Abstract
The hydroxy-pyrazole and 3-hydroxy-oxindole motifs have been utilised in several pharma and agrochemical leads but are distinctly underrepresented in the scientific literature due to the limited routes of preparation. We have developed a one-pot procedure for their synthesis starting from simple isatins. The method employs cheap and easy-to-handle building blocks and allows easy isolation.
Keywords:
pyrazole; oxindole; isatin; Pfitzinger reaction; Knorr pyrazole synthesis; quinoline; 15N-MASNMR 1. Introduction
The pyrazole moiety is commonly found in several pharmaceuticals and agrochemicals [1]. In particular, hydroxy-pyrazole units are described as antiviral (HIV-1 non-nucleoside reverse transcriptase inhibitors, 1) [2], anti-diabetic (Na-glucose cotransporter inhibitors, 3) [3,4,5], anti-cancer (Hsp90 inhibitor, 2) [6], anti-thrombotic (P2Y1 antagonist 4) [7], and antibacterial agents (5) [8,9] (examples in Figure 1).
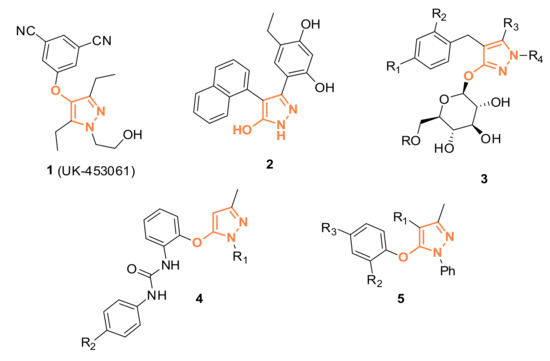
Figure 1.
Examples of hydroxy-pyrazole derivatives described in literature.
The 3-hydroxy-2-oxindole moiety is another related and widely represented motif present in several natural products (9,10,12) [10,11,12,13,14]. This scaffold has recently been employed in several areas of pharmacology (examples in Figure 2), and it has been described in the context of anti-cancer (tryptophan 2,3-dioxygenase inhibitor, 6) [15], anticonvulsant (8) [16], cytotoxic (11) [17] and growth hormone releasing agents (i.e., SM-130686, 7) [18,19].
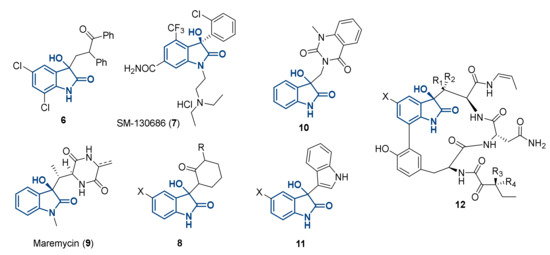
Figure 2.
Examples of 3-hydroxy-2-oxindoles derivatives described in literature.
Several methodologies for the generation of oxindole systems have been reported with one of these employing isatin [20]. Isatin (13) is an endogenous compound whose structure appears many times in natural products which have wide-ranging pharmacological activities. It has thus been widely utilised as a starting material in the preparations of a range of drug candidates [21].
Our research aim was to devise and validate a simple preparation comprising of the above cited pharmacophores, 3-hydroxy-pyrazole and 3-hydroxy-2-oxindole, based upon presenting the cores in a unique 3D architecture which could be easily tailored to further any medicinal chemistry exploration. To the best of our knowledge, structures such as compound 15 have not previously been made, and therefore, it would be of great interest for exploring these new areas of chemical space (Scheme 1).
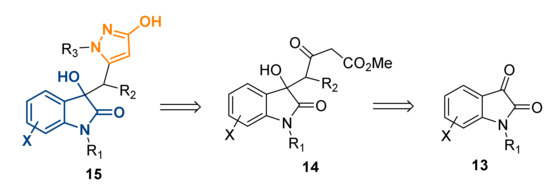
Scheme 1.
Proposed retrosynthetic synthesis of compound 15 starting from isatin (13).
2. Results and Discussion
2.1. Optimisation of the Synthesis Method
To achieve this aim we first investigated a base-catalysed aldol reaction as described by Liu et al. [22]. We decided to screen different reaction conditions, including evaluation of bases, in order to optimise the reaction for isatin 13 (Scheme 2 and Table 1).
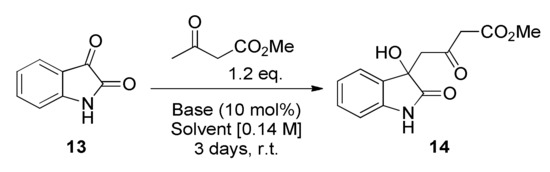
Scheme 2.
General conditions applied for the preparation of 14.

Table 1.
Screening of different bases for the synthesis of 14.
Piperidine was found to be the most effective catalyst allowing the desired compound to be obtained in 87% isolated yield (Entry 8). Whilst pursuing this work, Zhang et al. [23] reported the use of 1,8-diazabicyclo(5.4.0)undece-7-ene (DBU) in tetrahydrofuran (THF) as the preferred solvent for such a reaction. We had previously attempted to use DBU in MeOH, which had been disappointing (Entry 2). Unfortunately, changing the solvent to THF, although consuming the isatin, generated the desired product in only a modest 46% isolated yield (Entry 3). The by-products were difficult to isolate and were found unstable. Therefore, progressing with our catalyst of choice, piperidine, we investigated an expanded range of solvents. Additionally, inspired by the effect of water on 1,6-Michael additions as described by Kashinath et al., we also investigated water. However, for this reaction only a 15% conversion was detected, probably due to the low solubility of the starting materials in the aqueous media (Entry 11) [24]. No mixture with water was screened. Overall, MeOH still remained the best solvent; nonetheless, less polar solvents such as AcOEt and iPrOH also gave promising results (Entries 10 and 15). Attempts to avoid chromatography, by isolating and purifying the product by recrystallization, failed. Having determined effective conditions for the preparation of intermediate 14, we next examined the pyrazole formation using different hydrazine sources starting with hydrazine monohydrate.
To facilitate the transformation, intermediate 14 was mixed with excess hydrazine monohydrate and stirred at ambient temperature (Scheme 3). After 4 days, a precipitate was formed, which was isolated and identified as the quinoline 16, which is formed via a Pfitzinger type reaction [25,26], occurring via either water or hydrazine attack on the position 2 of the oxindole intermediate 14 (green route, Scheme 3).
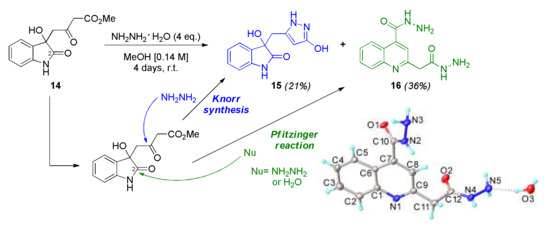
Scheme 3.
Initial reaction conditions employed for the preparation of 15. When hydrazine attacks the carbonyl, the pyrazole 15 is formed (Knorr synthesis), whereas the addition of water or hydrazine into the amide group (marked position 2) triggers the Pfitzinger reaction. The X-ray structure of product 16 is shown in the bottom right of the picture.
From this initial result we also investigated other hydrazine sources (Table 2). As our ultimate challenge was to develop a one-pot process, we examined the second step as a telescoped transformation of 13 to 15. Although the THF solution of hydrazine gave good results (Entry 3), we elected for safety considerations to keep using hydrazine monohydrate, as it is more stable, much cheaper and more widely available [27]. The better outcomes obtained from the hydrazine solution may be attributable to a dilution effect as diluted hydrazine is added in the mixture.
