
Effective Enantiodiscrimination in Electroanalysis Based on a New Inherently Chiral 1,1′-Binaphthyl Selector Directly Synthesizable in Enantiopure Form


 ,
,
Abstract

1. Introduction
2. Results and Discussion
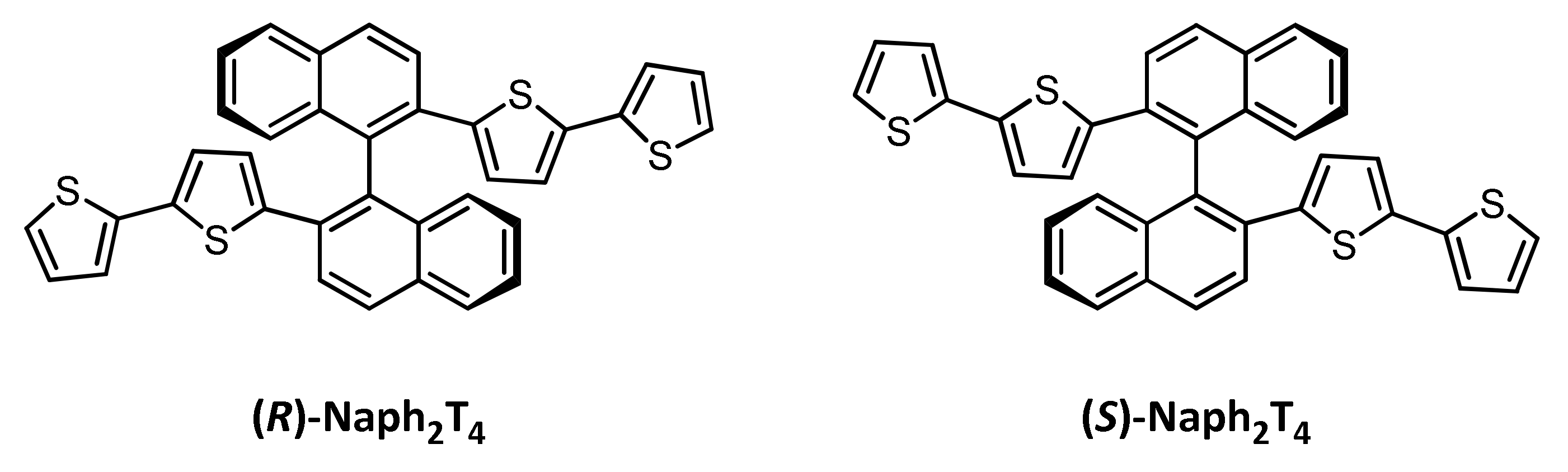
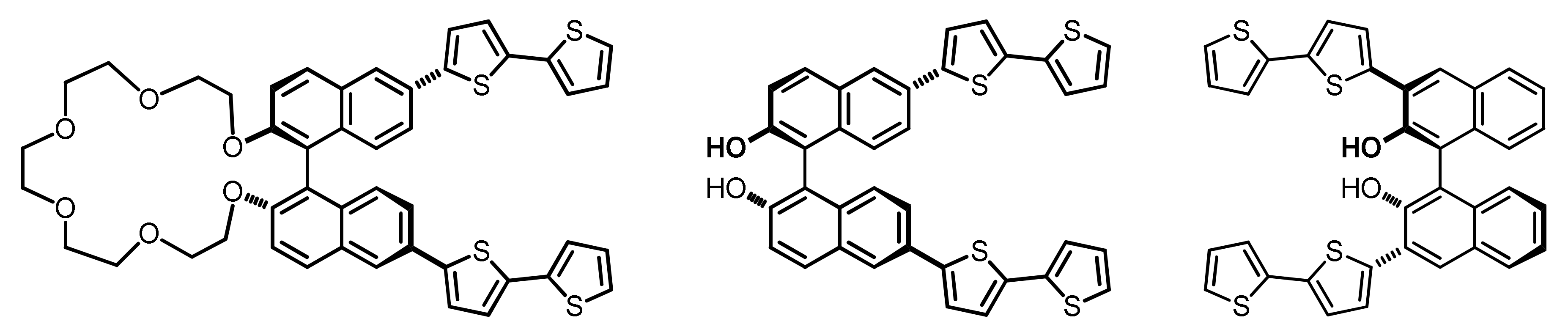
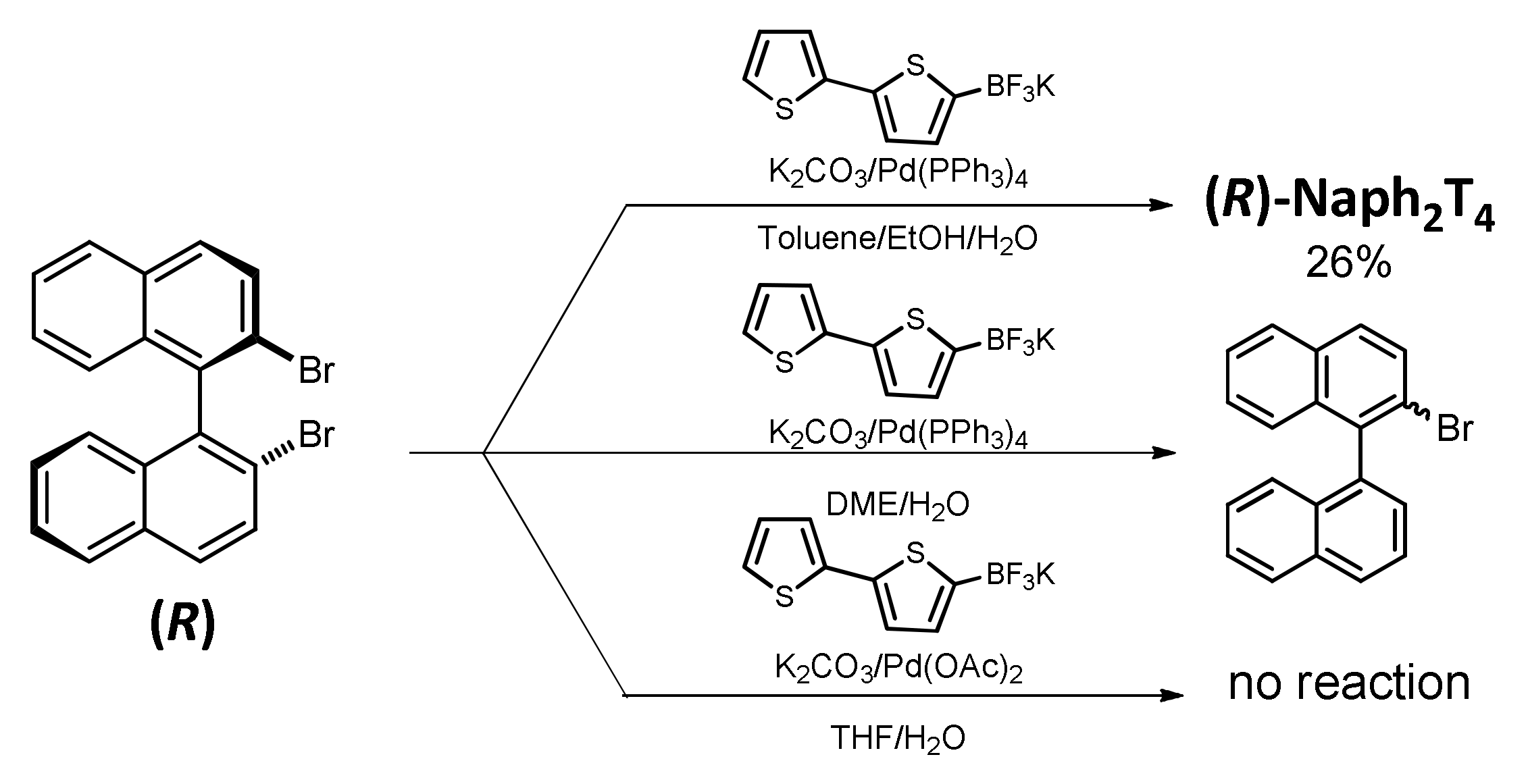
2.1. Monomer Synthesis
2.2. Computational Structural Study
2.2.1. Racemization Barriers
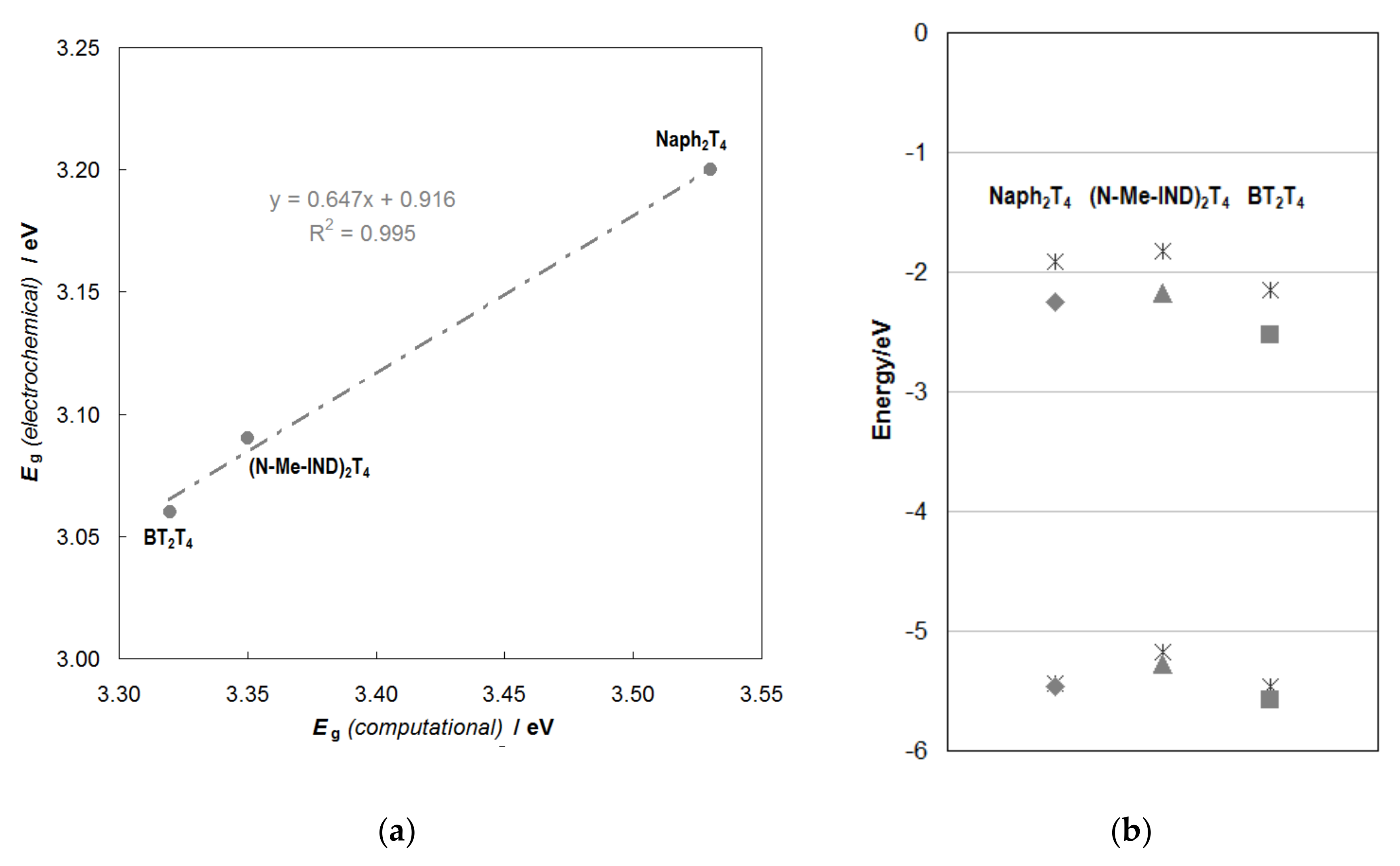
