Flexibility and Preorganization of Fluorescent Nucleobase-Pyrene Conjugates Control DNA and RNA Recognition

 , , , and
, , , and 
Abstract
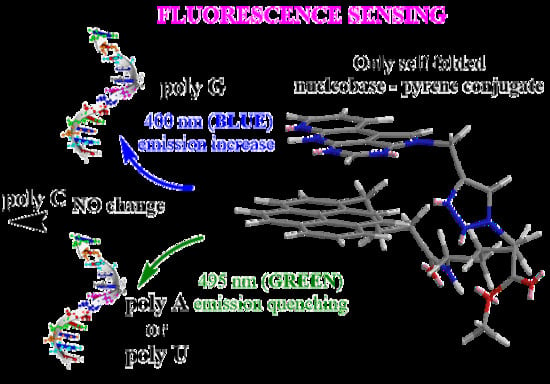
1. Introduction
2. Results and Discussion
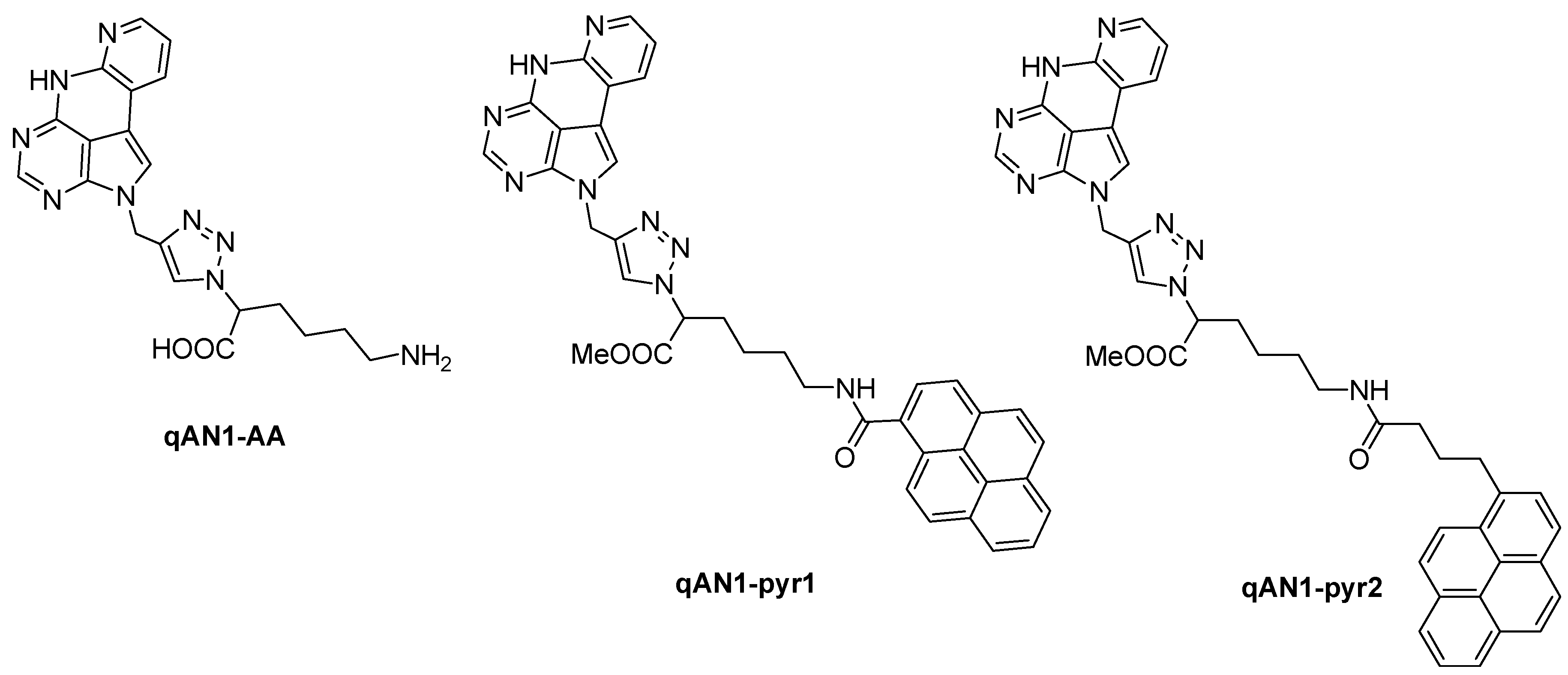
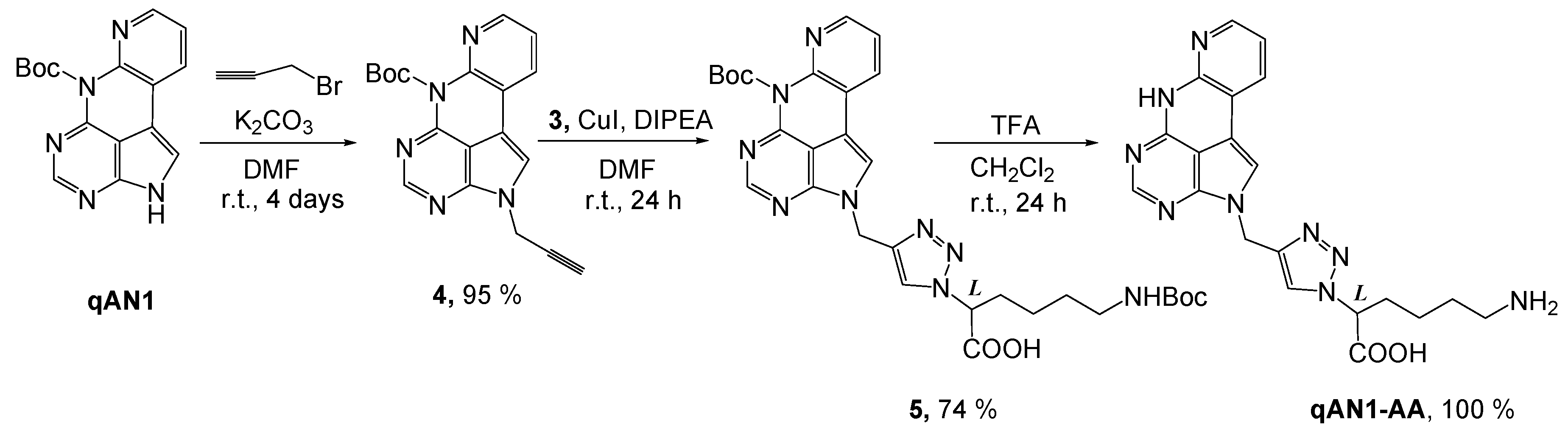
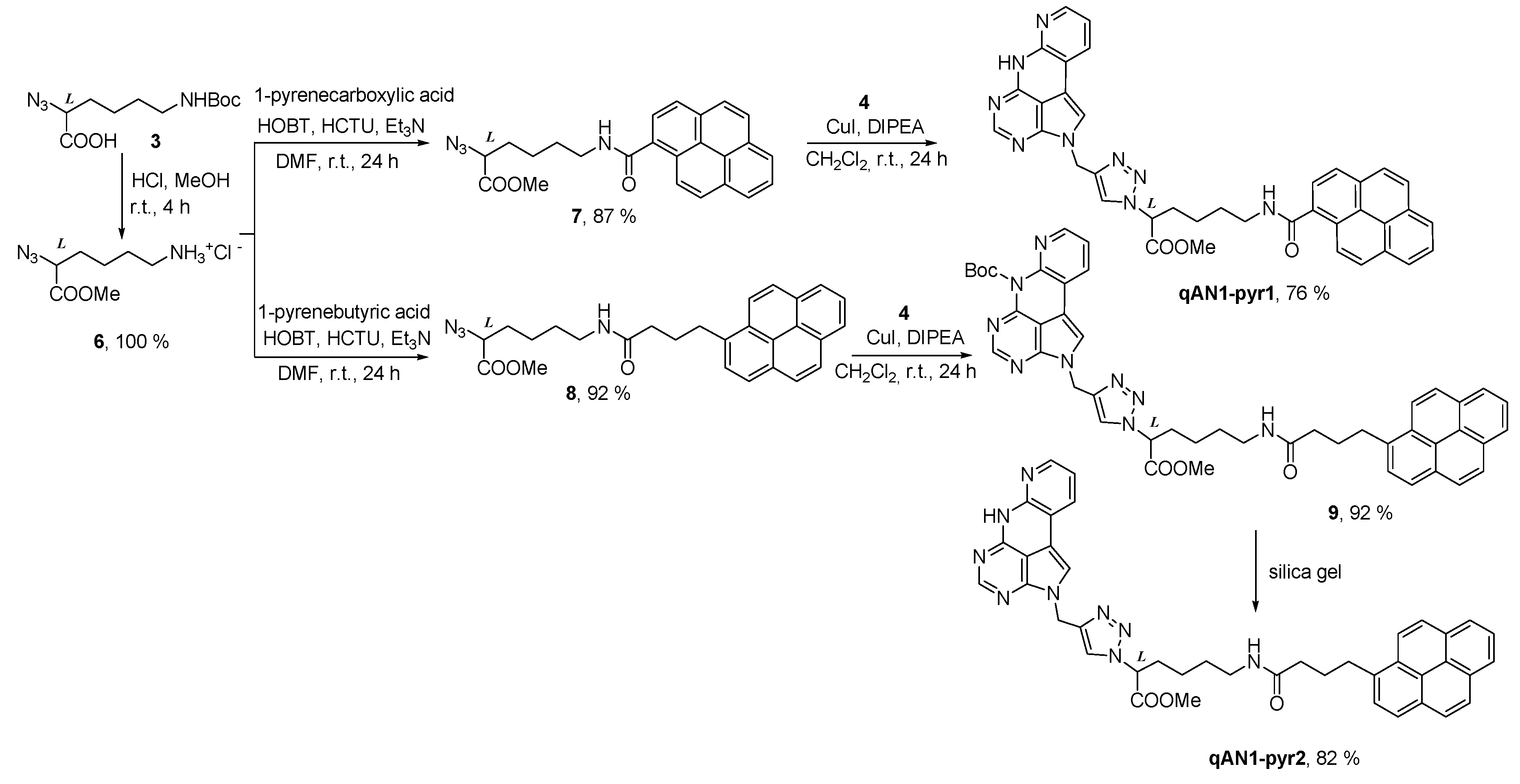
2.1. Chemistry
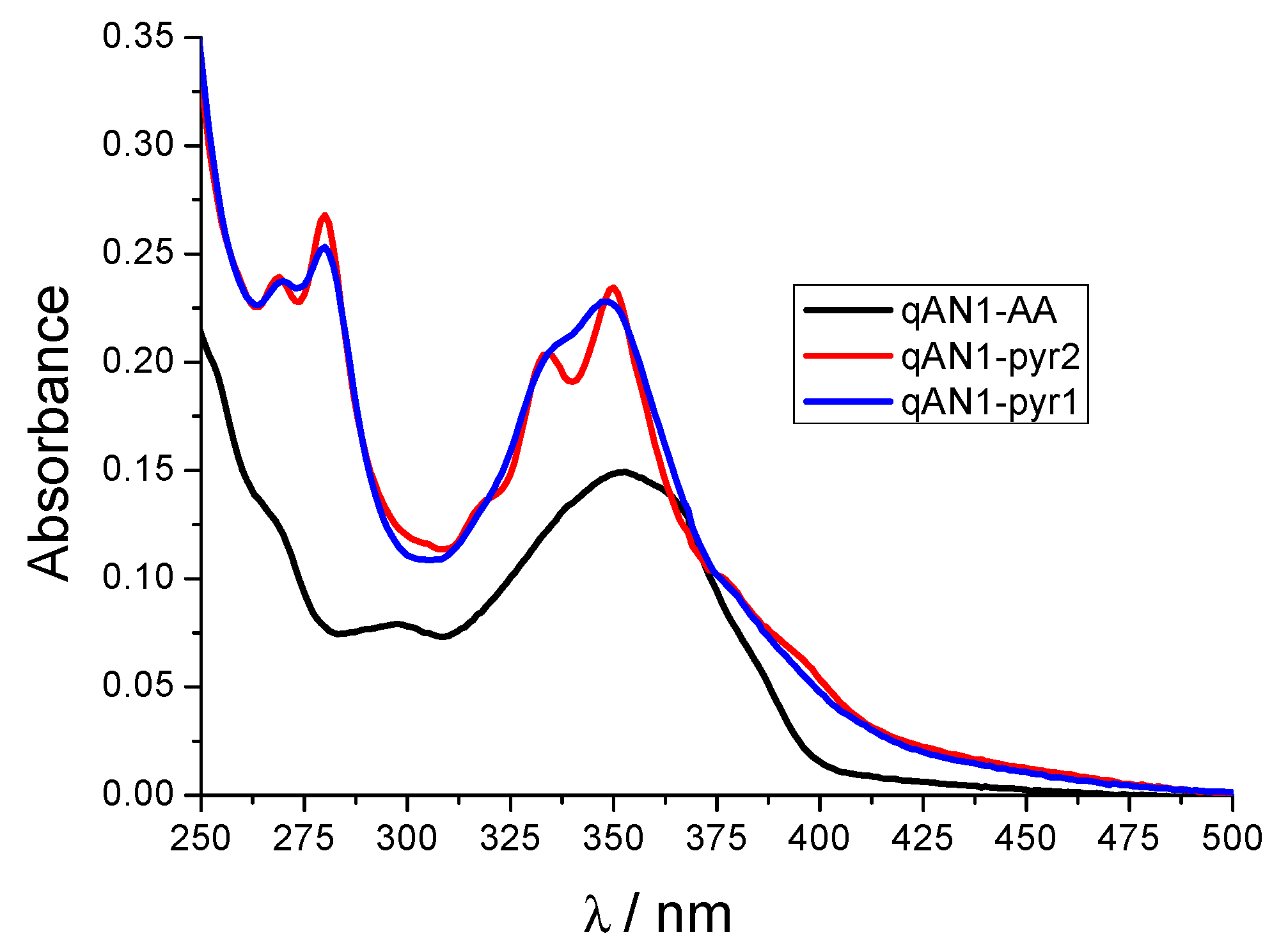
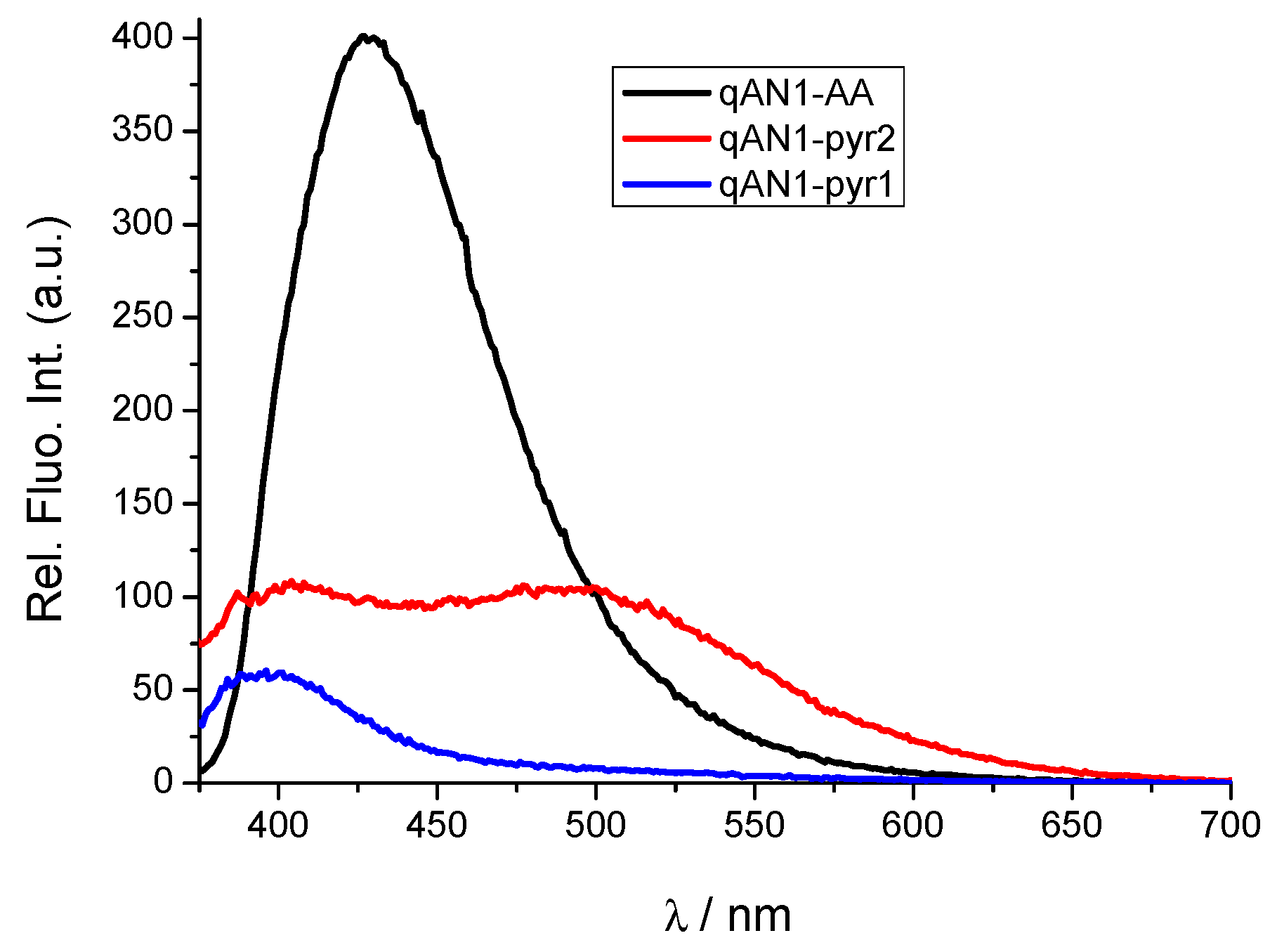
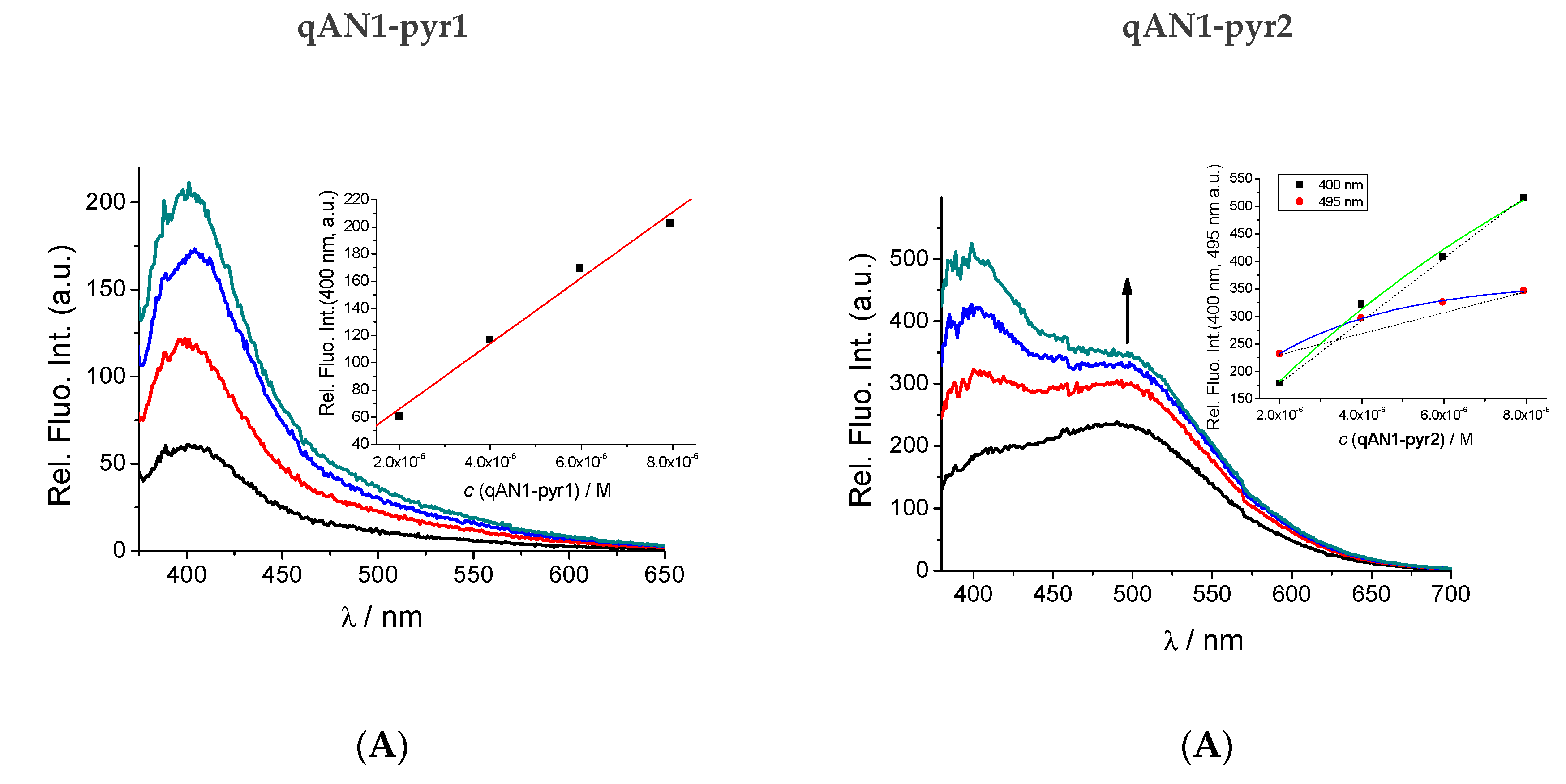
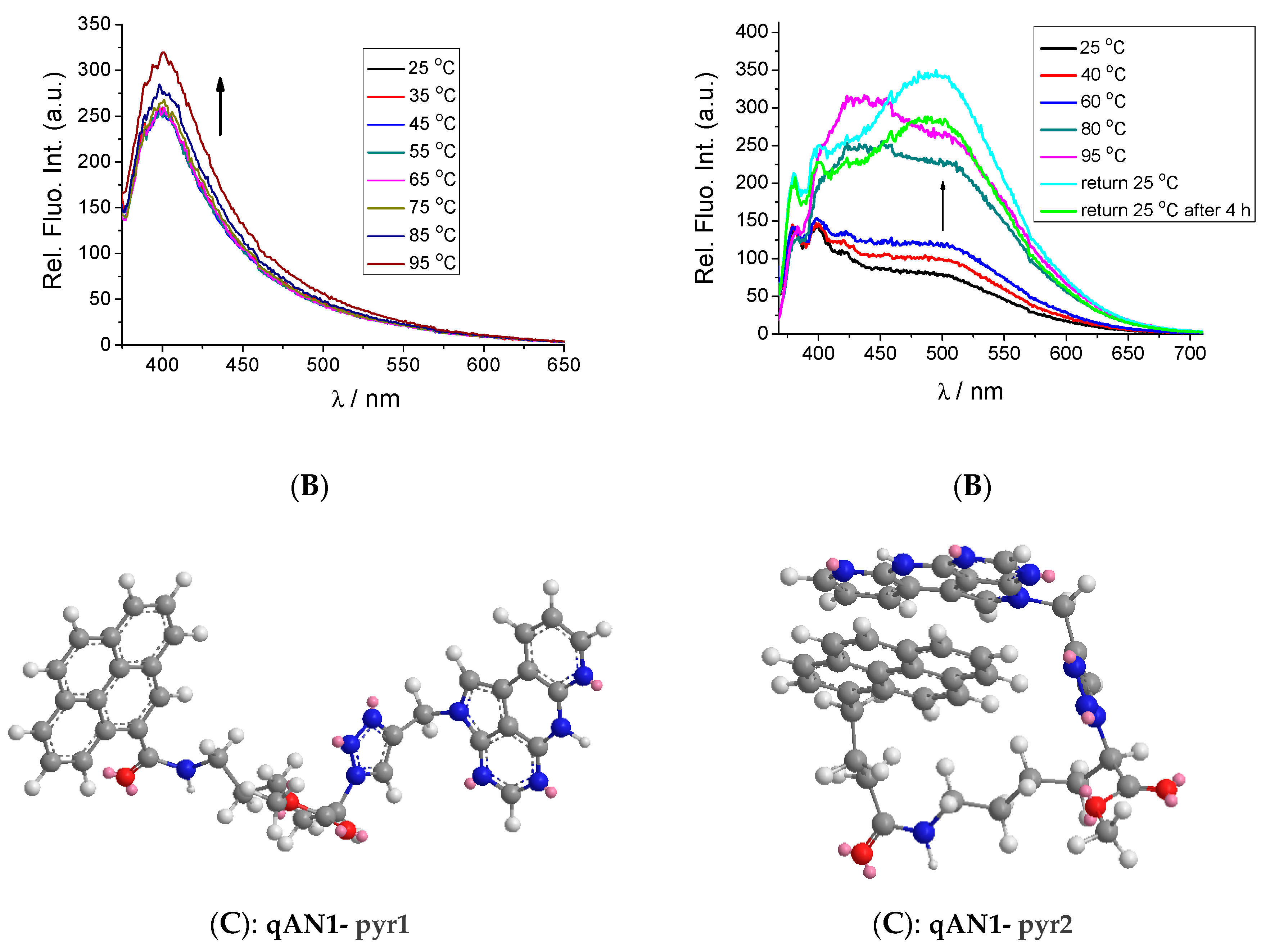
2.2. Characterization of qAN1-Derivatives in Aqueous Solutions
2.3. Interactions with ds-DNA, ds-RNA and ss-RNA
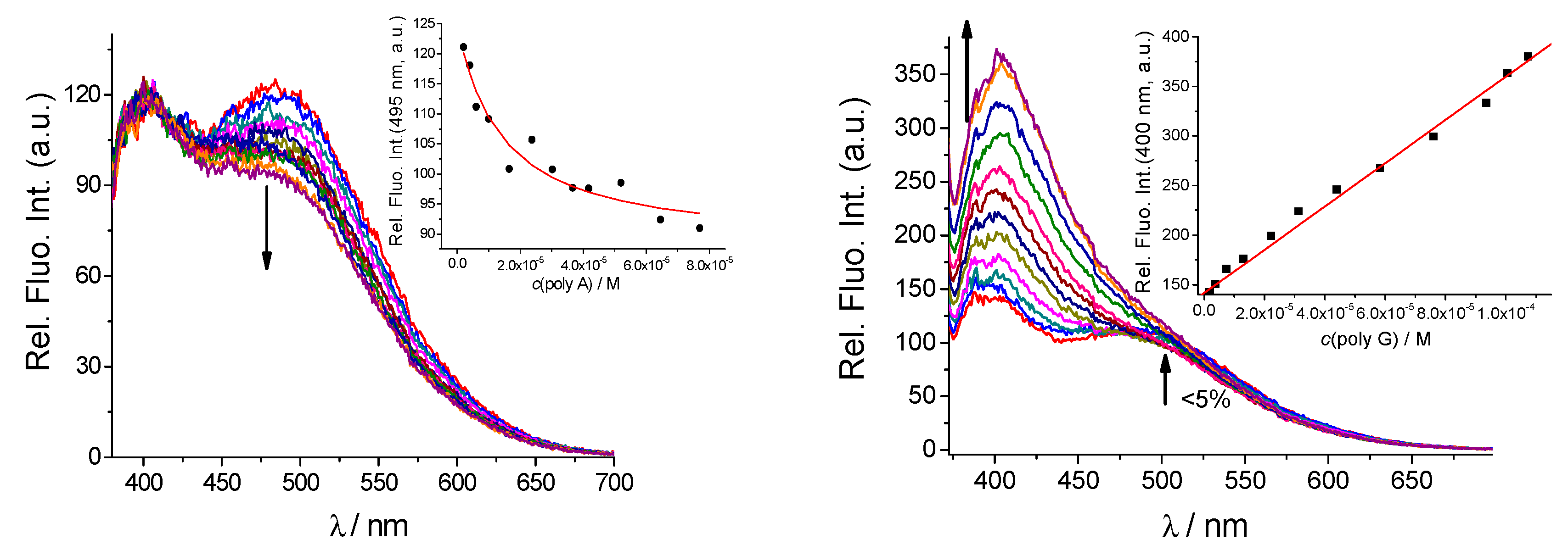
2.3.1. Fluorimetric Titrations
2.3.2. Circular Dichroism (CD) Experiments
3. Conclusions
4. Materials and Methods
4.1. General Information
4.2. Synthesis
4.3. Study of DNA/RNA Interactions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sinkeldam, R.W.; Greco, N.J.; Tor, Y. Fluorescent analogs of biomolecular building blocks: Design, properties, and applications. Chem. Rev. 2010, 110, 2579–2619. [Google Scholar] [CrossRef] [PubMed]
- Wilhelmsson, L.M. Fluorescent nucleic acid base analogues. Q. Rev. Biophys. 2010, 43, 159–183. [Google Scholar] [CrossRef] [PubMed]
- Matarazzo, A.; Hudson, R.H.E. Fluorescent adenosine analogs: A comprehensive survey. Tetrahedron 2015, 71, 1627–1657. [Google Scholar] [CrossRef]
- Xu, W.; Chan, K.M.; Kool, E.T. Fluorescent nucleobases as tools for studying DNA and RNA. Nat. Chem. 2017, 9, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Bood, M.; Sarangamath, S.; Wranne, M.S.; Grøtli, M.; Wilhelmsson, L.M. Fluorescent nucleobase analogues for base–base FRET in nucleic acids: Synthesis, photophysics and applications. Beilstein J. Org. Chem. 2018, 14, 114–129. [Google Scholar] [CrossRef]
- Burns, D.D.; Teppang, K.L.; Lee, R.W.; Lokensgard, M.E.; Purse, B.W. Fluorescence turn-on sensing of DNA duplex formation by a tricyclic cytidine analogue. J. Am. Chem. Soc. 2017, 139, 1372–1375. [Google Scholar] [CrossRef]
