Abstract
α-Nitrocinnamate underwent the conjugate addition of an active methylene compound such as nitroacetate, 1,3-dicarbonyl compound, or α-nitroketone, and the following ring closure afforded functionalized heterocyclic frameworks. The reaction of cinnamate with nitroacetate occurs via nucleophilic substitution of a nitro group by the O-attack of the nitronate, which results in isoxazoline N-oxide. This protocol was applicable to 1,3-dicarbonyl compounds to afford dihydrofuran derivatives, including those derived from direct substitution of a nitro group caused by O-attack of enolate. It was found the reactivity was lowered by an electron-withdrawing group on the carbonyl moiety. When α-nitroketone was employed as a substrate, three kinds of products were possibly formed; of these, only isoxazoline N-oxide was identified. This result indicates that the substituting ability of nitronate is higher than that of enolate for the direct SN2 substitution of a nitro group.
1. Introduction
The nitro group is one of the important functional groups because of its unique chemical properties, which are useful in many compounds. The strong electron-withdrawing property of the nitro group reduces the electron density of the adjacent atom or double bond through both inductive and resonance effects. The increased electrophilicity facilitates nucleophilic addition to a nitroalkene, while the resulting anionic intermediate is stabilized by the nitro group. The nitro group also serves as a good leaving group. Nitroalkane undergoes E2 elimination of nitrous acid to afford C–C double bonds under basic [1,2,3,4] or acidic conditions [5,6]. Direct SN2 substitution is also sometimes observed [7], in which a nitrite ion is eliminated. The high acidity of an α-proton of the nitro group easily generates a nitronate, which possesses both nucleophilic [8,9] and electrophilic sites to serve as a 1,3-dipole [10,11]. Moreover, the nitro group is a precursor of amino and carbonyl groups by reduction and Nef-type reactions [12], respectively. The many properties of the nitro group have facilitated diverse applications. Recently, the complex/mixed properties of the nitro group have attracted considerable attention for the synthesis of multi-functionalized/polyfunctionalized compounds. In our previous work, α-nitrocinnamate served as a precursor of functionalized enynes via conjugate addition of an acetylide ion followed by elimination of a nitrous acid [1]. When α-chloro-α,β-unsaturated ketone is subjected to the reaction with cyano-aci-nitroacetate, intramolecular nucleophilic substitution of the chloro group by the nitronate ion occurs after Michael addition [13]. Based on these works, this study furthered this topic by studying the synthesis of functionalized heterocyclic compounds using a combination of conjugate addition of an active methylene compound and subsequent O-attack of the resulting nitronate/enolate which undergoes direct substitution of the nitro group. The substituting abilities of the nitronate/enolate were compared.
2. Results and Discussion
As a highly electron-deficient substrate, methyl α-nitrocinnamate (1a) was employed because of its easy availability via condensation of benzaldehyde and nitroacetate in the presence of piperidine hydrochloride with the removal of water as an azeotrope mixture. Indeed, the reaction of 1a with propylamine quantitatively proceeded at room temperature in acetonitrile to afford product 2a as a 1:1 mixture of diastereomers, which implies that two stereocenters were newly formed; however, the product was not an adduct of 1a and propylamine. Based on spectral data of the reaction mixture, compound 2a was confirmed to be an adduct of 1a with methyl nitroacetate generated in situ. This reaction is thought to proceed as shown in Scheme 1. After conjugate addition of propylamine to 1a, C–C bond cleavage forms nitroacetate [14]. Indeed, signals of 1-phenyl-N-propylmethanimine (3) were observed in the 1H NMR spectrum of the reaction mixture (see Supplementary Materials). The generated nitroacetate underwent the conjugate addition to another cinnamate 1a to afford product 2a. However, product 2a could not be isolated by column chromatography because of its instability on silica gel. On the other hand, when the mixture of 2a and 3 was left at room temperature without solvent for 7 days, ring closure proceeded to afford isoxazoline N-oxide 4a, in which the nitronate underwent the direct substitution of the nitro group to form an isoxazoline framework (Scheme 1).