2.2.2. Molecular Orbital Energies and Topologies from DFT Calculations
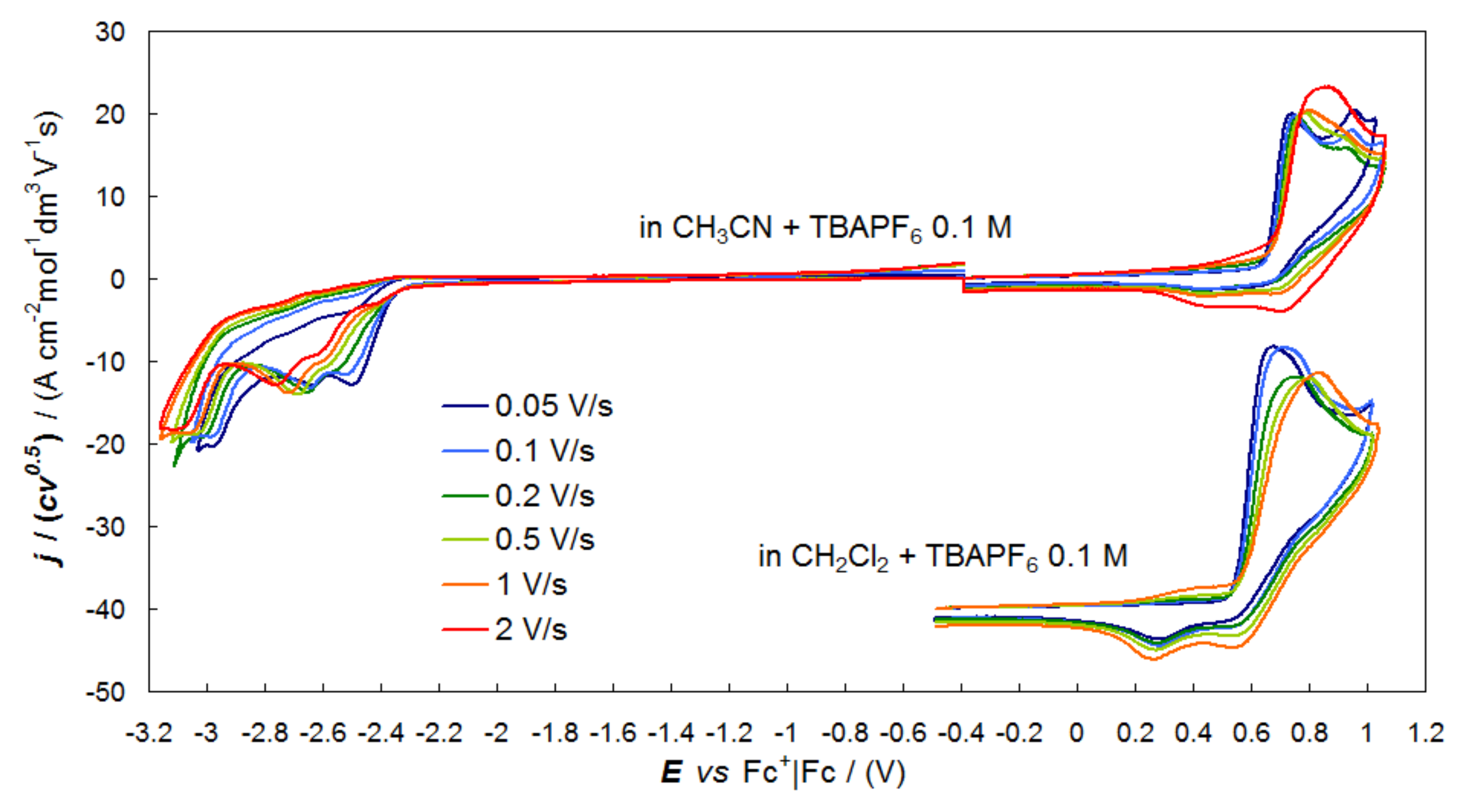
2.3. Electroactivity of the Naph2T4 Monomer
2.4. Electrodeposited Oligo-Naph2T4 Inherently Chiral Electroactive Films
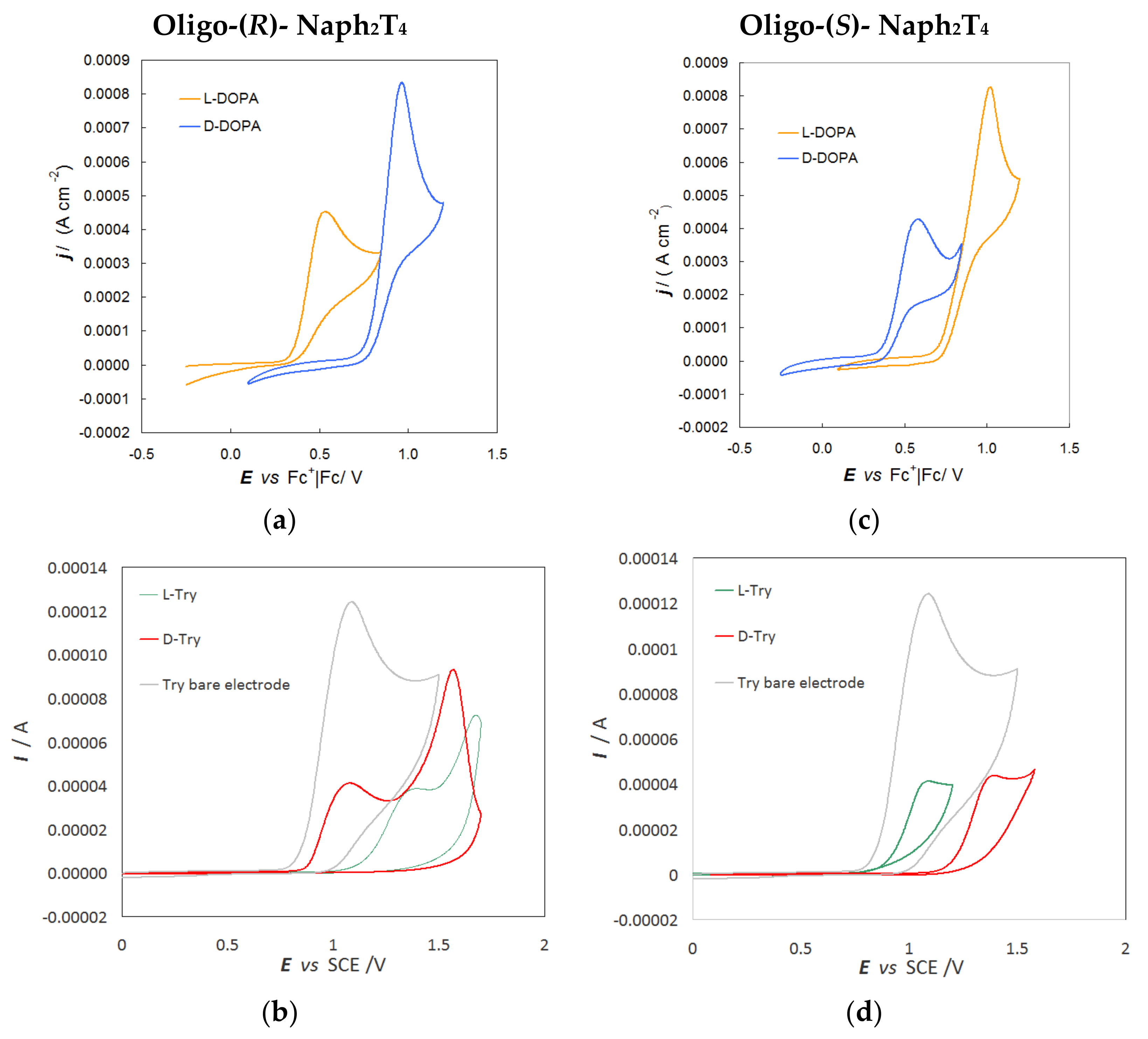
2.5. Enantiodiscrimination Voltammetry Tests
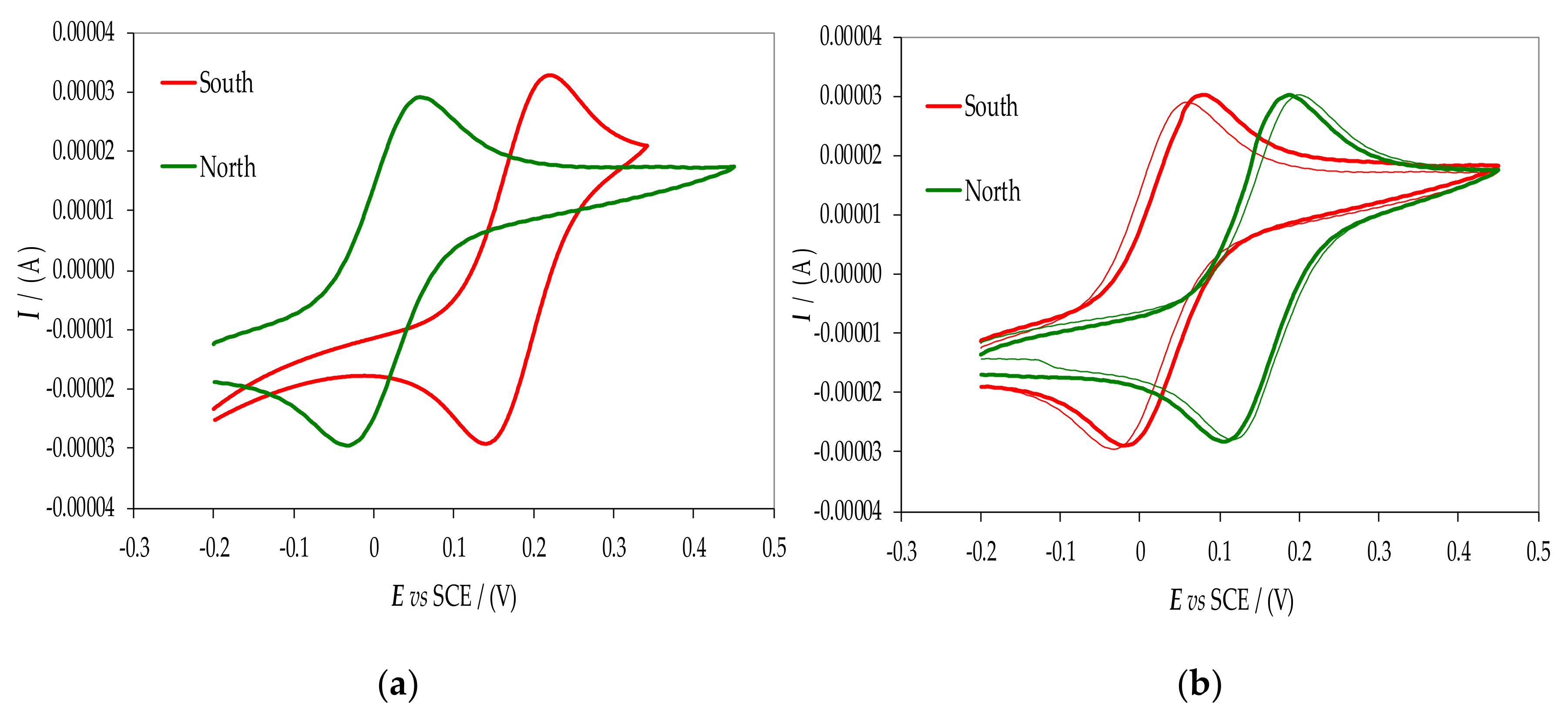
2.6. Magnetoelectrochemistry Experiment
- magnetic fields result in splitting of the α and β electron spin energies proportional to the magnetic field force, implying unbalance of α and β electrons;
