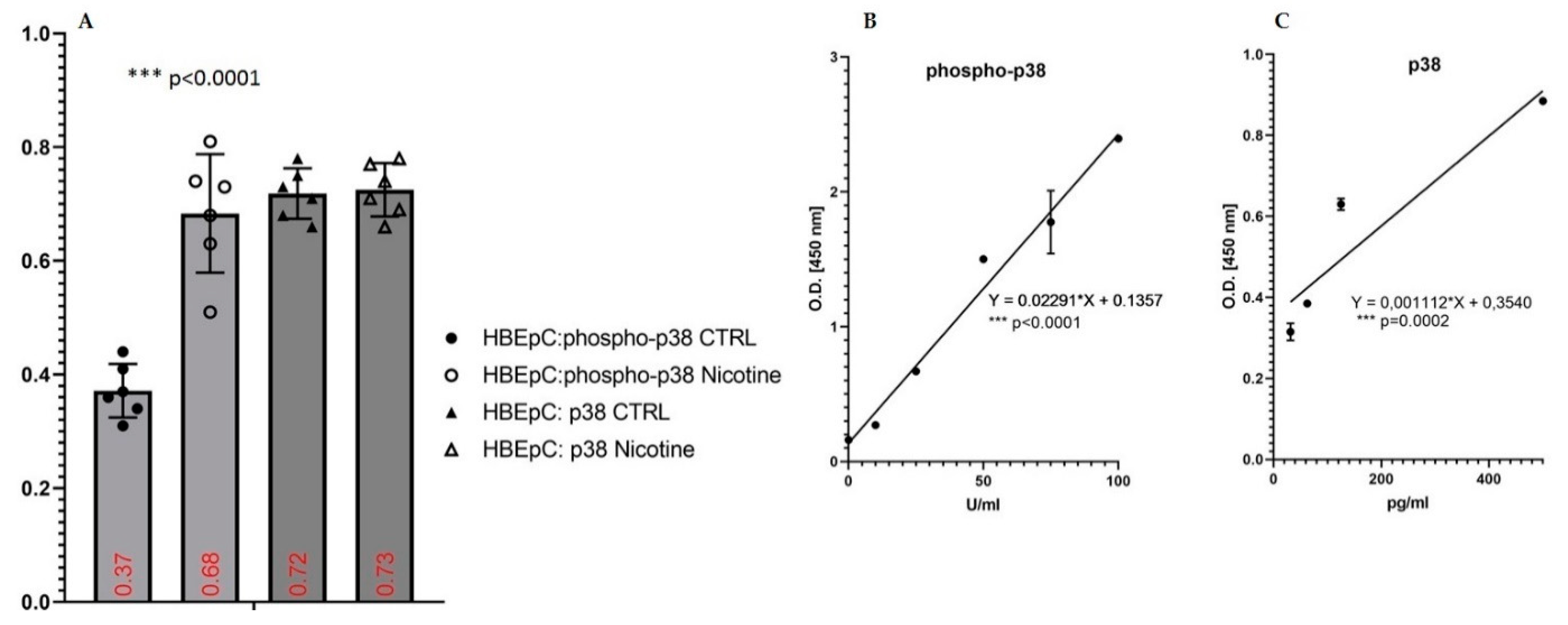
| ERK/MAPK, Phospho-p38 | Increase in HBEpC (this work and [35,47] and reviewed in [12]) | Activation of MAPK signaling [37] Activation of the p38/MAPK pathway during SARS-CoV-2 infection in ACE2-A549 and NHBE cells demonstrated that transcription factors regulated by the p38/MAPK pathway were among the most highly activated upon infection [74] SARS-CoV-2 activates p38 MAPK activity via suppression of NF-κB signaling pathway [75]. | SARS-CoV was shown to express proteins that directly upregulate p38 MAPK in vitro [76]. | |
| Cell Proliferation and Cell Cytotoxicity | ||||
| Cell proliferation | Increase in HBEpC and A549 cells (this work and [35,47] and reviewed in [12,77]) | Rapidly replicates in actively transcriptional cells causing major readjustments in cellular functions, including splicing, proteostasis and nucleotide