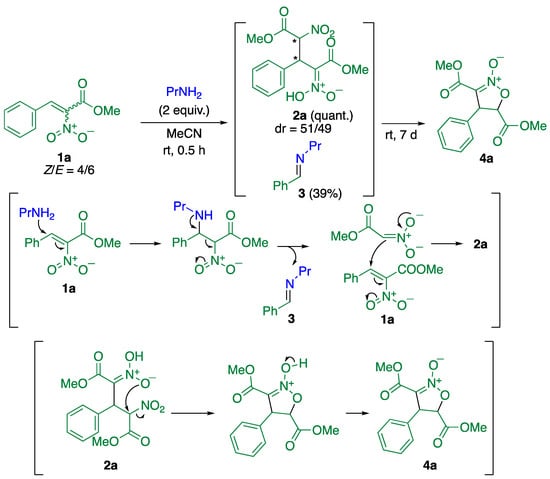
Scheme 1.
Reaction of α-nitrocinnamate 1a with propylamine and the subsequent ring closure, with plausible mechanisms.
There have been several studies of the formation of isoxazoline N-oxides from α-nitro-α,β-unsaturated esters with C–H acids such as secondary nitroalkane [15], (ethoxycarbonylmethyl)dimethylsulfonium salt [16], (ethoxycarbonylmethyl)ammonium salt [17], (ethoxycarbonylmethyl)pyridinium salt [18], α-halomalonate [19], and α-iodo aldehyde [20]. β,β-Dimethoxynitroethene is also usable as a nucleophile in this protocol [21]. Among these, only two methods employ a nitro group as a leaving group [15,21]. In these reactions, the nucleophilicity of the nitronate ion is relatively high. To the contrary, nucleophilicity of the nitronate in 2a is considered to be lower due to the electron-withdrawing ester functionality.
Thus, ethyl α-nitrocinnamate (1b) was allowed to react with ethyl nitroacetate in the presence of triethylamine. It was confirmed that the successive conjugate-addition/ring-closure reactions efficiently proceeded in one pot to afford isoxazoline N-oxide 4b [22] (Scheme 2). This result prompted the study of α-nitrocinnamate 1b reactions with other active methylene compounds such as 1,3-dicarbonyl compounds, because the nucleophilicity of the enolate ion is considered to be lower than that of the nitronate ion.

Scheme 2.
Synthesis of isoxazoline N-oxide 4b.
Although two studies on the reactions of 1b with β-keto esters were found in the literature [23,24], they were not conducted under same conditions (one reaction was conducted in the presence of tetrabutylammonium bromide). In order to compare the reactivity systematically and to generalize this protocol, the same reaction conditions should be used. Therefore, 1,3-dicarbonyl compounds 5a–f were subjected to reactions with 1b in the presence of triethylamine at 60 °C for 3 h in acetonitrile (Table 1). Cyclization efficiently proceeded to produce a furan derivative 6a [23] with 92% yield when ethyl acetoacetate was employed as a substrate (entry 1). The nucleophilicity of the enolate of the ketone functionality was higher than that of the ester functionality because of the electron-withdrawing inductive effect of the ethoxy group. Next, the acetyl group of 5a was replaced with a trifluoroacetyl group. In this case, decreasing the nucleophilicity of the enolate ion was more effective than increasing the acidity of the methylene group, which produced 5-(trifluoromethyl)-2,3-dihydrofuran 6b with a lower yield (entry 2). When benzoylacetate 5c was reacted under the same conditions, the cyclization occurred without significant effects from the steric hindrance of the phenyl group to furnish the corresponding dihydrofuran 6c (entry 3). Diketone 5d, acetylacetone, exhibited higher reactivity than keto esters 5a–c, and yielded 6d [24] quantitatively (entry 4). Cyclic diketone 5e, 1,3-cyclohexanedione, also underwent the reaction efficiently to produce bicyclic furan 6e, which was not influence by steric strain (entry 5) [23,25]. On the other hand, the formation of ester-substituted furan 6f was not detected when diester 5f was subjected to the reaction conditions because of the low nucleophilicity of the enolate ion (entry 6).