- a mere rigid translation of chemically and electrochemically reversible CV signals implies an energy difference;
- an electrode process on a bare electrode under a NS or SN magnetic field represents a couple of specular and therefore energetically equivalent situations;
- the same applies to the process in the absence of the external magnetic field but on the electrode modified with (R)- or (S)- chiral films (which, according to Naaman’s theory, correspond to specular "internal" chiral magnetic fields);
- instead, a combination of the "internal chiral" with the "external" magnetic field results in two couples of specular situations, reciprocally in diastereomeric (i.e., energetically different) conditions.
3. Materials and Methods
3.1. Synthesis
3.1.1. Synthesis of (R)-(+)-2,2’-dibromo-1,1’-binaphthalene
3.1.2. Synthesis of (R)-2,2’-bis(2,2’-bithiophen-5-yl)-1,1’-binaphthalene, (R)-Naph2T4
3.1.3. Synthesis of (S)-2,2’-bis(2,2’-bithiophen-5-yl)-1,1’-binaphthalene, (S)-Naph2T4
3.2. Computational Structural Study
3.3. Electrochemistry
- as working electrode, an ITO one, bare or modified with either oligo-BNT2T4 film antipode (by a single electrodeposition cycle in CH3CN +TBAPF6 medium);
- a Pt counter electrode
- an aqueous saturated calomel electrode (SCE) inserted in a compartment with porous frit as above described as a reference electrode.