Table 2.
Screening of different hydrazine sources for the synthesis of the pyrazole 15.
As Table 3 shows, the conversion to the oxindole 15 increased, and the quantity of quinoline 16 halved when the mixture was only reacted for 2 h (Entry 2). We later discovered that compound 15 completely decomposed after 10 days under the reaction conditions, whereas compound 16 was stable. We showed that the formation of compound 15 is partially reversible under the reaction conditions (15→14), and it cannot be attributed to the solvent. Consequently, compound 16 acts as a thermodynamic sink, and over time, its proportion increases in the reaction mixture. Overall, our best results were obtained when the hydrazine was slowly added to the mixture, with the desired material 15 being isolated via reverse-phase column chromatography in 38% yield (Table 3, entry 3).
Table 3.
Optimisation of the reaction conditions for the preparation of the pyrazole 15.
Having established a set of optimised conditions, we decided to screen other isatin derivatives and examine the different reactivity (Table 4). Despite optimisation, the purification step of the compounds 15a–j and 16a–j remained challenging in some cases. Consequently, we managed to isolate the compounds 15a–j, but not all of the corresponding quinoline 16 derivatives could be isolated cleanly. Attempts of crystallisations with different solvents (MeOH, EtOH, iPrOH, H2O, Et2O, THF, Toluene) brought about sticky materials as they incorporate solvent. The 5-, 6- and 7-chloro derivatives (respectively, 15d, 15g and 15h) were isolated in slightly better yields compared to 15 (Entries 4, 7 and 8). However, the highest conversions of 16 are detected when the 5-chloro and 5-fluoro substrates are used, attributed to the electronic effect of an electro-withdrawing group in para to the amide. This is consistent with the fact that an electro-donating group such as a methoxy in the 5-position reduces the quinoline formation (Entry 5). A similar electronic effect is also noticed when the chlorine is in the position 7, although the fluorine and the bulkier trifluoromethyl group do still form traces of the quinoline (Entries 8–10).
Table 4.
Application of the optimised conditions for the synthesis of the derivatives 15a–j.
When 4-fluoroisatin (13a) was exploited in the preparation method (Entry 1), the reaction occurred in very low conversions (only 30% of the isatin reacts). This outcome is attributed to the low electrophilicity of the isatin ketone which hampers the first step from occurring. This hypothesis is supported up by the 13C chemical shift of the carbonyl acquired in solid state (13C δ 179.15 ppm) compared with isatin 13 (13C δ 184.39 ppm), and a resultant higher stretching frequency of the same carbonyl (1739 cm−1 vs. 1721 cm−1) in Fourier transform infrared (FT-IR) spectra (see supporting materials). Indeed, substrate 13a shows only poor conversion to the corresponding aldol adduct 14a.
To our delight, when other halogens in position 4 were tested, the pyrazoles (15b–c) were isolated in high yields (Entries 2 and 3). We suggested a Thorpe–Ingold type effect due to the bulk of the halogen group, which lowers the ring opening of the oxindole ring and accelerates the pyrazole cyclization. This could be confirmed by the raise in conversions with increasing halogen size (from chloro to bromo). Compound 15c was crystallized and the structure of the crystal was resolved (Figure 3).
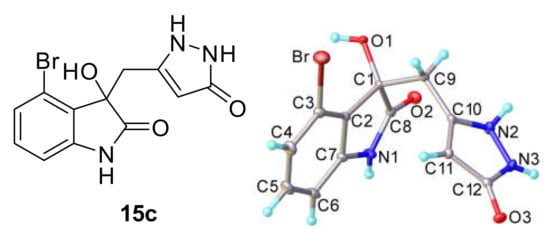
Figure 3.
X-ray structure of the compound 15c where the pyrazole is in its keto form.
In an attempt to reduce the competitive quinoline formation (compound 16) by making the nitrogen a worse leaving group, we N-benzyl-protected the nitrogen (Scheme 4). To our delight, the corresponding compound 15l was isolated in a superior yield (79%). This compound was also highly crystalline, and an X-ray structure was also obtained to validate the molecular connectivity.
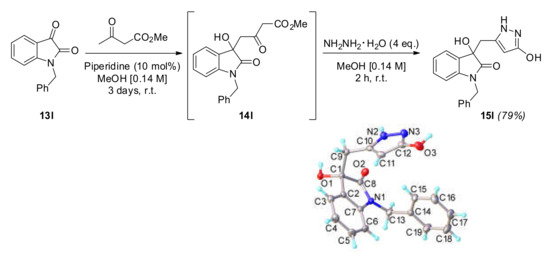
Scheme 4.
Methodology applied to the N-benzyl-protected isatin 13l. The X-ray structure of 15l is also pictured, where the pyrazole is in its enol form.
2.2. Observations of the Spectroscopic Data Obtained Illustrated for Compound 15
In order to confirm the structure of the molecules obtained, we exploited different spectroscopic methodologies. Recorded solution-state Nuclear Magnetic Resonance (NMR) spectra confirm the presence of two diastereotopic protons (δ 3.03 and 2.88 ppm) that by 2D NMR correlate with carbons present on the 2-oxindole group and on the pyrazole. The signals regarding the 3-hydoxypyrazole (13C δ 160.46, 138.04, 88.86 ppm) match those obtained from similar scaffolds by Ayoub et al. in 1981 [28]. In their report, the authors noticed a proton exchange process occurs in solution which involves the pyrazole’s protons and all the other labile protons present in the molecule. Solution-state 15N-NMR heteronuclear single quantum coherence (HSQC) and heteronuclear multiple bond coherence (HMBC) spectra were acquired and allowed the assignment of the 15N chemical shift of N1; however, due to the fast proton exchange, the experiments were unable to identify the other two nitrogen. N14 and N15 (15N δ −198, −215 ppm) were instead identified, along with the N1 (−248 ppm), when solid-state 15N- magic angle spinning NMR (15N-MASNMR) spectra were acquired. Furthermore, suppression of both the N14 and N15 signals in a cross-polarisation polarisation-inversion (CPPI) experiment is an indication of a proton exchange process occurring in the solid state (Figure 4).
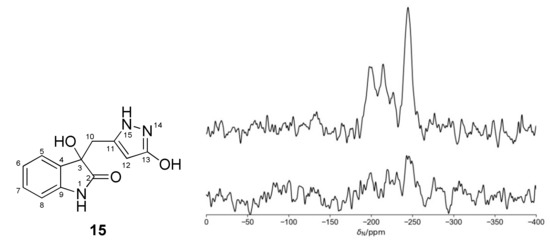
Figure 4.
Structure of the desired material 15 and its 15N- cross polarisation (CP) (top) and cross-polarisation polarisation-inversion (CPPI) (bottom) MASNMR spectra.
Unfortunately, all attempts to generate crystals of compound 15 produced amorphous powders. However, to our delight, we managed to crystallise certain derivatives of 15 and confirm the structures of the analogous products (e.g., 15c, 15l, Figure 5). In the solid state, the molecules 15 form closely packed lattices with extensive H-bonding interaction which would account for the ease of the proton exchange identified in the 15N-MASNMR analysis.