- Shin, D.; Sinkeldam, R.W.; Tor, Y. Emissive RNA alphabet. J. Am. Chem. Soc. 2011, 133, 14912–14915. [Google Scholar] [CrossRef]
- Rovira, A.R.; Fin, A.; Tor, Y. Chemical mutagenesis of an emissive RNA alphabet. J. Am. Chem. Soc. 2015, 137, 14602–14605. [Google Scholar] [CrossRef]
- Ward, D.C.; Reich, E.; Stryer, L. Fluorescence studies of nucleotides and polynucleotides. I. Formycin, 2-aminopurine riboside, 2,6-diaminopurine riboside, and their derivatives. J. Biol. Chem. 1969, 244, 1228–1237. [Google Scholar]
- Sandin, P.; Wilhelmsson, L.M.; Lincoln, P.; Powers, V.E.C.; Brown, T.; Albinsson, B. Fluorescent properties of DNA base analogue tC upon incorporation into DNA—Negligible influence of neighbouring bases on fluorescence quantum yield. Nucleic Acids Res. 2005, 33, 5019–5025. [Google Scholar] [CrossRef]
- Sandin, P.; Börjesson, K.; Li, H.; Mårtensson, J.; Brown, T.; Wilhelmsson, L.M.; Albinsson, B. Characterization and use of an unprecedentedly bright and structurally non-perturbing fluorescent DNA base analogue. Nucleic Acids Res. 2008, 36, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Börjesson, K.; Preus, S.; El-Sagheer, A.H.; Brown, T.; Albinsson, B.; Wilhelmsson, L.M. Nucleic acid base analog FRET-pair facilitating detailed structural measurements in nucleic acid containing systems. J. Am. Chem. Soc. 2009, 131, 4288–4293. [Google Scholar] [CrossRef] [PubMed]
- Dierckx, A.; Miannay, F.-A.; Gaied, N.B.; Preus, S.; Björck, M.; Brown, T.; Wilhelmsson, L.M. Quadracyclic adenine: A non-perturbing fluorescent adenine analogue. Chem. Eur. J. 2012, 18, 5987–5997. [Google Scholar] [CrossRef] [PubMed]
- Dumat, B.; Bood, M.; Wranne, M.S.; Lawson, C.P.; Larsen, A.F.; Preus, S.; Streling, J.; Gradén, H.; Wellner, E.; Grøtli, M.; et al. Second-generation fluorescent quadracyclic adenine analogues: Environment-responsive probes with enhanced brightness. Chem. Eur. J. 2015, 21, 403948. [Google Scholar] [CrossRef]