3.4. AFM Characterization of the Chiral Electrode Surfaces
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Arnaboldi, S.; Magni, M.; Mussini, P.R. Enantioselective selectors for chiral electrochemistry and electroanalysis: Stereogenic elements and enantioselection performance. Curr. Opin. Electrochem. 2018, 8, 60–72. [Google Scholar] [CrossRef]
- Arnaboldi, S.; Grecchi, S.; Magni, M.; Mussini, P.R. Electroactive chiral oligo- and polymer layers for electrochemical enantiorecognition. Curr. Opin. Electrochem. 2018, 7, 188–199. [Google Scholar] [CrossRef]
- Sannicolo’, F.; Arnaboldi, S.; Benincori, T.; Bonometti, V.; Cirilli, R.; Dunsch, L.; Kutner, W.; Longhi, G.; Mussini, P.R.; Panigati, M.; et al. Potential-Driven Chirality Manifestations and Impressive Enantioselectivity by Inherently Chiral Electroactive Organic Films. Angew. Chem. Int. Ed. 2014, 53, 2623–2627. [Google Scholar] [CrossRef] [PubMed]
- Sannicolo’, F.; Mussini, P.R.; Benincori, T.; Cirilli, R.; Abbate, S.; Arnaboldi, S.; Casolo, S.; Castiglioni, E.; Longhi, G.; Martinazzo, R.; et al. Inherently Chiral Macrocyclic Oligothiophenes: Easily Accessible Electrosensitive Cavities with Outstanding Enantioselection Performances. Chem. A Eur. J. 2014, 20, 15298–15302. [Google Scholar] [CrossRef]
- Benincori, T.; Appoloni, G.; Mussini, P.R.; Arnaboldi, S.; Cirilli, R.; Procopio, E.Q.; Panigati, M.; Abbate, S.; Mazzeo, G.; Longhi, G.; et al. Searching for Models Exhibiting High Circularly Polarized Luminescence: Electroactive Inherently Chiral Oligothiophenes. Chem. A Eur. J. 2018, 24, 11082–11093. [Google Scholar] [CrossRef] [PubMed]
- Benincori, T.; Gámez-Valenzuela, S.; Goll, M.; Bruchlos, K.; Malacrida, C.; Arnaboldi, S.; Mussini, P.R.; Panigati, M.; Navarrete, J.T.L.; Delgado, M.C.R.; et al. Electrochemical studies of a new, low-band gap inherently chiral ethylenedioxythiophene-based oligothiophene. Electrochim. Acta 2018, 284, 513–525. [Google Scholar] [CrossRef]
- Sannicolò, F.; Mussini, P.R.; Benincori, T.; Martinazzo, R.; Arnaboldi, S.; Appoloni, G.; Panigati, M.; Quartapelle Procopio, E.; Marino, V.; Cirilli, R.; et al. Inherently chiral spider-like oligothiophenes. Chem. Eur. J. 2016, 22, 10839–10847. [Google Scholar]
- Arnaboldi, S.; Benincori, T.; Penoni, A.; Vaghi, L.; Cirilli, R.; Abbate, S.; Longhi, G.; Mazzeo, G.; Grecchi, S.; Panigati, M.; et al. Highly Enantioselective “Inherently Chiral” Electroactive Materials Based on the 2,2′-Biindole Atropisomeric Scaffold. Chem. Sci. 2019, 10, 2708–2717. [Google Scholar] [CrossRef]
- Zi, G.; Xiang, L.; Zhang, Y.; Wang, Q.; Zhang, Z. Synthesis, structure, and activity of (PhCH2NH2)2CuCl2 for oxidative coupling of 2-naphthylamine. Appl. Organomet. Chem. 2007, 21, 177–182. [Google Scholar] [CrossRef]
- Kuhn, R.; Goldfinger, P. Stereochemistry of aromatic compounds. VIII. Optically active heterocyclic compounds and optically active azo dyes of the binaphthyl series. Justus Liebigs. Ann. Chem. 1929, 470, 183–200. [Google Scholar] [CrossRef]
- Jeong, D.-C.; Lee, H.; Yang, K.S.; Song, C. Effects of Substituent on Binaphthyl Hinge-Containing Conductive Polymers. Macromolecules 2012, 45, 9571–9578. [Google Scholar] [CrossRef]
- Kang, S.; Cha, I.; Han, J.G.; Song, C. Electroactive polymer sensors for chiral amines based on optically active 1,1′-binaphthyls. Mater. Express 2013, 3, 119–126. [Google Scholar] [CrossRef]
- Qian, P.; Matsuda, M.; Miyashita, T. Chiral molecular recognition in polymer Langmuir-Blodgett films containing axially chiral binaphthyl groups. J. Am. Chem. Soc. 1993, 115, 5624–5628. [Google Scholar] [CrossRef]
- Brown, K.J.; Berry, M.S.; Murdoch, J.R. Synthesis of optically active 2,2’-dihalo-1,1’-binaphthyls via stable diazonium salts. J. Org. Chem. 1985, 50, 4345–4349. [Google Scholar] [CrossRef]
- Molander, G.A.; Biolatto, B. Palladium-catalysed Suzuki-Miyaura cross-coupling reactions of potassium aryl- and heteroaryltrifluoroboronates. J. Org. Chem. 2003, 68, 4302–4314. [Google Scholar] [CrossRef] [PubMed]