biosynthesis [78]. Replication depends on the availability of cellular nucleotide pools [79]. Interactions between SARS-CoV-2 proteins and human proteins that are involved in several complexes and biological processes including DNA replication (NSP1) [80]. SARS-CoV-2 infects epithelial ACE2- and TMPRSS2-positive cells in the aero digestive and respiratory tracts that are metabolically-primed for glutamine synthesis and rapid replication [81]. | ||
| Cell Cytotoxicity | Decrease in HBEpC and A549 cells (this work and [35,47]) | |||
| Doubling time | Decrease in HBEpC and A549 cells (this work and[35,47]) | A significant increase in the fraction of cells in S phase and at the G2/M transition and a decrease in the fraction of cells in G0/G1 phase were observed [74]. | ||
| Ki67 | Increase in HBEpC (this work) | |||
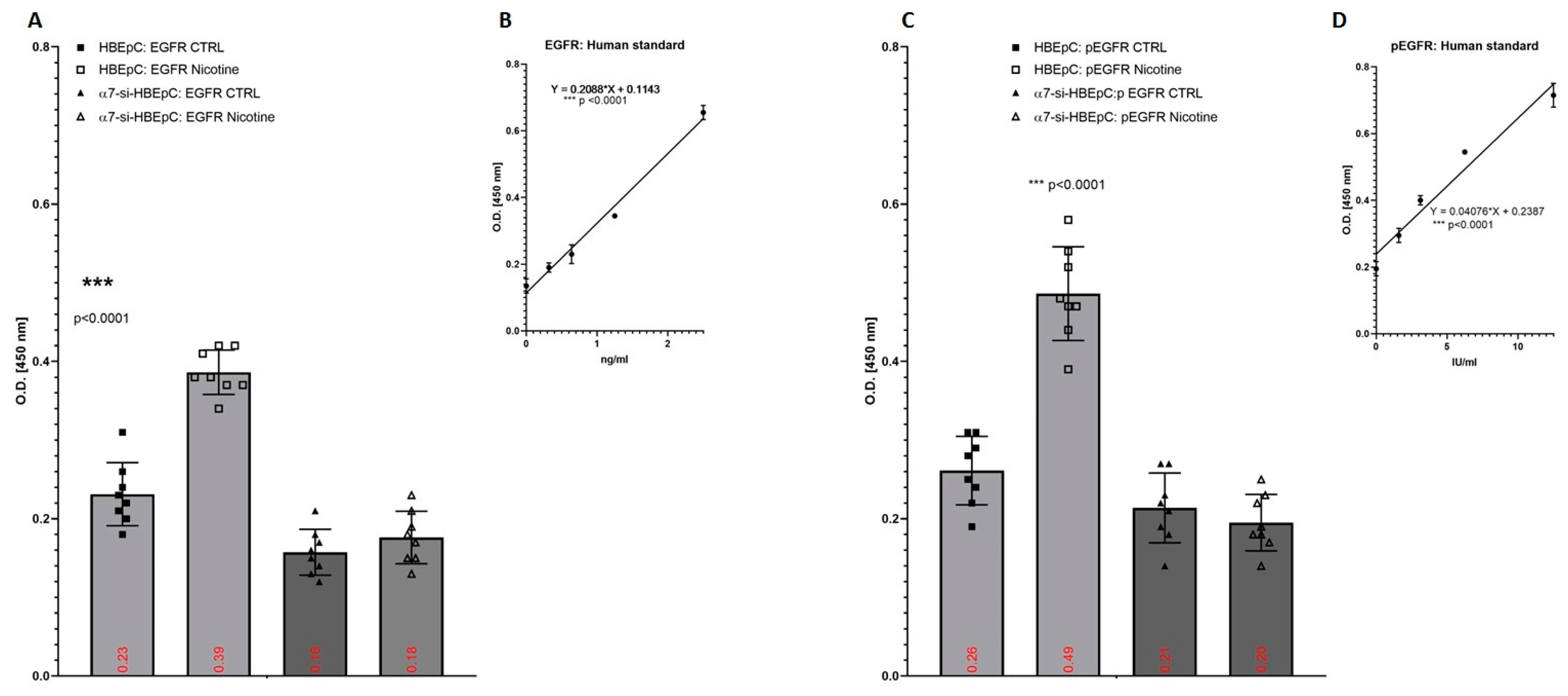