- Larsen, A.F.; Dumat, B.; Wranne, M.S.; Lawson, C.P.; Preus, S.; Bood, M.; Gradén, H.; Wilhelmsson, L.M.; Grøtli, M. Development of bright fluorescent quadracyclic adenine analogues: TDDFT-calculation supported rational design. Sci. Rep. 2015, 5, 12653–12664. [Google Scholar] [CrossRef]
- Wranne, M.S.; Füchtbauer, A.F.; Dumat, B.; Bood, M.; El-Sagheer, A.H.; Brown, T.; Gradén, H.; Grøtli, M.; Wilhelmsson, L.M. Toward complete sequence flexibility of nucleic acid base analogue FRET. J. Am. Chem. Soc 2017, 139, 9271–9280. [Google Scholar] [CrossRef]
- Lakowicz, J.R. Principles of Fluorescence Spectroscopy; Kluwer Academic/Plenum: New York, NY, USA, 1999. [Google Scholar]
- Winnik, F.M. Photophysics of preassociated pyrenes in aqueous polymer solutions and in other organized media. Chem. Rev. 1993, 93, 587–614. [Google Scholar] [CrossRef]
- Lehrer, S.S. Intramolecular pyrene excimer fluorescence: A probe of proximity and protein conformational change. Methods Enzymol. 1997, 278, 286–295. [Google Scholar]
- Hernandez-Folgado, L.; Schmuck, C.; Tomic, S.; Piantanida, I. A novel pyrene-guanidiniocarbonyl-pyrrole cation efficiently differentiates between ds-DNA and ds-RNA by two independent, sensitive spectroscopic methods. Bioorg. Med. Chem. Lett. 2008, 18, 2977–2981. [Google Scholar] [CrossRef]
- Orehovec, I.; Glavac, D.; Dokli, I.; Gredicak, M.; Piantanida, I. Impact of the supramolecular organisation of pyrene—Quinoline conjugates on their Interaction with ds—DNA. Croat. Chem. Acta 2017, 90, 603–611. [Google Scholar] [CrossRef]
- Kolb, H.C.; Sharpless, K.B. The growing impact of click chemistry on drug discovery. Drug Discov. Today 2003, 8, 1128–1137. [Google Scholar] [CrossRef]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. CuI-catalyzed alkyne–azide “click” cycloadditions from a mechanistic and synthetic perspective. Eur. J. Org. Chem. 2006, 2006, 51–68. [Google Scholar] [CrossRef]