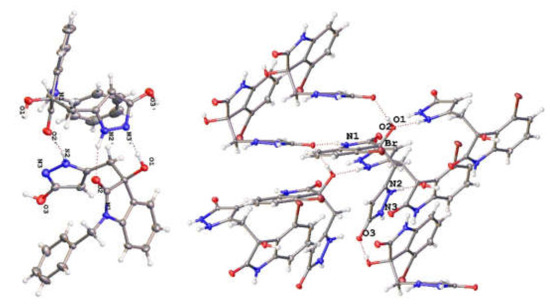
Figure 5.
Pictures showing the packaging in the crystal structure of 15l (left side) and 15c (right side). The H-bonding interactions between the molecules can be noticed (N2/N3 = N15/14).
3. Materials and Methods
Unless otherwise stated, all solvents were purchased from Fisher Scientific (Loughborough, Leicestershire, UK) and used without further purification. Substrates, their precursors and reagents were purchased from Alfa Aesar (Haverhill, MA, USA), Sigma Aldrich (Merck KGaA, Darmstadt, Germany), Fluorochem (Hadfield, Derbyshire, United Kingdom) or TCI (Tokyo, Tokyo, Japan).
1H-NMR spectra were recorded on either Bruker Avance-400, Varian VNMRS-700 or Varian VNMRS-500 instruments (Bruker UK Limited, Coventry, UK) and are reported relative to residual solvent DMSO-d6 (δ 2.50 ppm). 13C-NMR spectra were recorded on the same instruments and are reported relative to DMSO-d6 (δ 39.52 ppm). Data for 1H-NMR are reported as follows: chemical shift (δ/ppm) (multiplicity, coupling constant (Hz), integration). Multiplicities are reported as follows: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, brs = broad signal. Data for 13C-NMR are reported in terms of chemical shift (δC/ppm). DEPT-135, COSY, HSQC, HMBC, PSYCHE and NOESY experiments were used in structural assignments. Solid-state NMR experimental information is included in the Supplementary Information. Single crystals X-ray diffraction experiments were carried out on a Bruker D8 Venture diffractometer with PHOTON 100 CMOS area detector (Bruker UK Limited, Coventry, UK), using Mo-Kα (15c, 15l) or Cu-Kα (16) radiation from Incoatec IμS microsources with focusing mirrors. The crystals were cooled using a Cryostream 700 (Oxford Cryosystems, (Oxford, Oxfordshire, UK) open-flow N2 gas cryostat. The structures were solved by dual-space intrinsic phasing (SHELXT [29] program) and refined by full-matrix least squares using SHELXL [30] software on Olex2 platform [31].
IR spectra were obtained using a Perkin Elmer Spectrum Two UATR Two FT-IR Spectrometer (neat, ATR sampling, (Waltham, MA, USA)) with the intensities of the characteristic signals being reported as weak (w, <20% of tallest signal), medium (m, 21–70% of tallest signal) or strong (s, >71% of tallest signal).
Low resolution liquid chromatography mass spectrometry (LC-MS) was performed using a Waters TQD mass spectrometer (Waters Corp., Milford, MA, USA) and an Acquity UPLC BEH C18 1.7 µm column (2.1 mm × 50 mm) in ESI mode. ESI-HRMS was performed using a Waters QtoF Premier mass spectrometer (Waters Corp., Milford, MA, USA). For accurate mass measurements the deviation from the calculated formula is reported in mDa. Melting points were recorded on an Optimelt automated melting point system with a heating rate of 1 °C/min and are uncorrected. All the purifications were performed using a Teledyne CombiFlash® Rf+ (Teledyne ISCO, Lincoln, NE, USA) equipped with a RediSep Rf Gold 150 g high performance C18 cartridge. The purification was performed at a flow of 75 mL/min using a gradient from 15% methanol in water to 60% methanol in water in 20 min. The cartridge was then washed with 100% methanol and conditioned in 50% methanol in water for storage. The water from the sample was removed using a Labconco FreeZone 4.5 L freeze dry system (Labconco, Kansas City, MO, USA) connected with a Leybold Trivac® B D4B rotary cane vacuum pump (Leybold GmbH, Cologne, Germany). Organic solutions were concentrated under reduced pressure using a Buchi rotary evaporator and high vacuum was achieved using an Edwards RV5 pump and Schlenk line (BÜCHI Labortechnik AG, Postfach, Flawil, Switzerland).
3.1. General Procedure for the Synthesis of 14
To a heterogeneous mixture of isatin (735 mg, 5 mmol, 1 eq.) in MeOH (36 mL), was added methyl acetoacetate (650 μL, 6 mmol, 1.2 eq.) and piperidine (50 μL, 0.5 mmol, 0.1 eq.). The mixture was stirred at room temperature for 3 days. The solvent was then removed under reduced pressure to furnish 14 as a crude amorphous powder. The residue was purified using flash chromatography (eluent: 2.5% MeOH in dichloromethane) to isolate a pale orange amorphous solid (87% isolated yield).
3.2. General Procedure for the Synthesis of 15a–j
To a heterogeneous mixture of the appropriate isatin 13a–j (5 mmol, 1 eq.) in MeOH (36 mL), was added methyl acetoacetate (650 μL, 6 mmol, 1.2 eq.) and piperidine (50 μL, 0.5 mmol, 0.1 eq.). The mixture was stirred at room temperature for 3 days. Hydrazine monohydrate (970 μL, 20 mmol, 4 eq.) was added dropwise at room temperature. After stirring the solution for 2 h, the mixture was partly concentrated in vacuo and Celite® was added before all the solvent was removed. The residue was purified via reversed-phase chromatography as described above.
3.3. Characterisation of the Compounds 14 and 15a-j and 16 Derivatives
Methyl 4-(3-hydroxy-2-oxoindolin-3-yl)-3-oxobutanoate (14):1H NMR (700 MHz, DMSO-d6) δ 10.24 (s, 1H), 7.23 (d, J = 7.3 Hz, 1H), 7.18 (td, J = 7.7, 1.3 Hz, 1H), 6.91 (td, J = 7.3, 1.0 Hz, 1H), 6.78 (d, J = 7.7 Hz, 1H), 6.07 (s, 1H), 3.61 (s, 2H), 3.57 (s, 3H), 3.38 – 3.34 (m, 1H, overlapping with H2O), 3.09 (d, J = 17.0 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 200.27, 177.89, 167.36, 142.48, 131.20, 129.09, 123.79, 121.28, 109.48, 72.52, 51.76, 49.49, 49.20. IR (neat): ν (cm−1) = 3264 (OH, br), 2955 (CH, w), 1705 (C=O, s), 16,121 (CH, m), 1472 (OH, m), 1185 (C-O, m), 1013 (w), 753 (m). LC-MS Rt = 1.18 min m/z [M + H]+ = 264.2; HR-MS calculated for C13H14NO5 264.0872, found 264.0855 (Δ = −1.7 mDa). Melting point (°C): 100 °C decomposition.