| EGFR/pEGFR | Increase in HBEpC (this work and [82,83]) | Pathways of EGFR is influenced by SARS-CoV-2 [84]. | In human bronchial epithelial cells exposed to SARS-CoV, for 12, 24, and 48 h the expression of EGFR gene is high 12 h after the infection and then decreases after 24–48 h [85]. | |
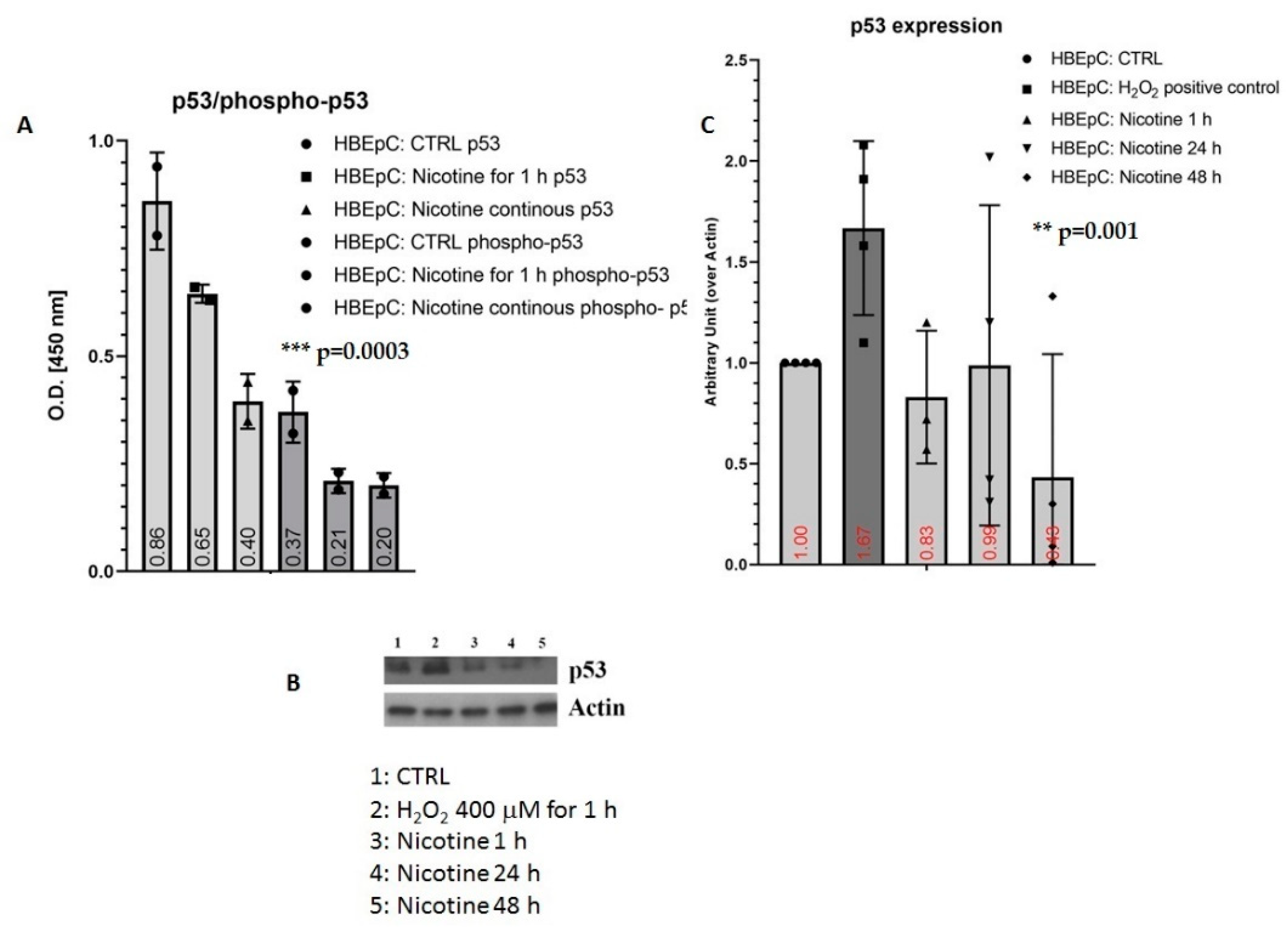
| p53/phospho-p53 | Decrease in HBEpC and A549 cells (this work and [35,47,86,87]) | p53 works as an anti-viral factor inhibiting viral replication thus in cells lacking p53 the rate of virus replication is higher than in cells expressing p53 [88]. | IAV infection is strictly p53 dependent: in p53-deficient mice IAV induces higher mortality, and higher viral load in the lungs than in p53 counterparts. Knockdown of p53 expression by RNA interference enhances IAV replication. All data suggest that the absence of p53 may delay the innate response, causing severe IAV-induced morbidity as observed in the p53KO mice (Reviewed in [89]) | |