- Huisgen, R. 1,3-dipolar cycloadditions. Past and future. Angew. Chem. Int. Edit. 1963, 2, 565–598. [Google Scholar] [CrossRef]
- Padwa, A. (Ed.) 1,3-Dipolar Cycloaddition Chemistry; Wiley: New York, NY, USA, 1984; pp. 1–176. [Google Scholar]
- Trost, B.M. (Ed.) Comprehensive Organic Synthesis; Pergamon: Oxford, UK, 1991; Volume 4, pp. 1069–1109. [Google Scholar]
- Moorhouse, A.D.; Santos, A.M.; Gunaratnam, M.; Moore, M.; Neidle, S.; Moses, J.E. Stabilization of G-quadruplex DNA by highly selective ligands via click chemistry. J. Am. Chem. Soc. 2006, 128, 15972–15973. [Google Scholar] [CrossRef]
- Lee, L.V.; Mitchell, M.L.; Huang, S.-J.; Fokin, V.V.; Sharpless, K.B.; Wong, C.-H. A potent and highly selective inhibitor of human alpha-1,3-fucosyltransferase via click chemistry. J. Am. Chem. Soc. 2003, 125, 9588–9589. [Google Scholar] [CrossRef]
- Wu, P.; Feldman, A.K.; Nugent, A.K.; Hawker, C.J.; Scheel, A.; Voit, B.; Pyun, J.; Frechet, J.M.J.; Sharpless, K.B.; Fokin, V.V. Efficiency and fidelity in a click-chemistry route to triazole dendrimers by the copper(I)-catalyzed ligation of azides and alkynes. Angew. Chem. Int. Edit. 2004, 43, 3928–3932. [Google Scholar] [CrossRef]
- Wu, P.; Malkoch, M.; Hunt, J.N.; Vestberg, R.; Kaltgrad, E.; Finn, M.G.; Fokin, V.V.; Sharpless, K.B.; Hawker, C. Multivalent, bifunctional dendrimers prepared by click chemistry. J. Chem. Commun. 2005, 46, 5775–5777. [Google Scholar] [CrossRef]
- Rozkiewicz, D.I.; Janczewski, D.; Verboom, W.; Ravoo, B.J.; Reinhoudt, D.N. “Click” chemistry by microcontact printing. Angew. Chem. Int. Edit. 2006, 45, 5292–5296. [Google Scholar] [CrossRef]
- Wang, Q.; Chan, T.R.; Hilgraf, R.; Fokin, V.V.; Sharpless, K.B.; Finn, M.G. Bioconjugation by copper(I)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 3192–3193. [Google Scholar] [CrossRef]