Table 1.
Synthesis of functionalized 2,3-dihydrofuran 6.
In the reaction of cinnamate 1b and diketone 5d, the conjugate addition of enolate ion of 5d to 1b afforded adduct intermediate 8d, from which 2,3-dihydrofuran 6d was formed by the substitution of the nitro group. In other words, 2,3-dihydrofuran 6d was also synthesized if intermediate 8d was formed [24]. Indeed, when diacetylated styrene 7 was reacted with ethyl nitroacetate in the presence of triethylamine under the same conditions, a high yield of dihydrofuran 6d was obtained (Scheme 3).

Scheme 3.
Synthesis of 2,3-dihydrofuran 6d from either diacetylated styrene 7 or cinnamate 1b.
Next, α-nitroketone 9 was employed as a nucleophile able to produce both an enolate and a nitronate ion, which facilitated the comparison of the substituting ability of these anions directly (Scheme 4). In this case, adduct 10 is thought to have formed intermediately, from which three possible structures 11–13 could be produced. Dihydrofuran 11 was formed by the attack of enolate (Path a), and isoxazoline N-oxide 12 is formed by the attack of nitronate derived from nitroketone (Path b). On the other hand, attack of the nitronate derived from nitrocinnamate affords isoxazoline N-oxide 13 (Path c).
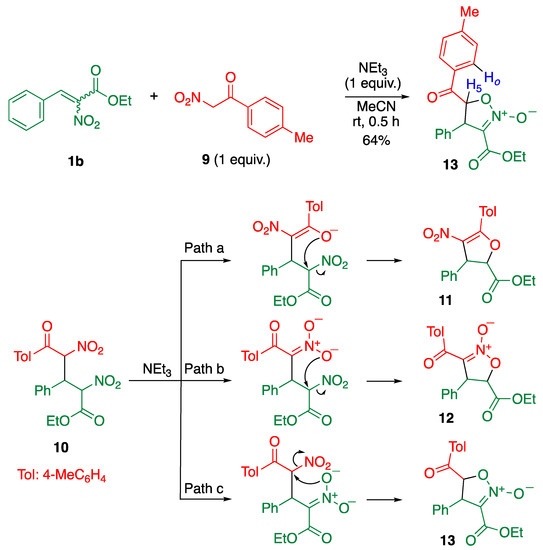
Scheme 4.
Reaction of cinnamate 1b with nitroketone 9 and three possible products 11–13.
When cinnamate 1b was allowed to react with nitroketone 9 in the presence of triethylamine, the reaction mixture became somewhat complex, so only one cyclic product was isolated as a major product. Since a lot of small signals were observed in the 1H NMR spectrum of the reaction mixture, it is difficult to know whether cyclic products were formed as minor products or not. In the 13C NMR spectrum of the major product, a signal corresponding to carbonyl carbon was observed at 191 ppm, which indicated that the product had a ketone functionality; thus, the possibility of 11 was excluded. In the 1H-1H NOESY 2D NMR spectrum, a correlation was observed between the proton at the 5-position of the isoxazoline ring and the ortho-proton of the benzoyl group, by which the product was determined to be isoxazoline N-oxide 13.
This result indicated that the nitronate ion substituted a nitro group via Path c. It is considered that the different reactivity of the two nitro groups was caused by the different electron-withdrawing ability of the ketone and the ester functionalities. The stronger electron-withdrawing toluoyl group increased the electrophilicity of the α-carbon and decreased the nucleophilicity of the nitronate ion, which facilitated the reaction via Path c leading to the predominant formation of 13.