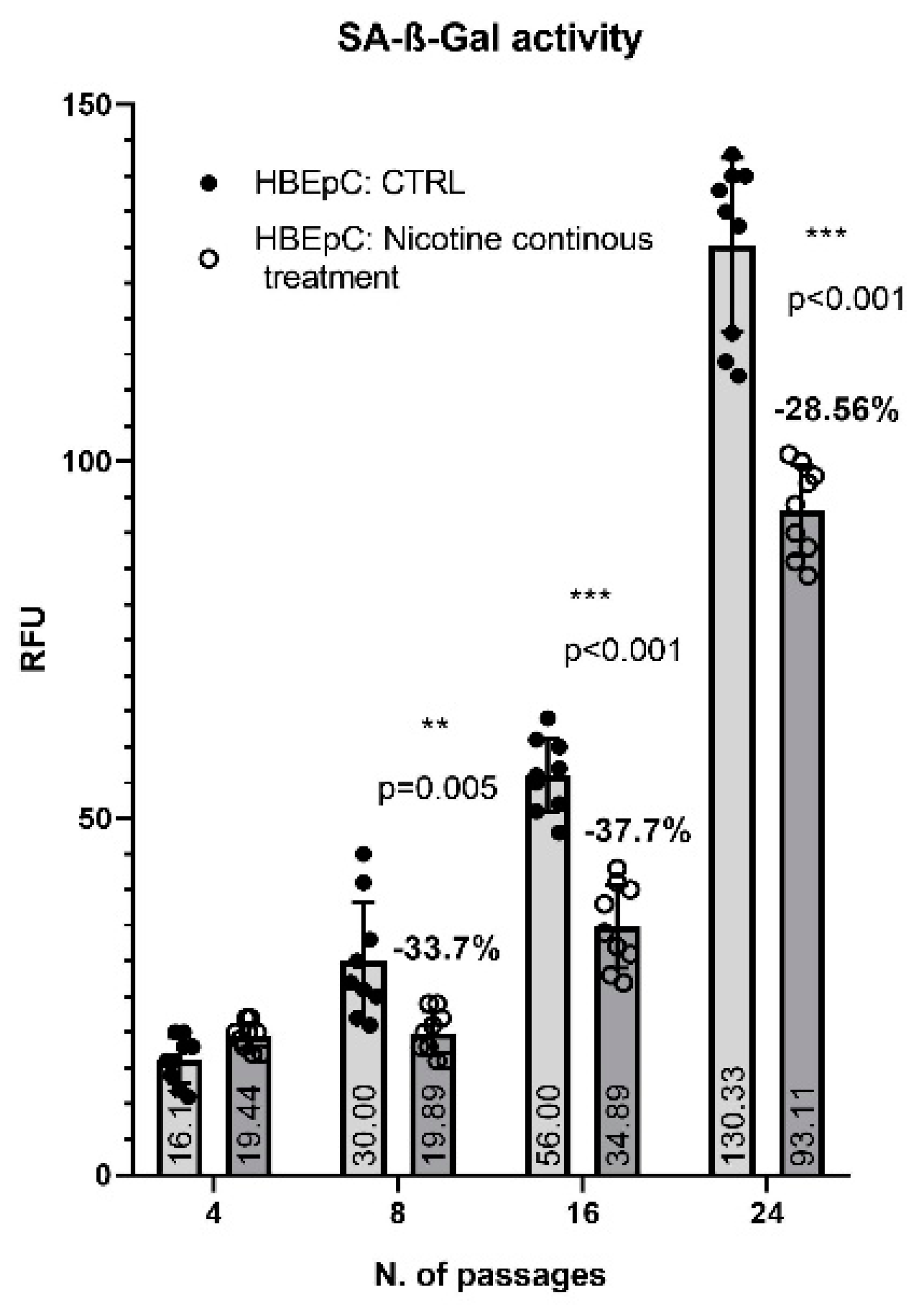
| SA-β-Gal activity | Delay in senescence (this work) | |||
| Mitochondrial Dysfunction | ||||
| Induction in HBEpC (this work and [90,91,92]) | Down regulation of genes in the mitochondrial and electron transport chain processes. Similar alterations are observed in infected human nasopharyngeal samples, used as control [93]. | |||
| ATP | Decrease in HBEpC (this work and [92]) | |||
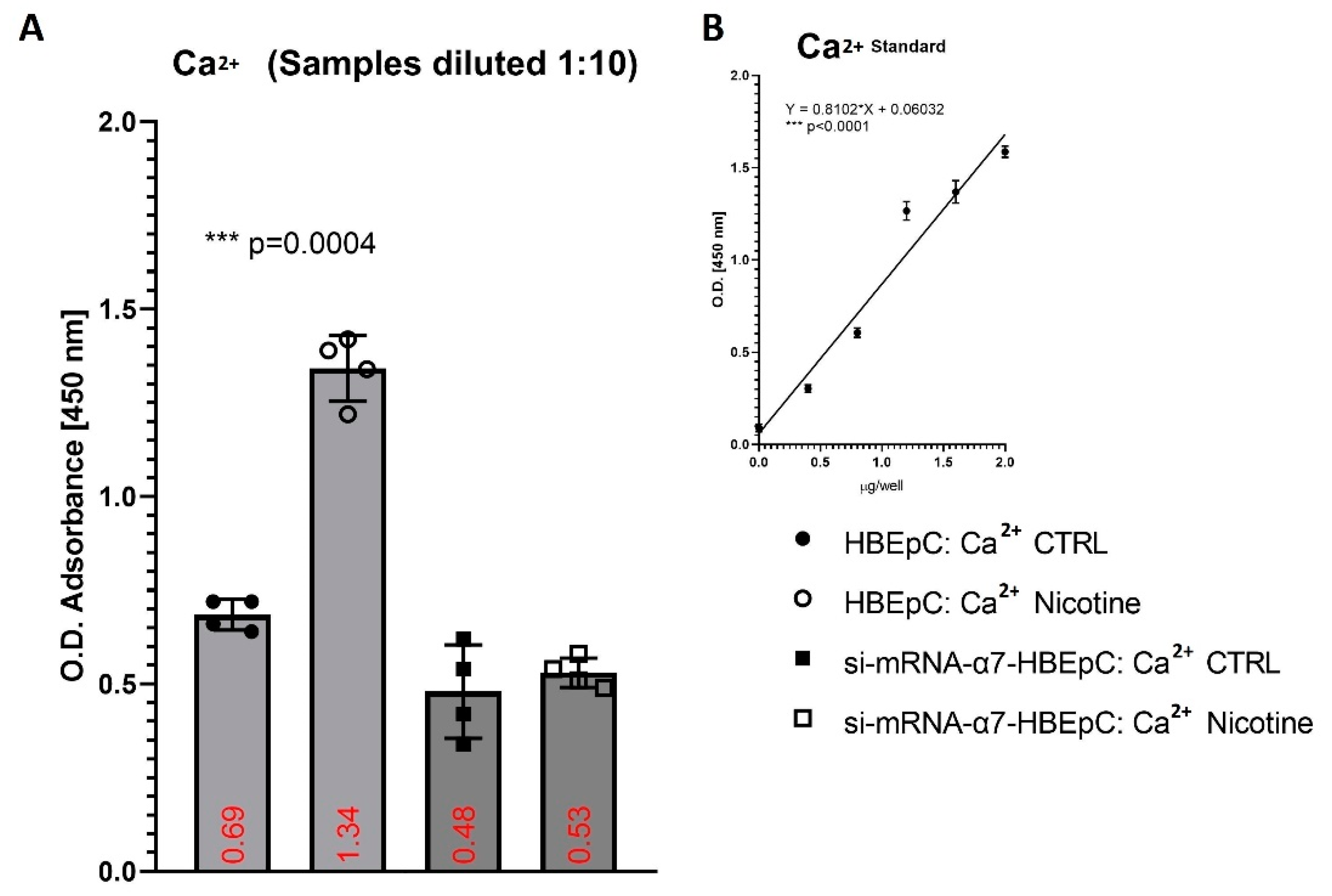
| Ca2+ | Increase basal Ca2+ in HBEpC (this work and [94,95]) | Over a two-fold increase in intracellular Ca2+ enhances the enter ability of the MERS-CoV, in the absence of Ca2+ fusion is attenuated, but not completely abrogated [96]. Depletion of intracellular Ca2+ completely abrogates SARS-CoV host cell