3-Hydroxy-3-[(3-hydroxy-1H-pyrazol-5-yl)methyl]indolin-2-one (15):1H NMR (700 MHz, DMSO-d6) δ 10.20 (brs, 2H), 7.17 (td, J = 7.7, 1.3 Hz, 1H), 7.11 (dd, J = 7.4, 1.3 Hz, 1H), 6.93 (td, J = 7.5, 1.0 Hz, 1H), 6.73 (d, J = 7.7 Hz, 1H), 4.80 (s, 1H), 3.05 (d, J = 14.0 Hz, 1H), 2.90 (d, J = 14.0 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 178.51, 160.48, 142.01, 138.04, 131.20, 129.12, 124.32, 121.45, 109.48, 88.86, 74.89, 34.38. 15N NMR (71 MHz, DMSO-d6) δ -246.15 (15N-1H HSQC shows correlation with the peak at δ 10.19 ppm in the 1H-NMR). 13C CPMAS MS-NMR (101 MHz) δ 179.98, 162.65, 142.72, 139.51, 132.79, 129.78, 127.43, 123.91, 110.57, 91.77, 76.77, 35.52. 15N CPMAS MS-NMR (40.556 MHz) δ −197.54, −214.53, −244.66. LC-MS (ESI+) Rt = 0.70 min m/z [M + H]+ = 246.7 HR-MS calculated for C12H12N3O3 246.0879, found 246.0878 (∆ = −0.1 mDa). IR (neat): ν (cm−1) = 3161 (NH, w), 2966 (CH, w), 1703 (C=O, s), 1595 (C=N, s), 1471 (s), 1340 (w), 1259 (m), 1078 (m), 1043 (s), 778 (w), 748 (w). Melting point (°C): 188 °C decomposition.
2-(2-Hydrazinyl-2-oxoethyl)quinoline-4-carbohydrazide (16):1H NMR (700 MHz, DMSO-d6) δ 9.90 (s, 1H), 9.37 (s, 1H), 8.10 (d, J = 8.4 Hz, 1H), 7.99 (d, J = 8.4 Hz, 1H), 7.78 (t, J = 7.7 Hz, 1H), 7.61 (t, J = 7.6 Hz, 1H), 7.50 (s, 1H), 4.67 (s, 2H), 4.29 (s, 2H), 3.78 (s, 2H). 13C NMR (176 MHz, DMSO-d6) δ 168.5, 166.3, 156.9, 147.8, 141.8, 130.3, 129.2, 127.1, 125.7, 123.7, 120.3, 44.1. IR (neat): ν (cm−1)= 3285 (NH, m), 3176 (NH, m), 1620 (C=O, s), 1533 (s), 1362 (m), 1174 (w), 1035.9 (w), 1005 (m), 879.4 (m), 758.4 (s), 699.1 (s). LC-MS: Rt= 2.41 min m/z [M + H]+ = 176.6; HR-MS calculated for C12H14N5O2 260.1147, found 260.1136 (Δ = −1.1 mDa). Melting point (°C): 220 °C decomposition. Crystal data: C12H15N5O3·H2O (M = 277.29 g/mol), monoclinic, space group P21/n (no. 14), a = 4.9407(5) Å, b = 14.0587(13) Å, c = 18.2746(16) Å, β = 97.040(5)°, V = 1259.8(2) Å3, Z = 4, T = 120 K, μ(Cu-Kα) = 0.91 mm−1, Dcalc = 1.462 g/cm3, 7750 reflections measured (2Θ ≤ 117°), 1719 unique (Rint = 0.086, Rσ = 0.080), R1 = 0.049 on 1183 data with I > 2σ(I), wR2 = 0.106 on all data, CCDC-1986566.
4-Fluoro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15a):1H NMR (700 MHz, DMSO-d6) δ 10.18 (brs, 3H), 7.22 (td, J = 8.1, 5.4 Hz, 1H), 6.73 (t, J = 8.8 Hz, 1H), 6.55 (d, J = 7.7 Hz, 1H), 4.65 (s, 1H), 3.19 (d, J = 14.0 Hz, 1H), 3.08 (d, J = 14.0 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 177.58, 159.75, 158.35, 144.36 (d, J = 9.6 Hz), 137.71, 131.45 (d, J = 8.5 Hz), 116.53 (d, J = 19.7 Hz), 109.01 (d, J = 20.6 Hz), 106.03, 87.87, 75.21, 33.30.13C CPMAS NMR (101 MHz) δ 179.99, 163.78, 161.32, 158.58, 143.68, 132.32, 115.96, 108.95, 91.29, 77.24, 34.06. 15N CPMAS NMR (40.556 MHz) δ −203.58, −221.81, −243.52. LC-MS (ESI+) Rt = 0.80 min m/z [M+H]+ = 264.2. HR-MS calculated for C12H11N3O3F 264.0784, found 264.0766 (∆ = −1.8 mDa). IR (neat): ν (cm−1) = 3103 (NH, w), 2748 (CH, w), 1709 (C=O, s), 1638 (C=N, s), 1567 (s), 1493 (m), 1464 (s), 1256 (w), 1229 (m), 1053 (m), 1033 (w), 1005 (w), 765 (s), 682 (w). Melting point (°C): 100 °C decomposition.
4-Chloro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15b):1H NMR (599 MHz, DMSO-d6) δ 10.32 (brs, 3H), 7.20 (t, J = 8.0 Hz, 1H), 6.95 (d, J = 8.2 Hz, 1H), 6.67 (d, J = 7.8 Hz, 1H), 6.27 (s, 1H), 4.55 (s, 1H), 3.47 (d, J = 14.0 Hz, 1H), 3.05 (d, J = 14.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 177.46, 160.57, 144.34, 137.42, 130.90, 130.54, 127.14, 122.35, 108.44, 87.63, 76.14, 32.08. 13C CPMAS NMR (101 MHz) δ 179.71, 164.07, 144.60, 134.63, 129.32, 123.84, 110.28, 92.38, 77.59, 33.72. 15N CPMAS NMR (40.556 MHz) δ −217.29, −224.15, −241.86. LC-MS (ESI+) Rt = 1.35 min m/z [M + H]+ = 280.5 HR-MS calculated for C12H11N3O3Cl 280.0489, found 280.0485 (∆= 0.4 mDa). IR (neat): ν (cm−1) = 3074 (NH, w), 2703 (CH, w), 1713 (C=O, s), 1589 (C=N, s), 1449 (s), 1415 (m), 1175 (m), 1083 (m), 948 (w), 765 (s), 657 (w). Melting point (°C): 135 °C decomposition.
4-Bromo-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15c):1H NMR (700 MHz, DMSO-d6) δ 10.32 (brs, 3H), 7.19 – 7.03 (m, 2H), 6.70 (dd, J = 6.6, 2.1 Hz, 1H), 6.24 (s, 1H), 4.51 (s, 1H), 3.56 (d, J = 14.0 Hz, 1H), 3.01 (d, J = 14.0 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 177.52, 160.25, 144.62, 137.61, 131.14, 128.77, 125.44, 119.06, 108.93, 87.68, 76.71, 31.92. 13C CPMAS NMR (101 MHz) δ 179.77, 164.39, 145.28, 134.65, 125.47, 111.34, 92.26, 78.21, 33.85. 15N CPMAS NMR (40.556 MHz) δ −217.48, −223.79, −243.05. LC-MS (ESI+) Rt= 1.45 min m/z [M + H]+ = 323.8 HR-MS calculated for C12H11N3O3Br 323.9984, found 323.9973 (∆= −1.1 mDa). IR (neat): ν (cm−1) = 3000 (NH, w), 2710 (CH, w), 1715 (C=O, s), 1585 (C=N, s), 1445 (s), 1413 (m), 1329 (w), 1169 (m), 1082 (m), 929 (w), 763 (s), 660 (s). Melting point (°C): 103 °C decomposition. Crystal data: C12H10BrN3O3 (M = 324.14 g/mol), orthorhombic, space group P212121 (no. 19), a = 6.9156(3) Å, b = 13.1025(6) Å, c = 13.3565(6) Å, V = 1210.25(9) Å3, Z = 4, T = 120 K, μ(MoKα) = 3.40 mm−1, Dcalc = 1.779 g/cm3, 32,088 reflections measured (2Θ ≤ 66.5°), 4654 unique (Rint = 0.036, Rσ = 0.027), R1 = 0.025 on 4238 reflections with I > 2σ(I), wR2 = 0.058 on all data. CCDC 1986565.