3. Experimental Section
3.1. General
All reagents were purchased from commercial sources and used without further purification. 1H and 13C NMR spectra were recorded on Bruker DPX-400 spectrometer (400 MHz and 100 MHz, respectively, Billerica, MA, USA) in CDCl3 using TMS as an internal standard. The 13C NMR assignments were performed via DEPT experiments. A Shimadzu IR spectrometer equipped with an ATR detector (Kyoto, Japan) was used to record infrared spectra. High-resolution mass spectra were obtained on an AB SCIEX Triplet TOF 4600 mass spectrometer (Framingham, MA, USA). Melting points were recorded on a Stanford Research Systems Optimelt automated melting point system (Sunnyvale, CA, USA) and were uncorrected.
3.2. Synthesis of Isoxazoline N-oxide 4b
To a solution of ethyl α-nitrocinnamate 1b (94.6 mg, 0.43 mmol) in acetonitrile (1.3 mL), ethyl nitroacetate (48 μL, 0.43 mmol) and triethylamine (60 μL, 0.43 mmol) were added, and the resultant mixture was stirred at room temperature for 30 min. After removal of the solvent under reduced pressure, the residual brown oil was dissolved in ethyl acetate (10 mL) and washed with water (10 mL × 4), and then dried over magnesium sulfate. After removal of the solvent, the residue was purified by column chromatography on silica gel to afford isoxazoline N-oxide 4b (eluted with hexane/EtOAc = 8/2, 121 mg, 0.41 mmol, 95%) as a yellow solid.
3,5-Bis(ethoxycarbonyl)-4-phenyl-2-isoxazoline 2-oxide (4b) [22]. Yellow solid, yield; 95%, m.p. 75–76 °C. 1H NMR (400 MHz, CDCl3) δ 7.40–7.30 (m, 5H), 4.93 (d, J = 2.8 Hz, 1H), 4.84 (d, J = 2.8 Hz, 1H), 4.35 (dq, J = 10.8, 7.2 Hz, 1H), 4.32 (dq, J = 10.8, 7.2 Hz, 1H), 4.21 (dq, J = 10.8, 7.2 Hz, 1H), 4.17 (dq, J = 10.8, 7.2 Hz, 1H), 1.35 (dd, J = 7.2, 7.2 Hz, 3H), 1.17 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.2 (C), 158.1 (C), 138.1 (C), 129.3 (CH), 128.7 (CH), 127.0 (CH), 109.0 (C), 78.8 (CH), 62.7 (CH2), 62.0 (CH2), 52.7 (CH), 14.1 (CH3), 13.9 (CH3).
3.3. Typical Procedure for Synthesis of 2,3-Dihydrofuran 6
To a solution of ethyl α-nitrocinnamate 1b (71.5 mg, 0.32 mmol) in acetonitrile (1.0 mL), ethyl acetoacetate 5a (41 μL, 0.32 mmol) and triethylamine (45 μL, 0.32 mmol) were added, and the resultant mixture was heated at 60 °C for 3 h. After removal of the solvent under reduced pressure, the residual orange oil was purified by column chromatography on silica gel to afford 2,3-dihydrofuran 6a (eluted with hexane/EtOAc = 1/1, 88 mg, 0.29 mmol, 92%) as a pale yellow oil. When other 1,3-dicarbonyl compounds 5 were used, the experiment was conducted in the same way.