entry [97]. | High levels of cytosolic Ca2+ concentrations and influx of Ca2+ into mitochondria sustain viral replication of different respiratory viruses such as RSV, MV and RV [97,98]. | |
| EMT | ||||
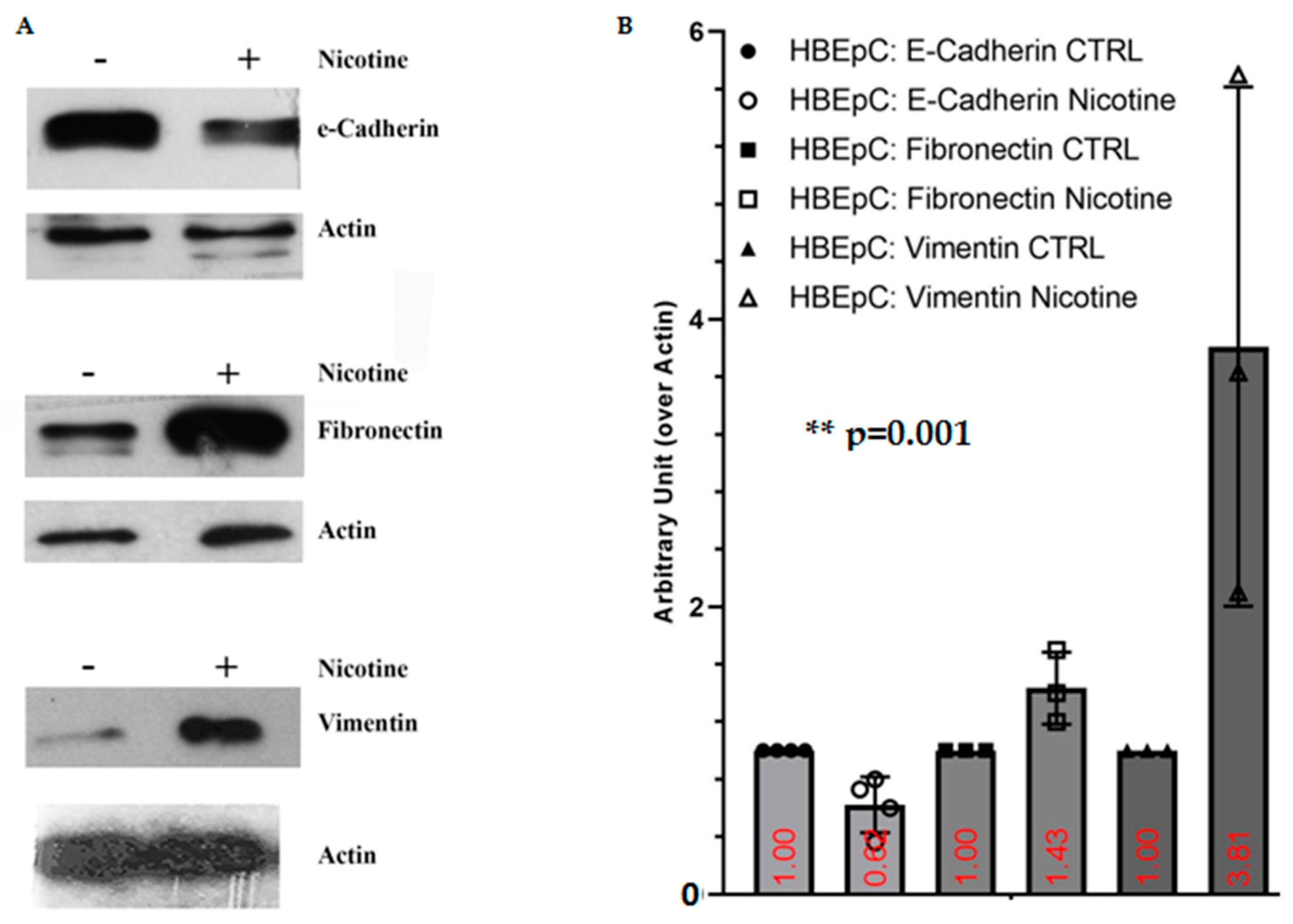
| Nicotine induced EMT in different epithelial cells (Reviewed in [12,83] and [99]) | SARS-CoV-2 infection shifts cells to a more mesenchymal phenotype, which is confirmed by downregulation of EPCAM expression following viral infection [81]. | |||
| E-Cadherin | Decrease of E-cadherin (this work and [99,100]) | |||
| Fibronectin | Increase FN (this work and [101,102]) | Bioinformatics analysis shows that FN interacts with ACE2, the mRNA and protein of this molecule are more expressed in lung epithelial cells after SARS-CoV-2 infection [103]. | ||