5-Chloro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15d):1H NMR (599 MHz, DMSO-d6) δ 10.30 (brs, 1H), 7.22 (dd, J = 8.3, 2.2 Hz, 1H), 7.12 (d, J = 2.3 Hz, 1H), 6.73 (d, J = 8.3 Hz, 1H), 6.28 (s, 1H), 4.84 (s, 1H), 3.06 (d, J = 14.0 Hz, 1H), 2.90 (d, J = 14.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.11, 140.87, 133.20, 128.88, 125.45, 124.48, 110.89, 88.83, 75.08, 24.39. 13C CPMAS NMR (101 MHz) δ 181.73, 163.08, 146.44, 140.53, 132.67, 128.35, 112.33, 90.37, 76.86, 36.96. 15N CPMAS NMR (40.556 MHz) −198.84, −211.43, −245.96. LC-MS (ESI+) Rt = 1.07 min m/z [M + H]+ = 280.6 HR-MS calculated for C12H11N3O3Cl 280.0489, found 280.0505 (∆ = 1.6 mDa). IR (neat): ν (cm−1) = 3173 (NH, w), 2627 (CH, w), 1670 (C=O, m), 1610 (C=N, s), 1480 (m), 1440 (m), 1249 (w), 1192 (w), 973 (w), 817 (w), 758 (s), 615 (s). Melting point (°C): 140 °C decomposition.
3-Hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)-5-methoxyindolin-2-one (15e):1H NMR (599 MHz, DMSO-d6) δ 10.02 (brs, 3H), 6.75 – 6.69 (m, 2H), 6.64 (d, J = 8.2 Hz, 1H), 4.86 (s, 1H), 3.68 (s, 3H), 3.04 (d, J = 14.1 Hz, 1H), 2.87 (d, J = 14.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.41, 160.48, 154.70, 137.96, 135.13, 132.37, 113.70, 111.30, 109.78, 88.96, 75.27, 55.37, 34.35. 13C CPMAS NMR (101 MHz) δ 180.57, 162.56, 156.41, 139.29, 133.63, 117.12, 112.15, 93.07, 77.07, 55.43, 34.66. 15N CPMAS NMR (40.556 MHz) δ −200.09, −221.84, −246.55. LC-MS (ESI+) Rt = 1.04 min m/z [M + H]+ = 276.6. HR-MS calculated for C13H14N3O4 276.0984, found 276.0980 (∆ = −0.4 mDa). IR (neat): ν (cm−1) = 3064 (NH, w), 2758 (CH, w), 1714 (C=O, s), 1637 (C=N, s), 1597 (s), 1466 (s), 1233 (m), 1053 (m), 765 (s), 537 (m). Melting point (°C): 101 °C decomposition.
5-Fluoro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15f): 1H NMR (700 MHz, DMSO-d6) δ 10.23 (brs, 3H), 7.03 – 6.99 (m, 1H), 6.93 (dd, J = 8.2, 2.7 Hz, 1H), 6.71 (dd, J = 8.5, 4.3 Hz, 1H), 6.29 (s, 1H), 4.86 (s, 1H), 3.05 (d, J = 14.0 Hz, 1H), 2.91 (d, J = 14.0 Hz, 1H). 13C NMR (176 MHz, DMSO-d6) δ 178.43, 160.47 (d, J = 33.2 Hz), 158.56, 157.21, 138.11 (d, J = 1.7 Hz), 137.64, 132.93 (d, J = 7.6 Hz), 115.30 (d, J = 23.2 Hz), 112.08 (d, J = 24.3 Hz), 110.22 (d, J = 7.9 Hz), 88.88, 75.28, 34.27. 13C CPMAS NMR (101 MHz) δ 180.04, 163.66, 159.53, 143.98, 138.00, 130.76, 119.82, 111.70, 92.78, 77.29, 35.66. 15N CPMAS NMR (40.556) δ −216.10, −225.14, −242.50. LC-MS (ESI+) Rt = 0.80 min m/z [M + H]+ = 264.4 HR-MS calculated for C12H11N3O3F 264.0784, found 264.0763 (∆ = −2.1 mDa). IR (neat): ν (cm−1) = 3250 (NH, w), 2965 (w), 2862 (CH, w), 1708 (C=O, m), 1588 (C=N, m), 1485 (s), 1363 (w), 1187 (m), 1077 (w), 1033 (w), 825 (m), 601 (s). Melting point (°C): 110 °C decomposition.
6-Chloro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15g):1H NMR (599 MHz, DMSO-d6) δ 10.31 (brs, 3H), 7.10 (d, J = 7.9 Hz, 1H), 6.98 (dd, J = 7.9, 1.9 Hz, 1H), 6.74 (d, J = 1.9 Hz, 1H), 6.23 (s, 1H), 4.83 (s, 1H), 3.04 (d, J = 14.0 Hz, 1H), 2.90 (d, J = 14.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.38, 143.52, 133.28, 130.06, 125.78, 121.16, 109.52, 88.84, 74.61, 39.10, 34.19. 13C CPMAS NMR (101 MHz) δ 180.52, 163.52, 142.20, 136.09, 127.70, 111.70, 92.75, 75.70, 35.04. 15N CPMAS NMR (40.556 MHz) δ −199.82, −209.90, −242.92. LC-MS (ESI+) Rt = 1.48 min m/z [M + H]+ = 280.6 HR-MS calculated for C12H11N3O3Cl 280.0489, found 280.0498 (∆ = 0.9 mDa). IR (neat): ν (cm−1) = 3147 (NH, w), 2771 (CH, w), 1711 (C=O, s), 1592 (C=N, s), 1482 (m), 1447 (w), 1331 (w), 1068 (m), 917 (w), 738 (w), 593 (w). Melting point (°C): 120 °C decomposition.
7-Chloro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15h):1H NMR (400 MHz, DMSO-d6) δ 7.25 (dd, J = 8.1, 1.1 Hz, 1H), 7.07 (dd, J = 7.3, 1.2 Hz, 1H), 6.96 (dd, J = 8.1, 7.4 Hz, 1H), 4.80 (s, 1H), 3.05 (d, J = 14.1 Hz, 1H), 2.92 (d, J = 14.0 Hz, 1H). 13C NMR (101 MHz, DMSO-d6) δ 178.45, 144.15, 139.69, 133.18, 129.10, 122.95, 122.88, 113.68, 88.71, 75.53, 46.10. 13C CPMAS NMR (101 MHz) δ 180.70, 162.73, 145.14, 139.39, 131.75, 130.50, 123.84, 115.65, 90.98, 77.51, 35.83. 15N CPMAS NMR (40.556 MHz) −200.62, −216.52, −247.87. LC-MS (ESI+) Rt = 1.33 min m/z [M + H]+ = 280.5 HR-MS calculated for C12H11N3O3Cl 280.0489, found 280.0501 (∆ = 1.2 mDa). IR (neat): ν (cm−1) = 3137 (NH, m), 2752 (CH, w), 1713 (C=O, s), 1620 (C=N, s), 1562 (s), 1474 (m), 1454 (m), 1223 (w), 1171 (m), 1138, (s), 782 (s), 736 (s). Melting point (°C): 134 °C decomposition.