2,4-Bis(ethoxycarbonyl)-2,3-dihydro-5-methyl-3-phenylfuran (6a) [23]. Pale yellow oil, yield; 92%. 1H NMR (400 MHz, CDCl3) δ 7.34–7.30 (m, 2H), 7.27–7.22 (m, 3H), 4.83 (d, J = 4.8 Hz, 1H), 4.41 (dq, J = 4.8, 1.2 Hz, 1H), 4.30 (dq, J = 10.8, 7.2 Hz, 1H), 4.26 (dq, J = 10.8, 7.2 Hz, 1H), 4.04 (dq, J = 10.8, 7.2 Hz, 1H), 3.98 (dq, J = 10.8, 7.2 Hz, 1H), 2.40 (d, J = 1.2 Hz, 3H), 1.32 (dd, J = 7.2, 7.2 Hz, 3H), 1.07 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.1 (C), 168.4 (C), 164.9 (C), 142.6 (C), 128.6 (CH), 127.2 (CH), 127.1(CH), 106.4 (C), 85.8 (CH), 61.8 (CH2), 59.6 (CH2), 52.8 (CH), 14.2 (CH3), 14.1 (CH3), 14.1 (CH3); IR (ATR/cm−1) 1755, 1701, 1651, 1207, 1088, 1038; HRMS (ESI/TOF) calcd. for [M + H]+ C17H21O5: 305.1384, found: 305.1384.
2,4-Bis(ethoxycarbonyl)-5-trifluoromethyl-2,3-dihydro-3-phenylfuran (6b). Yellow oil, yield; 43%. 1H NMR (400 MHz, CDCl3) δ 7.4–7.2 (m, 5H), 4.99 (d, J = 4.8 Hz, 1H), 4.62 (dq, J = 4.8, 2.4 Hz, 1H), 4.32 (dq, J = 11.6, 7.2 Hz, 1H), 4.30 (dq, J = 11.6, 7.2 Hz, 1H), 4.12 (dq, J = 10.8, 7.2 Hz, 1H), 4.05 (dq, J = 10.8, 7.2 Hz, 1H), 1.33 (dd, J = 7.2, 7.2 Hz, 3H), 1.13 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 168.5 (C), 161.2 (C), 151.2 (C, q, J = 40.0 Hz), 139.8 (C), 129.1 (CH), 128.1 (CH), 127.2 (CH), 118.0 (C, q, J = 271.0 Hz), 113.0 (C, q, J = 3.0 Hz), 86.2 (CH), 62.3 (CH2), 61.1 (CH2), 53.8 (CH), 14.1 (CH3), 13.7 (CH3); IR (ATR/cm−1) 1759, 1728, 1200, 1157, 1111; HRMS (ESI/TOF) calcd. for [M + H]+ C17H18F3O5: 359.1101, found: 359.1092.
2,4-Bis(ethoxycarbonyl)-2,3-dihydro-1,3-diphenylfuran (6c). Colorless oil, yield; 62%. 1H NMR (400 MHz, CDCl3) δ 7.98–7.95 (m, 2H), 7.48–7.42 (m, 3H), 7.42–7.35 (m, 4H), 7.35–7.28 (m, 1H), 4.96 (d, J = 4.4 Hz, 1H), 4.62 (d, J = 4.4 Hz, 1H), 4.34 (dq, J = 10.8, 7.2 Hz, 1H), 4.31 (dq, J = 10.8, 7.2 Hz, 1H), 4.01 (dq, J = 10.8, 7.2 Hz, 1H), 3.98 (dq, J = 10.8, 7.2 Hz, 1H), 1.36 (dd, J = 7.2, 7.2 Hz, 3H), 1.03 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 170.2 (C), 165.4 (C), 164.1 (C), 142.5 (C), 130.1 (CH), 129.8 (CH), 129.2 (C), 128.8 (CH), 127.7 (CH), 127.4 (CH), 127.2 (CH), 106.7 (C), 85.0 (CH), 61.8 (CH2), 59.9 (CH2), 54.1 (CH), 14.2 (CH3), 13.9 (CH3); IR (ATR/cm−1) 1751, 1697, 1628, 1203, 1076, 752, 694; HRMS (ESI/TOF) calcd. for [M + H]+ C22H23O5: 367.1540, found: 367.1540.