- Speers, A.E.; Adam, G.C.; Cravatt, B.F. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3 + 2] cycloaddition. J. Am. Chem. Soc. 2003, 125, 4686–4687. [Google Scholar] [CrossRef]
- Speers, A.E.; Cravatt, B.F. Profiling enzyme activities in vivo using click chemistry methods. Chem. Biol. 2004, 11, 535–546. [Google Scholar] [CrossRef] [PubMed]
- Burley, G.A.; Gierlich, J.; Mofid, M.R.; Nir, H.; Tal, S.; Eichen, Y.; Carell, T. Directed DNA metallization. J. Am. Chem. Soc. 2006, 128, 1398–1399. [Google Scholar] [CrossRef] [PubMed]
- Chittepu, P.; Sirivolu, V.R.; Seela, F. Nucleosides and oligonucleotides containing 1,2,3-triazole residues with nucleobase tethers: Synthesis via the azide-alkyne ‘click’ reaction. Bioorg. Med. Chem. 2008, 16, 8427–8439. [Google Scholar] [CrossRef] [PubMed]
- Efthymiou, T.; Gong, W.; Desaulniers, J.-P. Chemical architecture and applications of nucleic acid derivatives containing 1,2,3-triazole functionalities synthesized via click chemistry. Molecules 2012, 17, 12665–12703. [Google Scholar] [CrossRef] [PubMed]
- Devi1, G.; Ganesh, K.N. Synthesis of TzNA oligomers by “click” reaction on solid phase and stabilization of derived triplexes with DNA. Artif. DNA PNA XNA 2010, 1, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Ban, Ž.; Žinić, B.; Vianello, R.; Schmuck, C.; Piantanida, I. Nucleobase–guanidiniocarbonyl-pyrrole conjugates as novel fluorimetric sensors for single stranded RNA. Molecules 2017, 22, 2213. [Google Scholar] [CrossRef]
- Piecyk, K.; Lukaszewicz, M.; Darzynkiewicz, E.; Jankowska-Anyszka, M. Triazole-containing monophosphate mRNA cap analogs as effective translation inhibitors. RNA 2018, 20, 1539–1547. [Google Scholar] [CrossRef]
- Goddard-Borger, E.D.; Stick, R.V. An efficient, inexpensive, and shelf-stable diazotransfer reagent: Imidazole-1-sulfonyl azide hydrochloride. Org. Lett. 2007, 9, 3797–3800. [Google Scholar] [CrossRef]