3-Hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)-7-(trifluoromethyl)indolin-2-one (15i):1H NMR (599 MHz, DMSO-d6) δ 10.00 (brs, 3H), 7.47 (d, J = 8.0 Hz, 1H), 7.35 (d, J = 7.3 Hz, 1H), 7.12 (t, J = 7.7 Hz, 1H), 6.37 (s, 1H), 4.82 (s, 1H), 3.10 (d, J = 14.1 Hz, 1H), 2.93 (d, J = 14.1 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.93, 160.32, 139.45, 137.62, 133.13, 128.23, 125.50 (q, J = 4.4 Hz), 123.95 (q, J = 271.7 Hz), 121.65, 110.50 (q, J = 32.6 Hz), 88.76, 73.66, 34.21. 13C CPMAS NMR (101 MHz) δ 181.20, 163.58, 142.93, 138.75, 132.50, 130.62, 126.20, 122.33, 112.60, 93.37, 72.54, 48.96, 32.16. 15N CPMAS NMR (40.556 MHz) δ −206.98, −217.10, −246.09. LC-MS (ESI+) Rt = 1.93 min m/z [M + H]+ = 314.3 HR-MS calculated for C13H11N3O3F3 314.0753, found 314.0758 (∆ = 0.5 mDa). IR (neat): ν (cm−1) = 3175 (NH, s), 2826 (CH, w), 1734 (C=O, s), 1594 (s), 1455 (s), 1325 (s), 1170 (s), 1119 (s), 1104 (s), 1038 (s), 801 (s), 752 (s), 711 (s), 612 (s). Melting point (°C): 115 °C decomposition.
7-Fluoro-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15j):1H NMR (599 MHz, DMSO-d6) δ 10.42 (brs, 3H), 7.10 (ddd, J = 10.4, 7.9, 1.5 Hz, 1H), 7.02 – 6.83 (m, 2H), 6.30 (brs, 1H), 4.78 (s, 1H), 3.06 (d, J = 14.1 Hz, 1H), 2.93 (d, J = 14.0 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 178.28, 160.33, 147.03, 145.43, 137.71, 134.26 (d, J = 3.2 Hz), 128.89 (d, J = 12.1 Hz), 122.39 (d, J = 5.6 Hz), 120.36 (d, J = 3.0 Hz), 116.17 (d, J = 17.2 Hz), 88.70, 75.10 (d, J = 2.8 Hz), 34.42. 13C CPMAS NMR (101 MHz) δ 178.95, 162.17, 148.70, 146.06, 142.92, 132.71, 129.95, 124.18, 119.87, 118.74, 90.85, 76.82, 35.85. 15N CPMAS NMR (40.556) δ −216.39, −223.03, −250.73. LC-MS (ESI+) Rt = 0.77 min m/z [M + H]+= 264.5. HR-MS calculated for C12H11N3O3F 264.0784, found 264.0764 (∆ = −2.0 mDa). IR (neat): ν (cm−1)= 3698 (NH, w), 3358 (NH, w), 2974 (CH, s), 1731 (C=O, s), 1647 (w), 1587 (C=N, s), 1557 (s), 1493 (s), 1469 (s), 1332 (w), 1192 (w), 1053 (s), 1033 (s), 1014 (s), 791 (s), 758 (s), 731 (s), 567 (s). Melting point (°C): 115 °C decomposition.
6-Chloro-2-(2-hydrazinyl-2-oxoethyl)quinoline-4-carbohydrazide (16d):1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 9.38 (s, 1H), 8.17 (d, J = 2.4 Hz, 1H), 8.02 (d, J = 9.0 Hz, 1H), 7.81 (dd, J = 9.0, 2.5 Hz, 1H), 7.58 (s, 1H), 4.71 (s, 2H), 4.30 (s, 2H), 3.78 (s, 2H). 13C NMR (101 MHz, DMSO) δ 167.91, 165.43, 157.31, 145.92, 140.34, 131.34, 131.00, 130.42, 124.16, 123.99, 121.03, 43.64. LC-MS (ESI+) Rt = 1.42 min m/z [M + H]+ = 294.3 HR-MS calculated for C12H13N5O2Cl 294.0758, found 294.0771 (∆ = 1.3 mDa). IR (neat): ν (cm−1) = 3295 (NH, s), 3274 (NH, s), 3047 (CH, w), 1639 (C=O, s), 1618 (C=O, m), 1587 (C=N, m), 1528 (m), 1373 (m), 1303 (w), 1211 (w), 1013 (m), 1011 (m), 836 (s), 690 (s), 606 (s), 565 (s), 491 (s). Melting point (°C): 221 °C decomposition.
6-Fluoro-2-(2-hydrazinyl-2-oxoethyl)quinoline-4-carbohydrazide (16f):1H NMR (700 MHz, DMSO-d6) δ 9.95 (s, 1H), 9.37 (s, 1H), 8.07 (dd, J = 9.3, 5.6 Hz, 1H), 7.86 (dd, J = 10.2, 2.9 Hz, 1H), 7.71 (td, J = 8.8, 2.9 Hz, 1H), 7.58 (s, 0H), 4.55 (brs, 1H), 3.78 (s, 1H). 13C NMR (176 MHz, DMSO-d6) δ 168.00, 165.52, 160.53, 159.14, 156.14, 144.72, 140.54, 131.71, 123.97 (d, J = 10.5 Hz), 120.85, 119.88 (d, J = 25.7 Hz), 108.81 (d, J = 23.6 Hz), 43.53. LC-MS (ESI+) Rt = 0.97 min m/z [M + H]+ = 278.2. HR-MS calculated for C12H13N5O2F 278.1053, found 278.1035 (∆ = −1.8 mDa). IR (neat): ν (cm−1) = 3275 (NH, s), 3196 (w), 3054 (CH, w), 1643 (C=O, s), 1610 (m), 1533 (s), 1470 (w), 1381 (w), 1227 (w), 1012 (w), 837 (w), 682 (w), 579 (w), 500 (w). Melting point (°C): 210 °C decomposition.
7-Chloro-2-(2-hydrazinyl-2-oxoethyl)quinoline-4-carbohydrazide (16g):1H NMR (400 MHz, DMSO-d6) δ 9.97 (s, 1H), 9.39 (s, 1H), 8.14 (d, J = 9.0 Hz, 1H), 8.05 (s, 1H), 7.67 (d, J = 9.0 Hz, 1H), 7.54 (s, 1H), 4.69 (brs, 2H), 4.31 (brs, 2H), 3.78 (s, 2H). 13C NMR (101 MHz, DMSO) δ 167.93, 165.49, 158.18, 147.92, 141.33, 134.54, 127.48, 121.98, 120.54, 43.68. LC-MS (ESI+) Rt = 1.45 min m/z [M + H]+ = 294.3 HR-MS calculated for C12H13N5O2Cl 294.0758, found 294.0771 (∆ = 1.3 mDa). IR (neat): ν (cm−1) = 3288.5 (NH, m), 3250.2 (NH, m), 3074.61 (CH, w), 1713.87 (C=O, s), 1643.3 (C=O, s), 1616.38 (s), 1590.61 (s), 1533.77 (s), 1493.34 (m), 1361.98 (m), 1257.41 (w), 1138.43 (w), 961.82 (m), 839.91 (m), 756.43 (m). Melting point (°C): 223 °C decomposition.