4-Ethanoyl-2-ethoxycarbonyl-2,3-dihydro-5-methyl-3-phenylfuran (6d) [24]. Yellow solid, yield; quant., m.p. 63–64 °C. 1H NMR (400 MHz, CDCl3) δ 7.37–7.33 (m, 2H), 7.30–7.23 (m, 3H), 4.72 (d, J = 4.8 Hz, 1H), 4.49 (dq, J = 4.8, 1.2 Hz, 1H), 4.31 (dq, J = 10.8, 7.2 Hz, 1H), 4.27 (dq, J = 10.8, 7.2 Hz, 1H), 2.44 (d, J = 1.2 Hz, 3H), 1.95 (s, 3H), 1.34 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 194.3 (C), 170.0 (C), 168.6 (C), 142.2 (C), 129.1 (CH), 127.6 (CH), 127.2 (CH), 115.1 (C), 86.0 (CH), 61.9 (CH2), 53.3 (CH), 29.6 (CH3), 14.9 (CH3), 14.2 (CH3); IR (ATR/cm−1) 1755, 1674, 1624, 1604, 1196, 1038; HRMS (ESI/TOF) calcd. for [M + H]+ C16H18O4Na: 297.1097, found: 297.1099.
5,6-Cyclohexa-2-ethoxycarbonyl-2,3-dihydro-3-phenylfuran-4-one (6e) [25]. Yellow oil, yield; quant. 1H NMR (400 MHz, CDCl3) δ 7.34–7.31 (m, 2H), 7.27–7.21 (m, 3H), 4.96 (d, J = 4.8 Hz, 1H), 4.46 (br d, J = 4.8 Hz, 1H), 4.32 (dq, J = 10.8, 7.2 Hz, 1H), 4.27 (dq, J = 10.8, 7.2 Hz, 1H), 2.68–2.65 (m, 2H), 2.44–2.31 (m, 2H), 2.19–2.10 (m, 2H), 1.33 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 194.3 (C), 177.4 (C), 169.5 (C), 141.1 (C), 128.9 (CH), 127.4 (CH), 127.0 (CH), 115.8 (C), 88.0 (CH), 62.0 (CH2), 49.8 (CH), 36.8 (CH2), 23.9 (CH2), 21.7 (CH2), 14.2 (CH3); IR (ATR/cm−1) 1751, 1639, 1396, 1219, 748; HRMS (ESI/TOF) calcd. for [M + H]+ C17H19O4: 287.1278, found: 287.1278.
3-Ethoxycarbonyl-4,5-dihydro-5-(4-methylbenzoyl)-4-phenylisoxazoline 2-oxide (13). Yellow oil, yield; 64%. 1H NMR (400 MHz, CDCl3) δ 7.81 (d, J = 8.0 Hz, 2H), 7.42–7.36 (m, 5H), 7.29 (d, J = 8.0 Hz, 2H), 5.66 (d, J = 3.6 Hz, 1H), 5.12 (d, J = 3.6 Hz, 1H), 4.19 (dq, J = 10.8, 7.2 Hz, 1H), 4.14 (dq, J = 10.8, 7.2 Hz, 1H), 2.43 (s, 3H), 1.13 (dd, J = 7.2, 7.2 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 191.3 (C), 158.4 (C), 146.0 (C), 138.6 (C), 130.9 (C), 129.9 (CH), 129.5 (CH), 129.5 (CH), 128.8 (CH), 127.6 (CH), 109.8 (C), 81.7 (CH), 62.0 (CH2), 51.8 (CH), 22.0 (CH3), 14.0 (CH3); IR (KBr/cm−1) 1736, 1697, 1628, 1606, 1228, 740; HRMS (ESI/TOF) calcd. for [M + H]+ C20H20NO5: 354.1336 found: 354.1337.
4. Conclusions
2,3-Dihydrofurans and isoxazoline N-oxides were synthesized from α-nitrocinnamate 1 and active methylene compounds by conjugate addition and the subsequent O-attack. Via a series of reactions using several substrates, the nitro group increased the electrophilicity of the α-carbon and served as a good leaving group. The nitro group also served as a good nucleophile when it was converted to nitronate ion, which is more reactive than the enolate ion of ketone or ester functionalities. These results will be useful information for researchers studying synthetic chemistry using the multi-functionalities of a nitro group.