- Dourtoglou, V.; Gross, B.; Lambropoulou, V.; Zioudrou, C. O-benzotriazolil-N,N,N′,N′-tetramethyluronium hexafluorophosphate as coupling reagent for the synthesis of peptides of biological interest. Synthesis 1984, 7, 572–574. [Google Scholar] [CrossRef]
- Ferrara, C.G.; Chara, O.; Grigera, J.R. Aggregation of non-polar solutes in water at different pressures and temperatures: The role of hydrophobic interaction. J. Chem. Phys. 2012, 137, 135104. [Google Scholar] [CrossRef]
- Scatchard, G. The attractions of proteins for small molecules and ions. Ann. N.Y. Acad. Sci. 1949, 51, 660–672. [Google Scholar] [CrossRef]
- McGhee, J.D.; Hippel, P.H.V. Theoretical aspects of DNA-protein interactions: Co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol. 1974, 86, 469–489. [Google Scholar] [CrossRef]
- Mergny, J.L.; Lacroix, L. Analysis of thermal melting curves. Oligonucleotides 2003, 13, 515–537. [Google Scholar] [CrossRef] [PubMed]
- Gröger, K.; Baretić, D.; Piantanida, I.; Marjanović, M.; Kralj, M.; Grabar, M.; Tomić, S.; Schmuck, C. Guanidiniocarbonyl-pyrrole-aryl conjugates as nucleic acid sensors: Switch of binding mode and spectroscopic responses by introducing additional binding sites into the linker. Org. Biomol. Chem. 2011, 9, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Weil, J.; Min, T.; Yang, C.; Wang, S.; Sutherland, C.; Sinha, N.; Kang, C. Stabilization of thei-motif by intramolecular adenine-adenine-thymine base triple in thestructure of d(ACCCT). Acta Cryst. 1999, 55, 422–429. [Google Scholar]
- Rodger, A.; Norden, B. Circular Dichroism and Linear Dichroism; Oxford University Press: New York, NY, USA, 1997; Chapter 2. [Google Scholar]
- Šmidlehner, T.; Piantanida, I.; Pescitelli, G. Polarization spectroscopy methods in the determination of interactions of small molecules with nucleic acids—Tutorial. Beilstein J. Org. Chem. 2018, 14, 84–105. [Google Scholar] [CrossRef]