| Vimentin | Increase Vimentin (this work and [104]) | Important role for Vimentin in SARS-CoV virus entry through interaction with its S protein [105] in the SARS-CoV-permissive cell line Vero E6. In these cells the expression of extracellular Vimentin is upregulated after virus interaction and enhances its delivery to ACE2. | ||
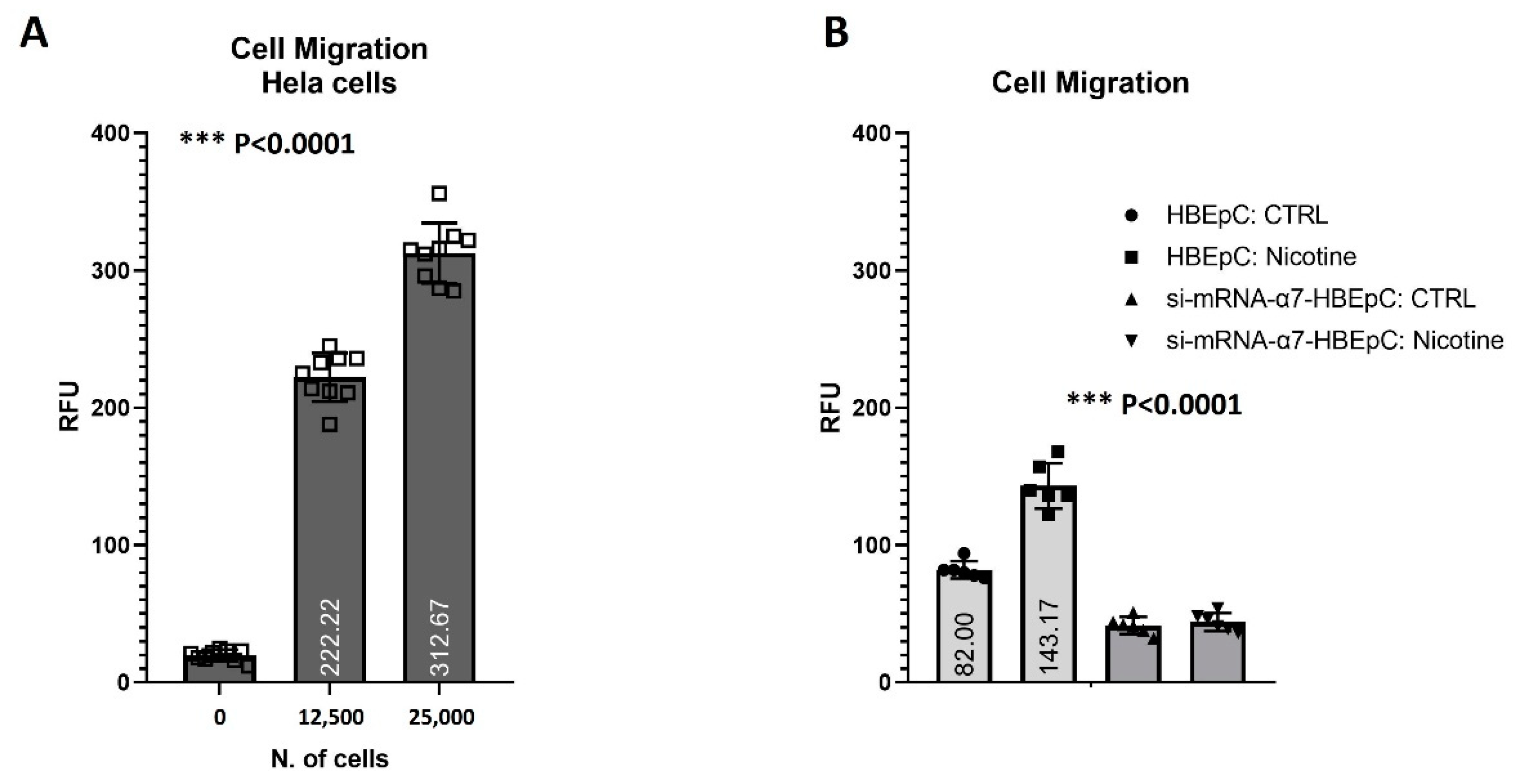
| Cell Migration | Increase cell migration (this work and [12]). | |||
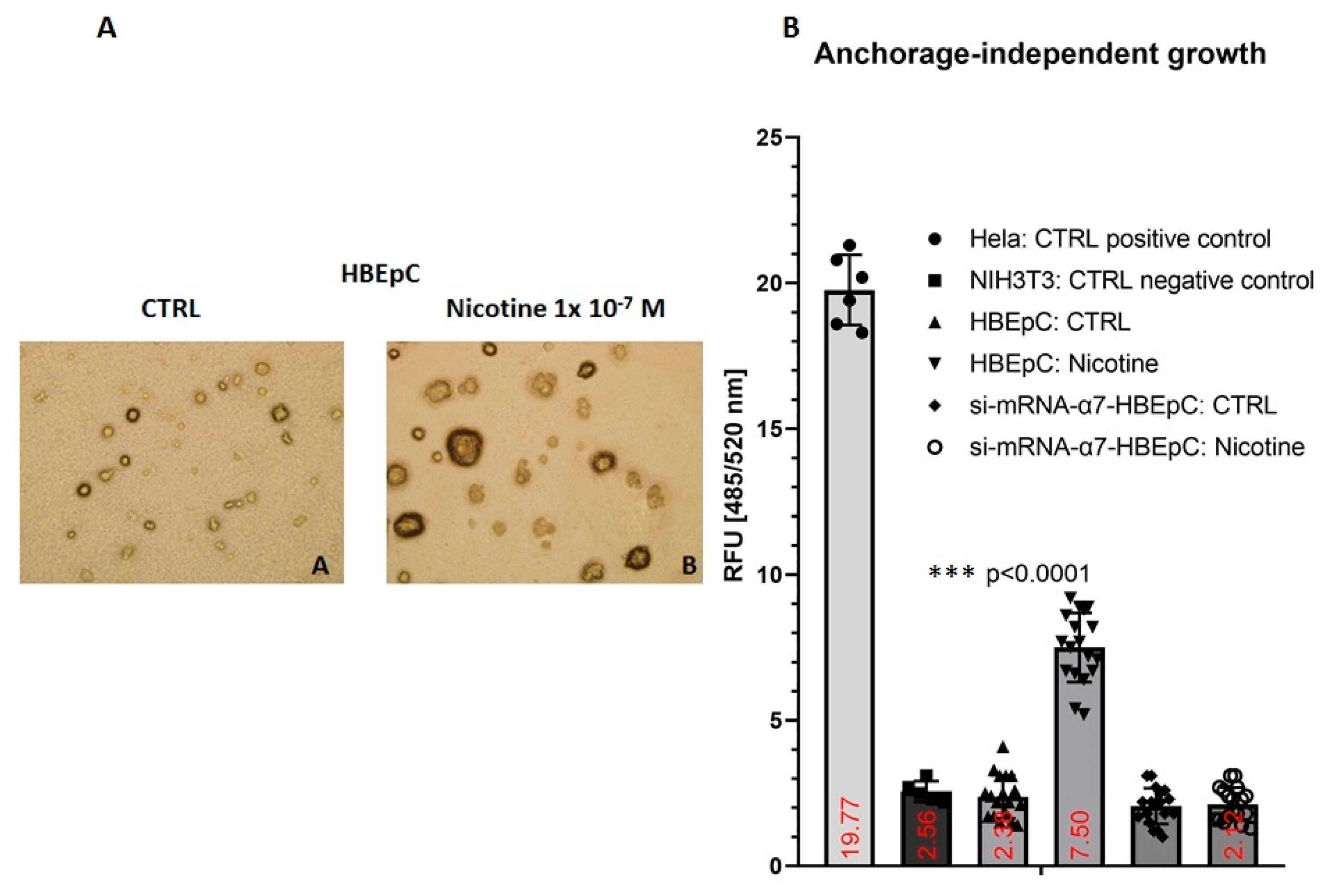
| Anchorage independent growth | Increase slowly (this work and [106]) | |||
| Neo-Angiogenesis | ||||
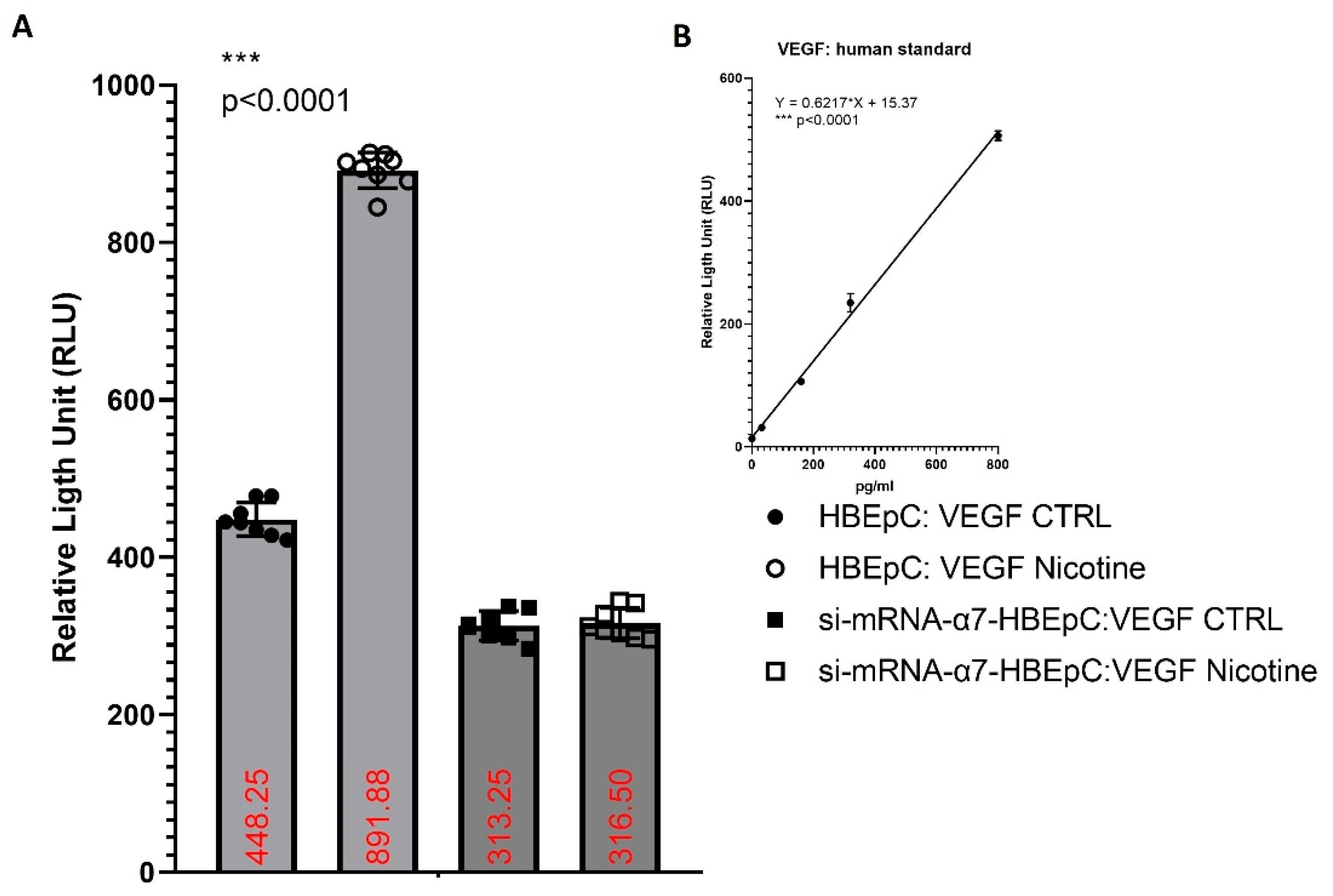
| Increase (this work and [83,106]) | SARS-CoV-2 infection show the presence of IA as well as of conventional SA [107]. | Viruses, such as CMV or HCV may regulate angiogenesis directly or indirectly, may activate vessels through endothelial cell tropism and/or producing chemokines and/or growth factors (i.e., VEGF) creating a pro-angiogenic microenvironment [108]. | ||