Supplementary Materials
The following are available online. 1H and 13C NMR spectra of 2a, 4, 6 and 13.
Author Contributions
Y.M. did experiments and wrote a draft; S.Y. and N.N. analyzed data and discussed with Y.M.; all authors contributed to the revision. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Asahara, H.; Sofue, A.; Kuroda, Y.; Nishiwaki, N. Alkynylation and cyanation of alkenes using diverse properties of a nitro group. J. Org. Chem. 2018, 83, 13691–13699. [Google Scholar] [CrossRef]
- Nishiwaki, N. Chemistry of nitroquinolones and synthetic application to unnatural 1-methyl-2-quinolone derivatives. Molecules 2010, 15, 5174–5195. [Google Scholar] [CrossRef]
- Hao, F.; Nishiwaki, N. Recent progress in nitro-promoted direct functionalization of pyridones and quinolones. Molecules 2020, 25, 673. [Google Scholar] [CrossRef]
- Chiurchiù, E.; Gabrielli, S.; Ballini, R.; Palmieri, A. A new valuable synthesis of polyfunctionalized furans starting from β-nitroenones and active methylene compounds. Molecules 2019, 24, 4575. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Gabrielli, S.; Ballini, R. Efficient two-step sequence for the synthesis of 2,5-disubstituted furan derivatives from functionalized nitroalkanes: successive amberlyst A21- and amberlyst 15-catalyzed processes. Chem. Commun. 2010, 46, 6165–6167. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, A.; Gabrielli, S.; Parlapiano, M.; Ballini, R. One-pot synthesis of alkyl pyrrole-2-carboxylates starting from β-nitroacrylates and primary amines. RSC Adv. 2015, 5, 4210–4213. [Google Scholar] [CrossRef]
- Mo, Y.; Liu, S.; Liu, Y.; Ye, L.; Shi, Z.; Zhao, Z.; Li, X. Highly stereoselective synthesis of 2,3-dihydrofurans via a cascade michael addition-alkylation process: A nitro group as the leaving group. Chem. Commun. 2019, 55, 6285–6288. [Google Scholar] [CrossRef]
- Noble, A.; Anderson, J.C. Nitro-mannich reaction. Chem. Rev. 2013, 113, 2887–2939. [Google Scholar] [CrossRef]
- Sukhorukov, A.Y. C-H reactivity of the a-position in nitrones and nitronates. Adv. Synth. Catal. 2020, 362, 724–754. [Google Scholar] [CrossRef]
- Baiazitov, R.; Denmark, S.E. Tandem [4 + 2]/[3 + 2] cycloadditions. In Methods and Applications of Cycloaddition Reactions in Organic Syntheses; Nishiwaki, N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2014; pp. 471–550. [Google Scholar]
- Nishiwaki, N.; Kumegawa, Y.; Iwai, K.; Yokoyama, S. Development of safely handleable synthetic equivalent of cyanonitrile oxide by 1,3-dipolar cycloaddition of nitroacetonitrile. Chem. Commun. 2019, 55, 7903–7905. [Google Scholar] [CrossRef]
- Ballini, R.; Petrini, M. The nitro to carbonyl conversion (nef reaction): New perspectives for a classical transformation. Adv. Synth. Catal. 2015, 357, 2371–2402. [Google Scholar] [CrossRef]
- Iwai, K.; Asahara, H.; Nishiwaki, N. Synthesis of functionalized 3-cyanoisoxazoles using a dianionic reagent. J. Org. Chem. 2017, 82, 5409–5415. [Google Scholar] [CrossRef] [PubMed]