- Demeunynck, M.; Bailly, C.; Wilson, W.D. Small Molecule DNA and RNA Binders: From Synthesis to Nucleic Acid Complexes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2004. [Google Scholar]
- Cantor, C.R.; Schimmel, P.R. Biophysical Chemistry; WH Freeman and Co.: San Francisco, CA, USA, 1980; Volume 3. [Google Scholar]
- Egli, M.; Saenger, W. Principles of Nucleic Acid Structure; Springer: New York, NY, USA, 1983. [Google Scholar]
- Martelli, A.; Constant, J.-F.; Demeunynck, M.; Lhomme, J.; Dumy, P. Design of site specific DNA damaging agents for generation of multiply damaged sites. Tetrahedron 2002, 58, 4291–4298. [Google Scholar] [CrossRef]
- Grabar Branilović, M.; Tomić, S.; Tumir, L.-M.; Piantanida, I. The bis-phenanthridinium system flexibility and position of covalently bound uracil finely tunes the interaction with polynucleotides. Mol. Biosyst. 2013, 9, 2051–2062. [Google Scholar] [CrossRef]
- Saftić, D.; RadićStojković, M.; Žini#x107;, B.; Glavaš-Obrovac, L.; Jukić, M.; Piantanida, I.; Tumir, L.M. Impact of linker between triazolyl-uracil and phenanthridine on recognition of DNA and RNA. Recognition of uracil—Containing RNA. New J. Chem. 2017, 41, 13240–13252. [Google Scholar]
- Tumir, L.M.; Šupljika, F.; Piantanida, I. Bisphenanthridinium—Adenine conjugates as fluorescent and CD reporters for fine structural differences in ds-DNA/RNA and ss-RNA structures. Supramol. Chem. 2016, 28, 267–274. [Google Scholar] [CrossRef]
- Ban, Ž.; Žinić, B.; Matković, M.; Tomašić Paić, A.; Crnolatac, I.; Piantanida, I. Pyrrolocytosine-pyrene conjugates as fluorescent and CD probes for the fine sensing of ds-polynucleotide secondary structure and specific recognition of poly G. New J. Chem. 2019, 43, 8204–8214. [Google Scholar] [CrossRef]
- Chaires, J.B.; Dattagupta, N.; Crothers, D.M. Studies on interaction of anthracycline antibiotics and deoxyribonucleic acid: Equilibrium binding studies on interaction of daunomycin with deoxyribonucleic. Biochemistry 1982, 21, 3933–3940. [Google Scholar] [CrossRef] [PubMed]