- Demchuk, O.M.; Yoruk, B.; Blackburn, T.; Snieckus, V. A Mixed Naphthyl-Phenyl Phosphine Ligand Motif for Suzuki, Heck, and Hydrodehalogenation Reactions. Synlett 2006, 2006, 2908–2913. [Google Scholar] [CrossRef]
- Yin, J.; Buchwald, S.L. A Catalytic Asymmetric Suzuki Coupling for the Synthesis of Axially Chiral Biaryl Compounds. J. Am. Chem. Soc. 2000, 122, 12051–12052. [Google Scholar] [CrossRef]
- Barder, T.E.; Walker, S.D.; Martinelli, J.R.; Buchwald, S.L. Catalysts for Suzuki-Miyaura coupling processes: Scope and studies of the effect of ligand structure. J. Am. Chem. Soc. 2005, 127, 4685–4696. [Google Scholar] [CrossRef]
- Demchuk, O.M.; Kaplon, K.; Mazur, L.; Strzelecka, D.; Pietrusiewicz, K.M. Readily available catalysts for demanding Suzuki and Miyaura couplings under mild conditions. Tetrahedron 2016, 72, 6668–6677. [Google Scholar] [CrossRef]
- Meca, L.; Řeha, D.; Havlas, Z. Racemization Barriers of 1,1‘-Binaphthyl and 1,1‘-Binaphthalene-2,2‘-diol: A DFT Study. J. Org. Chem. 2003, 68, 5677–5680. [Google Scholar] [CrossRef]
- Patel, D.; Woods, R.M.; Breitbach, Z.S.; Berthod, A.; Armstrong, D.W. Thermal racemization of biaryl atropisomers. Tetrahedron Asymmetry 2017, 28, 1557–1561. [Google Scholar] [CrossRef]
- Tan, J.; Paton, R.S. Frontier molecular orbital effects control the hole-catalyzed racemization of atropisomeric biaryls. Chem. Sci. 2018, 10, 2285–2289. [Google Scholar] [CrossRef] [PubMed]
- Tkachenko, N.V.; Scheiner, S. Optical Stability of 1,1′-Binaphthyl Derivatives. ACS Omega 2019, 4, 6044–6049. [Google Scholar] [CrossRef] [PubMed]
- Gennaro, A.; Isse, A.A.; Giussani, E.; Mussini, P.R.; Primerano, I.; Rossi, M. Relationship between supporting electrolyte bulkiness and dissociative electron transfer at catalytic and non-catalytic electrodes. Electrochimica Acta 2013, 89, 52–62. [Google Scholar] [CrossRef]
- Sannicolo’, F.; Rizzo, S.; Benincori, T.; Kutner, W.; Noworyta, K.; Sobczak, J.; Bonometti, V.; Falciola, L.; Mussini, P.R.; Pierini, M. An effective multipurpose building block for 3D electropolymerisation: 2,2′-Bis(2,2′-bithiophene-5-yl)-3,3′-bithianaphthene. Electrochimica Acta 2010, 55, 8352–8364. [Google Scholar] [CrossRef]
- Benincori, T.; Capaccio, M.; De Angelis, F.; Falciola, L.; Muccini, M.; Mussini, P.R.; Ponti, A.; Toffanin, S.; Traldi, P.; Sannicolo’, F. Spider-Like Oligothiophenes. Chem. A Eur. J. 2008, 14, 459–471. [Google Scholar] [CrossRef]
- Heinze, J.; Frontana-Uribe, B.A.; Ludwigs, S. Electrochemistry of Conducting Polymers—Persistent Models and New Concepts†. Chem. Rev. 2010, 110, 4724–4771. [Google Scholar] [CrossRef]
- Nojima, Y.; Hasegawa, M.; Hara, N.; Imai, Y.; Mazaki, Y. Stereogenic cyclic oligonaphthalenes displaying ring size-dependent handedness of circularly polarized luminescence (CPL). Chem. Commun. 2019, 55, 2749–2752. [Google Scholar] [CrossRef]
- Longhi, G.; Abbate, S.; Mazzeo, G.; Castiglioni, E.; Mussini, P.R.; Benincori, T.; Martinazzo, R.; Sannicolo’, F. Structural and Optical Properties of Inherently Chiral Polythiophenes: A Combined CD-Electrochemistry, Circularly Polarized Luminescence, and TD-DFT Investigation. J. Phys. Chem. C 2014, 118, 16019–16027. [Google Scholar] [CrossRef]
- Mondal, P.C.; Fontanesi, C.; Waldeck, D.H.; Naaman, R. Spin-Dependent Transport through Chiral Molecules Studied by Spin-Dependent Electrochemistry. Accounts Chem. Res. 2016, 49, 2560–2568. [Google Scholar] [CrossRef]
- Benincori, T.; Arnaboldi, S.; Magni, M.; Grecchi, S.; Cirilli, R.; Fontanesi, C.; Mussini, P.R. Highlighting spin selectivity properties of chiral electrode surfaces from redox potential modulation of an achiral probe under an applied magnetic field. Chem. Sci. 2019, 10, 2750–2757. [Google Scholar] [CrossRef] [PubMed]