2-(2-Hydrazinyl-2-oxoethyl)-8-(trifluoromethyl)quinoline-4-carbohydrazide (16i):1H NMR (599 MHz, DMSO-d6) δ 9.97 (s, 1H), 9.38 (s, 1H), 8.38 (dd, J = 8.5, 1.4 Hz, 1H), 8.21 (dd, J = 7.5, 1.4 Hz, 1H), 7.76 (dd, J = 8.5, 7.5 Hz, 1H), 7.66 (s, 1H), 4.70 (brs, 1H), 4.30 (brs, 1H), 3.84 (s, 2H). 13C NMR (151 MHz, DMSO-d6) δ 168.18, 165.79, 158.16, 143.93, 142.17, 130.78, 128.95 (q, J = 5.3 Hz), 126.44, 126.23, 124.50 (q, J = 272 Hz), 124.22, 121.36, 44.27. LC-MS (ESI+) Rt = 1.65 min m/z [M + H]+ = 328.3 HR-MS calculated for C13H13N5O2F3 328.1021, found 328.1028 (∆ = 0.7 mDa). IR (neat): ν (cm−1) = 3295.15 (br, NH), 1670.75 (m, C=O), 1644.92 (s, C=O), 1596.49 (s), 1530.03 (s), 1363.51 (m), 1297.84 (s), 1162.63 (m), 1130.38 (s), 1051.34 (m), 1005.59 (m), 769.31 (m). Melting point (°C): 225 °C decomposition.
1-Benzyl-3-hydroxy-3-((3-hydroxy-1H-pyrazol-5-yl)methyl)indolin-2-one (15l): 1H NMR (599 MHz, DMSO-d6) δ 10.97 (brs, 2H), 7.30–7.24 (m, 3H), 7.24–7.20 (m, 1H), 7.17 (td, J = 7.7, 1.3 Hz, 1H), 7.06 – 6.99 (m, 3H), 6.66 (d, J = 7.8 Hz, 1H), 6.37 (s, 1H), 4.90 (d, J = 16.0 Hz, 1H), 4.71 – 4.65 (m, 2H), 3.18 (d, J = 13.9 Hz, 1H), 3.04 (d, J = 13.9 Hz, 1H). 13C NMR (151 MHz, DMSO-d6) δ 176.80, 160.57, 142.55, 137.73, 135.88, 130.69, 129.16, 128.57, 127.11, 126.81, 124.07, 122.25, 108.99, 89.00, 74.93, 42.46, 34.66. 13C CPMAS NMR (101 MHz) δ 178.20, 177.34, 164.17, 159.74, 143.92, 141.99, 136.37, 134.72, 131.23, 129.81, 128.03, 126.61, 125.39, 124.44, 123.57, 109.93, 91.69, 90.57, 77.74, 76.23, 44.55, 41.28, 35.56, 34.18. 15N CPMAS NMR (40.556) δ −134.38, −138.40, −199.90, −202.03, −237.79, −239.63. LC-MS (ESI+) Rt = 1.55 min m/z [M + H]+ = 335.8. HR-MS calculated for C19H18N3O3 336.1348, found 336.1337 (∆ = −1.1 mDa). IR (neat): ν (cm−1) = 3228 (NH, w), 2940 (CH, W), 1699 (C=O, s), 1616 (C=N, s), 1569 (w), 1489 (m), 1469 (s), 1355 (s), 1259 (w), 1179 (m), 1074 (s), 1000 (m), 773 (s), 748 (s), 732 (s), 693 (s). Melting point (°C): 166 °C decomposition. Crystal data: C19H17N3O3 (M = 335.35 g/mol), monoclinic, space group P21/n (no. 14), a = 8.5554(6), b = 29.118(2), c = 13.1745(9) Å, β = 92.172(3)°, V = 3279.6(4) Å3, Z = 8, T = 120 K, μ(Mo-Kα) = 0.09 mm−1, Dcalc = 1.358 g/cm3, 48,703 reflections measured (2Θ ≤ 50°), 5792 unique (Rint = 0.067, Rσ = 0.049), R1 = 0.041 on 4146 reflections with I > 2σ(I), wR2 = 0.091 on all data, CCDC-1986567.
4. Conclusions
We developed a methodology for the synthesis of a new scaffold comprising of two key pharmacophores units. The procedure is simple and employs readily available hydrazine monohydrate and other cheap building blocks, such as methyl acetoacetate and isatin. A library of 12 compounds was prepared in order to study the versatility of the method which appears to be applicable to a range of isatin starting materials in which the 3-oxo group is active for aldol reactions. Furthermore, we acquired different spectroscopic analyses such as 15N-MASNMR, 15N-NMR and SXRD to confirm the structure of the products. To the best our knowledge, no 15N-MASNMR spectra on 3-hydroxypyrazole scaffolds had previously been performed. The compounds generated are currently under investigation for specific biological activity and will be reported separately.
Supplementary Materials
The following are available online, 1H and 13C-NMR spectra, 13C and 15N MASNMR spectra, mass spectra, infrared spectra and XRD crystallographic data of compounds. Crystallographic data in CIF format have been deposited with Cambridge Structural Database (CCDC-1986565 to 1986567) and are available online.
Author Contributions
Conceptualization, I.R.B.; Formal analysis, G.G and D.C.A.; Investigation, G.G.; Supervision, I.R.B..; Writing—original draft, I.R.B, G.G. and D.C.A; Writing—review & editing, I.R.B, G.G. and D.C.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Acknowledgments
We thank the Juan A. Aguilar-Malavia and Andrei Batsanov of Durham University for assistance given in solving and characterising the structures. We also thank David C. Apperley for his patience in acquiring the 13C and 15N-MASNMR spectra.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Karrouchi, K.; Radi, S.; Ramli, Y.; Taoufik, J.; Mabkhot, Y.N.; Al-Aizari, F.A.; Ansar, M. Synthesis and Pharmacological Activities of Pyrazole Derivatives: A Review. Molecules 2018, 23, 134. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Zhan, P.; De Clercq, E.; Liu, X. Strategies for the Design of HIV-1 Non-Nucleoside Reverse Transcriptase Inhibitors: Lessons from the Development of Seven Representative Paradigms. J. Med. Chem. 2012, 55, 3595–3613. [Google Scholar] [CrossRef] [PubMed]
- Ohsumi, K.; Matsueda, H.; Hatanaka, T.; Hirama, R.; Umemura, T.; Oonuki, A.; Ishida, N.; Kageyama, Y.; Maezono, K.; Kondo, N. Pyrazole-O-glucosides as novel Na(+)-glucose cotransporter (SGLT) inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 2269–2272. [Google Scholar] [CrossRef]
- Pinnetti, S.; Streicher, R.; Thomas, L.; Dugi, K. Pharmaceutical Composition Comprising a Pyrazole-O-Glucoside Derivative. U.S. Patent 12/521,644, 16 December 2008. [Google Scholar]
- Fushimi, N.; Fujikura, H.; Shiohara, H.; Teranishi, H.; Shimizu, K.; Yonekubo, S.; Ohno, K.; Miyagi, T.; Itoh, F.; Shibazaki, T.; et al. Structure–activity relationship studies of 4-benzyl-1H-pyrazol-3-yl β-d-glucopyranoside derivatives as potent and selective sodium glucose co-transporter 1 (SGLT1) inhibitors with therapeutic activity on postprandial hyperglycemia. Bioorg. Med. Chem. 2012, 20, 6598–6612. [Google Scholar] [CrossRef] [PubMed]