- Tumir, L.-M.; Piantanida, I.; Novak, P.; Žinić, M. Interactions of novel phenanthridinium-nucleobase conjugates with complementary and non-complementary nucleotides in aqueous media. J. Phys. Org. Chem. 2002, 15, 599–607. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
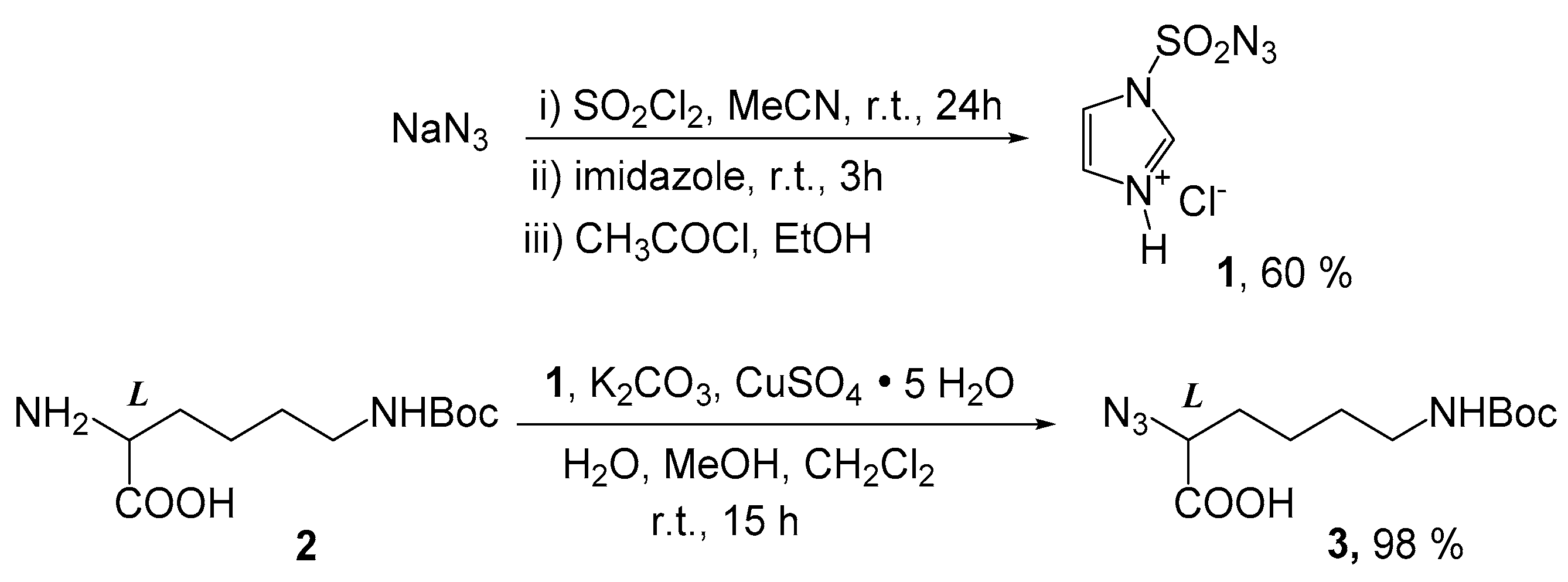{kind=link}
{kind=link}
{kind=link}
| qAN1-AA logKs/I/I0 b | qAN1-pyr1 logKs/I/I0 b | qAN1-pyr2 logKs/I/I0 b | |
|---|---|---|---|
| ct-DNA | >0.95 d | 5.7/1.3 | 5.9/0.7 |
| poly (dAdT)2 | >0.95 d | 5.0/3.3 | 5.7/0.5 |
| poly A-poly U | 1 d | 5.5/1.3 | >0.95 d |
| poly G | >0.95 d | 5 e/1.1 | <3/>2 c |
| poly A | 1 d | 5 e/1.1 | 5.2/0.7 |
| poly C | 1 d | 5 e/1.2 | >0.95 d |
| poly U | 1 d | 5 e/1.1 | 4.7/0.6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ban, Ž.; Matić, J.; Žinić, B.; Foller Füchtbauer, A.; Wilhelmsson, L.M.; Piantanida, I. Flexibility and Preorganization of Fluorescent Nucleobase-Pyrene Conjugates Control DNA and RNA Recognition. Molecules 2020, 25, 2188. https://doi.org/10.3390/molecules25092188
Ban Ž, Matić J, Žinić B, Foller Füchtbauer A, Wilhelmsson LM, Piantanida I. Flexibility and Preorganization of Fluorescent Nucleobase-Pyrene Conjugates Control DNA and RNA Recognition. Molecules. 2020; 25(9):2188. https://doi.org/10.3390/molecules25092188
Chicago/Turabian StyleBan, Željka, Josipa Matić, Biserka Žinić, Anders Foller Füchtbauer, L. Marcus Wilhelmsson, and Ivo Piantanida. 2020. "Flexibility and Preorganization of Fluorescent Nucleobase-Pyrene Conjugates Control DNA and RNA Recognition" Molecules 25, no. 9: 2188. https://doi.org/10.3390/molecules25092188
APA StyleBan, Ž., Matić, J., Žinić, B., Foller Füchtbauer, A., Wilhelmsson, L. M., & Piantanida, I. (2020). Flexibility and Preorganization of Fluorescent Nucleobase-Pyrene Conjugates Control DNA and RNA Recognition. Molecules, 25(9), 2188. https://doi.org/10.3390/molecules25092188