- Arnaboldi, S.; Cauteruccio, S.; Grecchi, S.; Benincori, T.; Marcaccio, M.; Biroli, A.O.; Longhi, G.; Licandro, E.; Mussini, P.R. Thiahelicene-based inherently chiral films for enantioselective electroanalysis. Chem. Sci. 2018, 10, 1539–1548. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides†. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Schrödinger LLC. MacroModel, Version 11.2; Schrödinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Schrodinger LLC. Small-Molecule Drug Discovery Suite 2016-4; Schrodinger, LLC: New York, NY, USA, 2016. [Google Scholar]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [PubMed]
- Rassolov, V.; Ratner, M.A.; Pople, J.A.; Redfern, P.C.; Curtiss, L.A. 6-31G* basis set for third-row atoms. J. Comput. Chem. 2001, 22, 976–984. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision D.01; Gaussian, Inc.: Wallingford, UK, 2013. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 (S)-(N-Me-IND)2T4 |  (S)-BT2T4 |  (S)-Naph2T4 | ||||
|---|---|---|---|---|---|---|
| Level of Theory | TS1 ΔΔG≠ (ΔΔE≠) b (kcal/mol) | TS2 ΔΔG≠ (ΔΔE≠) b (kcal/mol) | TS1 ΔΔG≠ (ΔΔE≠) (kcal/mol) | TS2 ΔΔG≠ (ΔΔE≠) (kcal/mol) | TS1 ΔΔG≠ (ΔΔE≠) (kcal/mol) | TS2 ΔΔG≠ (ΔΔE≠) (kcal/mol) |
| PM6 | 24.77 (20.75) | 28.56 (24.06) | 25.32 (21.25) | 25.95 (22.42) | 36.17 (33.30) | 35.22 (32.92) |
| B3LYP/6-31G(d,p) | 30.14 (26.90) | 36.46 (33.38) | 38.55 (34.76) | 39.78 (35.94) | 46.15 (43.95) | 46.64 (44.76) |
| B3LYP-D3/6-31G(d,p) | 28.77 (26.32) | 30.90 (27.58) | 35.07 (33.78) | 34.53 (32.53) | 45.49 (45.43) | 46.61 (46.41) |
| B3LYP/TZVP | 30.68 (27.19) | 36.49 (33.47) | 39.04 (35.50) | 40.42 (36.32) | 46.09 (43.95) | 47.02 (44.76) |
| B3LYP/6 − 311G + (3df,3pd) // B3LYP/6-31G(d,p) a | - (27.38) | - (33.17) | - (35.42) | - (36.63) | - (43.77) | - (44.54) |
| Sample | Rms Surface Roughness (nm) |
|---|---|
| oligo-(R)-Naph2T4 CH2Cl2 | 6.1 ± 2.8 (mean ± SD; n = 30) |
| oligo-(R)-Naph2T4 CH3CN | 14.5 ± 6.7 (mean ± SD; n = 30) |
| Monomer | Dihedral Angle | ΔE FcA/mV | ΔE DOPA/mV |
|---|---|---|---|
| Naph2T4 | 78 | 70 | 450 |
| BT2T4 | 101 | 45 | 100 |
| (N-Me-IND)2T4 | 117.61 | 270 | 200 |
 | ||||||
|---|---|---|---|---|---|---|
| # | ΔE * (kcal/mol) | δ (Degree) | φ1 (Degree) | φ2 (Degree) | θ1 (Degree) | θ2 (Degree) |
| 1 | 0 | −75.38 | −50.98 | −50.66 | 169.60 | 170.06 |
| 2 | 3.17 | −75.69 | −50.13 | −52.68 | 29.54 | 149.99 |
| 3 | 3.40 | −76.82 | −51.29 | −51.82 | −33.15 | 172.30 |
| 4 | 6.54 | 75.86 | 52.00 | 50.05 | 36.64 | −36.37 |
| 5 | 6.63 | 78.68 | 51.31 | −112.95 | 179.78 | 167.70 |
| 6 | 7.73 | −78.28 | −52.43 | −52.40 | −32.50 | −32.39 |
| 7 | 9.08 | 78.22 | 51.81 | −113.73 | 36.58 | 168.14 |
| 8 | 9.74 | −78.46 | 113.13 | −51.04 | 35.59 | −176.53 |
| 9 | 9.97 | 78.91 | 51.60 | −111.99 | −30.66 | 167.25 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bonetti, G.; Arnaboldi, S.; Grecchi, S.; Appoloni, G.; Massolo, E.; Rossi, S.; Martinazzo, R.; Orsini, F.; Mussini, P.R.; Benincori, T. Effective Enantiodiscrimination in Electroanalysis Based on a New Inherently Chiral 1,1′-Binaphthyl Selector Directly Synthesizable in Enantiopure Form. Molecules 2020, 25, 2175. https://doi.org/10.3390/molecules25092175
Bonetti G, Arnaboldi S, Grecchi S, Appoloni G, Massolo E, Rossi S, Martinazzo R, Orsini F, Mussini PR, Benincori T. Effective Enantiodiscrimination in Electroanalysis Based on a New Inherently Chiral 1,1′-Binaphthyl Selector Directly Synthesizable in Enantiopure Form. Molecules. 2020; 25(9):2175. https://doi.org/10.3390/molecules25092175
Chicago/Turabian StyleBonetti, Giorgia, Serena Arnaboldi, Sara Grecchi, Giulio Appoloni, Elisabetta Massolo, Sergio Rossi, Rocco Martinazzo, Francesco Orsini, Patrizia R. Mussini, and Tiziana Benincori. 2020. "Effective Enantiodiscrimination in Electroanalysis Based on a New Inherently Chiral 1,1′-Binaphthyl Selector Directly Synthesizable in Enantiopure Form" Molecules 25, no. 9: 2175. https://doi.org/10.3390/molecules25092175
APA StyleBonetti, G., Arnaboldi, S., Grecchi, S., Appoloni, G., Massolo, E., Rossi, S., Martinazzo, R., Orsini, F., Mussini, P. R., & Benincori, T. (2020). Effective Enantiodiscrimination in Electroanalysis Based on a New Inherently Chiral 1,1′-Binaphthyl Selector Directly Synthesizable in Enantiopure Form. Molecules, 25(9), 2175. https://doi.org/10.3390/molecules25092175