| VEGF | Increase VEGF (this work and [83,109]) | Bioinformatics analysis shows that VEGF interacts with ACE2, the mRNA and protein of this molecule are more expressed in lung epithelial cells after SARS-CoV-2 infection [103]. | ||
| Effects Induced by Nicotine in SARS-CoV-2 Infection | ||||
| Increase SARS-CoV-2 replication in A549 cells [47]. | ||||
| Increase the transcription of SARS-CoV-2 viral proteins in A549 cells [47]. | ||||
| Increase SARS-CoV-2 cytopathic effect in A549 cells [47]. | ||||
Sample Availability: Samples of the compounds (nicotine) are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lupacchini, L.; Maggi, F.; Tomino, C.; De Dominicis, C.; Mollinari, C.; Fini, M.; Bonassi, S.; Merlo, D.; Russo, P. Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity. Molecules 2021, 26, 101. https://doi.org/10.3390/molecules26010101
Lupacchini L, Maggi F, Tomino C, De Dominicis C, Mollinari C, Fini M, Bonassi S, Merlo D, Russo P. Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity. Molecules. 2021; 26(1):101. https://doi.org/10.3390/molecules26010101
Chicago/Turabian StyleLupacchini, Leonardo, Fabrizio Maggi, Carlo Tomino, Chiara De Dominicis, Cristiana Mollinari, Massimo Fini, Stefano Bonassi, Daniela Merlo, and Patrizia Russo. 2021. "Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity" Molecules 26, no. 1: 101. https://doi.org/10.3390/molecules26010101
APA StyleLupacchini, L., Maggi, F., Tomino, C., De Dominicis, C., Mollinari, C., Fini, M., Bonassi, S., Merlo, D., & Russo, P. (2021). Nicotine Changes Airway Epithelial Phenotype and May Increase the SARS-COV-2 Infection Severity. Molecules, 26(1), 101. https://doi.org/10.3390/molecules26010101