- Foley, K.P.; Du, Z. Method for Treating Proliferative Disorders Associated with Mutations in C-Met. U.S. Patent 9,108,933, 18 August 2015. [Google Scholar]
- Pfefferkorn, J.A.; Choi, C.; Winters, T.; Kennedy, R.; Chi, L.; Perrin, L.A.; Lu, G.; Ping, Y.-W.; McClanahan, T.; Schroeder, R.; et al. P2Y1 receptor antagonists as novel antithrombotic agents. Bioorg. Med. Chem. Lett. 2008, 18, 3338–3343. [Google Scholar] [CrossRef] [PubMed]
- Song, M.-X.; Zheng, C.-J.; Deng, X.-Q.; Sun, L.-P.; Wu, Y.; Hong, L.; Li, Y.-J.; Liu, Y.; Wei, Z.-Y.; Jin, M.-J.; et al. Synthesis and antibacterial evaluation of rhodanine-based 5-aryloxy pyrazoles against selected methicillin resistant and quinolone-resistant Staphylococcus aureus (MRSA and QRSA). Eur. J. Med. Chem. 2013, 60, 376–385. [Google Scholar] [CrossRef] [PubMed]
- Jardosh, H.; Sangani, C.B.; Patel, M.P.; Patel, R.G. One step synthesis of pyrido[1,2-a]benzimidazole derivatives of aryloxypyrazole and their antimicrobial evaluation. Chin. Chem. Lett. 2013, 24, 123–126. [Google Scholar] [CrossRef]
- Tang, Y.-Q.; Sattler, I.; Thiericke, R.; Grabley, S.; Feng, X.-Z. ChemInform Abstract: Maremycins C (I) and D (II), New Diketopiperazines, and Maremycins E (III) and F (IV), Novel Polycyclic spiro-Indole Metabolites Isolated from Streptomyces sp. Eur. J. Org. Chem. 2010, 33, 261–267. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, C.; Lin, H.; Liu, Y.; Li, Y.; Zhao, Y.; Li, P.; Liu, J. Discovery of the Potential Biomarkers for Discrimination between Hedyotis diffusa and Hedyotis corymbosa by UPLC-QTOF/MS Metabolome Analysis. Molecules 2018, 23, 1525. [Google Scholar] [CrossRef]
- Qian, P.; Zhang, Y.-B.; Yang, Y.-F.; Xu, W.; Yang, X. Pharmacokinetics Studies of 12 Alkaloids in Rat Plasma after Oral Administration of Zuojin and Fan-Zuojin Formulas. Molecules 2017, 22, 214. [Google Scholar] [CrossRef]
- Koguchi, Y.; Kohno, J.; Nishio, M.; Takahashi, K.; Okuda, T.; Ohnuki, T.; Komatsubara, S. TMC-95A, B, C, and D, novel proteasome inhibitors produced by Apiospora montagnei Sacc. TC 1093. Taxonomy, production, isolation, and biological activities. J. Antibiot. 2000, 53, 105–109. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.; Yang, Z.-Q.; Kwok, B.H.B.; Koldobskiy, M.; Crews, C.M.; Danishefsky, S.J. Total Synthesis of TMC-95A and -B via a New Reaction Leading toZ-Enamides. Some Preliminary Findings as to SAR. J. Am. Chem. Soc. 2004, 126, 6347–6355. [Google Scholar] [CrossRef] [PubMed]
- Pantouris, G.; Loudon-Griffiths, J.; Mowat, C.G. Insights into the mechanism of inhibition of tryptophan 2,3-dioxygenase by isatin derivatives. J. Enzym. Inhib. Med. Chem. 2016, 31, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Raj, M.; Veerasamy, N.; Singh, V.K. Highly enantioselective synthesis of 3-cycloalkanone-3-hydroxy-2-oxindoles, potential anticonvulsants. Tetrahedron Lett. 2010, 51, 2157–2159. [Google Scholar] [CrossRef]
- Prathima, P.S.; Rajesh, P.; Rao, V.J.; Kailash, U.S.; Sridhar, B.; Rao, M.M. “On water” expedient synthesis of 3-indolyl-3-hydroxy oxindole derivatives and their anticancer activity in vitro. Eur. J. Med. Chem. 2014, 84, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Tokunaga, T.; Hume, W.E.; Umezome, T.; Okazaki, K.; Ueki, Y.; Kumagai, K.; Hourai, S.; Nagamine, J.; Seki, H.; Taiji, M.; et al. Oxindole derivatives as orally active potent growth hormone secretagogues. J. Med. Chem. 2001, 44, 4641–4649. [Google Scholar] [CrossRef] [PubMed]
- Tomita, D.; Yamatsugu, K.; Kanai, M.; Shibasaki, M. Enantioselective Synthesis of SM-130686 Based on the Development of Asymmetric Cu(I)F Catalysis to Access 2-Oxindoles Containing a Tetrasubstituted Carbon. J. Am. Chem. Soc. 2009, 131, 6946–6948. [Google Scholar] [CrossRef]
- Bergman, J. Oxindoles. Advances in Heterocyclic Chemistry 2015, 117, 1–81. [Google Scholar] [CrossRef]
- Medvedev, A.E.; Buneeva, O.; Glover, V. Biological targets for isatin and its analogues: Implications for therapy. Boil. Targets Ther. 2007, 1, 151–162. [Google Scholar]
- Liu, H.; Wu, H.-Y.; Luo, Z.; Shen, J.; Kang, G.; Liu, B.; Wan, Z.; Jiang, J. Regioselectivity-Reversed Asymmetric Aldol Reaction of 1,3-Dicarbonyl Compounds. Chem. A Eur. J. 2012, 18, 11899–11903. [Google Scholar] [CrossRef]
- Zhang, D.; Chen, Y.; Cai, H.; Yin, L.; Zhong, J.; Man, J.; Zhang, Q.-F.; Bethi, V.; Tanaka, F. Direct Catalytic Asymmetric Synthesis of Oxindole-Derived δ-Hydroxy-β-ketoesters by Aldol Reactions. Org. Lett. 2019, 22, 6–10. [Google Scholar] [CrossRef]
- Nagaraju, S.; Satyanarayana, N.; Paplal, B.; Vasu, A.K.; Kanvah, S.; Kashinath, D. Synthesis of functionalized isoxazole–oxindole hybrids via on water, catalyst free vinylogous Henry and 1,6-Michael addition reactions. RSC Adv. 2015, 5, 81768–81773. [Google Scholar] [CrossRef]
- Shvekhgeimer, M.G.-A. The Pfitzinger Reaction. Eur. J. Org. Chem. 2004, 35, 257–294. [Google Scholar] [CrossRef]
- Ramann, G.A.; Cowen, B. Recent Advances in Metal-Free Quinoline Synthesis. Molecules 2016, 21, 986. [Google Scholar] [CrossRef] [PubMed]
- Health and Safety Executive. EH40/2005 Workplace Exposure Limits Limits for Use with the Control of Substances, 4th ed.; The Stationery Office: London, UK, 2020. [Google Scholar]
- Ayoub, M.T.; Shandala, M.Y.; Bashi, G.M.G.; Pelter, A. The conversion of 5,6-dihydro-4-methoxy-2-pyrones into 3-alkyl-5-hydroxypyrazoles. J. Chem. Soc. Perkin Trans. 1 1981, 697. [Google Scholar] [CrossRef]
- Dolomanov, O.; Bourhis, L.J.; Gildea, R.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT - integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are available from the authors. |



© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).