- Kallitsakis, M.G.; Tancini, P.D.; Dixit, M.; Mpourmpakis, G.; Lykakis, I.N. Mechanistic studies on the Michael addition of amines and hydrazines to nitrostyrenes: Nitroalkane elimination via a retro-aza-henry-type process. J. Org. Chem. 2018, 83, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Melot, J.M.; Texier-Boullet, F.; Foucaud, A. Alumina supported potassium fluoride promoted reaction of nitroalkanes with electrophilic alkenes. synthesis of 4,5-dihydrofurans and isoxazoline n-oxides. Tetrahedron 1988, 44, 2215–2224. [Google Scholar] [CrossRef]
- Zhu, C.-Y.; Sun, X.-L.; Deng, X.-M.; Zheng, J.-C.; Tang, Y. Synthesis of isoxazoline N-oxides and its application in the formal synthesis of dehydroclausenamide. Tetrahedron 2008, 64, 5583–5589. [Google Scholar] [CrossRef]
- Zhu, C.-Y.; Deng, X.-M.; Sun, X.-L.; Zheng, J.-C.; Tang, Y. Highly enantioselective synthesis of isoxazoline N-oxides. Chem. Commun. 2008, 738–740. [Google Scholar] [CrossRef]
- Chernysheva, N.B.; Maksimenko, A.S.; Andreyanov, F.A.; Kislyi, V.P.; Strelenko, Y.A.; Khrustalev, V.N.; Victor, V. Synthesis of 3,4-diaryl-5-carboxy-4,5-dihydroisoxazole 2-oxides as valuable synthons for anticancer molecules. Tetrahedron 2017, 73, 6728–6735. [Google Scholar] [CrossRef]
- Le Menn, J.C.; Sarrazin, J.; Tallec, A. Formation of isoxazoline N-oxides and dihydrofurans by cyclocondensation of bromo- or chloromalonate carbanion with michael acceptors. Bull. Soc. Chim. Fr. 1991, 562–565. [Google Scholar]
- Shi, Z.; Tan, B.; Leong, W.; Wen, Y.; Zeng, X.; Lu, M.; Zhong, G. Catalytic asymmetric formal [4 + 1] annulation leading to optically active cis-isoxazoline N-oxides. Org. Lett. 2010, 12, 5402–5405. [Google Scholar] [CrossRef]
- Chen, X.; Peng, Y.; Yu, W.; Zhang, X.; Shao, X.; Xu, X.; Li, Z. Condition-based selective synthesis of 3,4,5-trisubstituted isoxazoline N-oxides, 4,5-dihydroisoxazoles and isoxazoles. ChemistrySelect 2018, 3, 6344–6348. [Google Scholar] [CrossRef]
- Rouf, A.; Sahin, E.; Tanyeli, C. Divergent synthesis of polysubstituted isoxazoles, isoxazoline N-oxides, and dihydroisoxazoles by a one-pot cascade reaction. Tetrahedron 2017, 73, 331–337. [Google Scholar] [CrossRef]
- Zhang, Y.-R.; Luo, F.; Huang, X.-J.; Xie, J.-W. Water-compatible cascade reaction: An efficient route to substituted 2,3-dihydrofurans. Chem. Lett. 2012, 41, 777–779. [Google Scholar] [CrossRef]
- Chuang, C.-P.; Chen, K.-P.; Hsu, Y.-L.; Tsai, A.-I.; Liu, S.-T. α-nitro carbonyl compounds in the synthesis of 2,3-dihydrofurans. Tetrahedron 2008, 64, 7511–7516. [Google Scholar] [CrossRef]
- Berestovitskaya, V.M.; Baichurin, R.I.; Baichuria, L.V.; Fel’gendler, A.V.; Aboskalova, N.I. Geminally activated β-nitrostyrenes in reactions with cyclic β-diketones. Russ. J. Gen. Chem. 2013, 83, 1755–1763. [Google Scholar] [CrossRef]
Sample Availability: Not available. |

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).