
The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations

 , ,
, ,  ,
, 
Abstract
1. Introduction
2. Results and Discussion
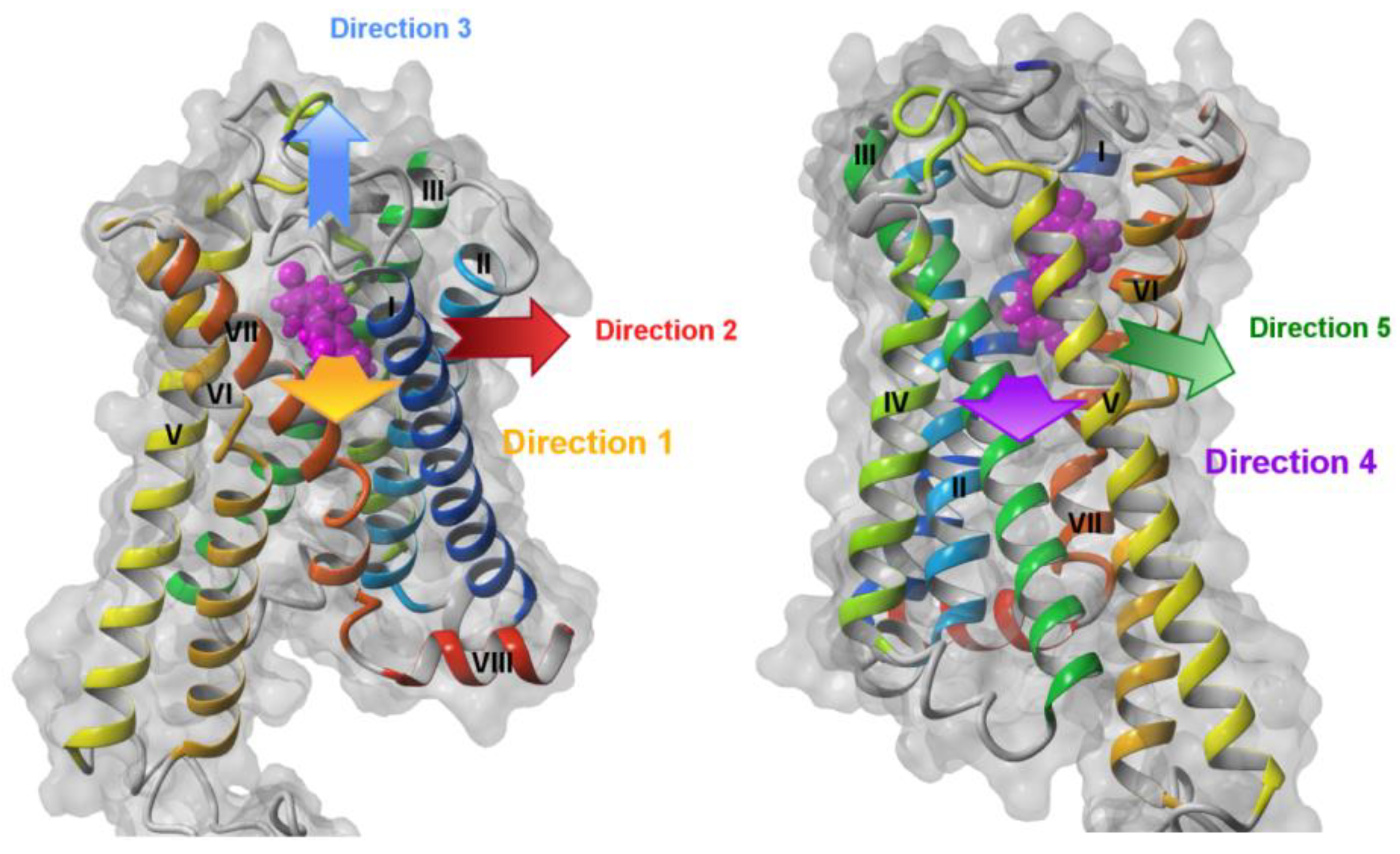
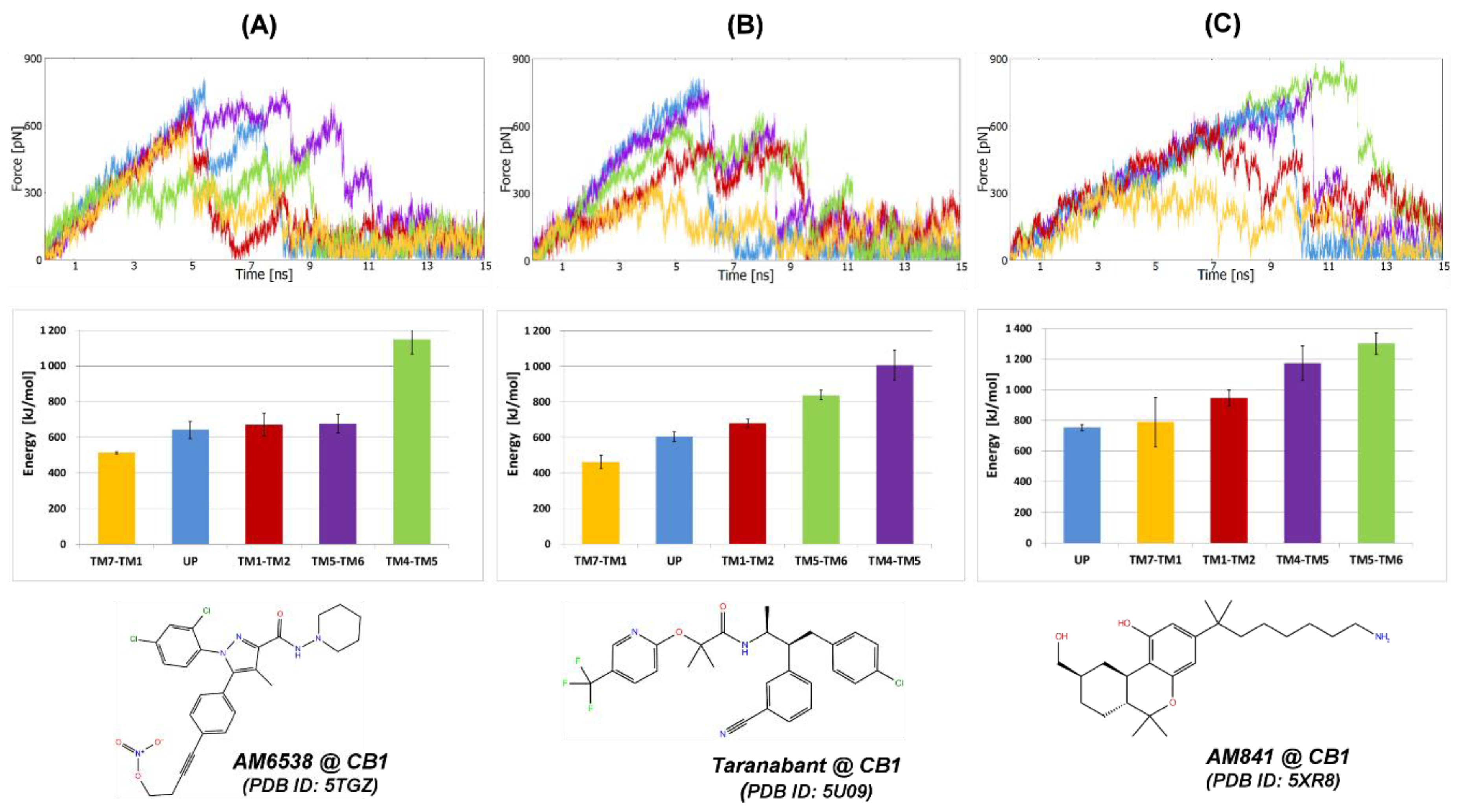
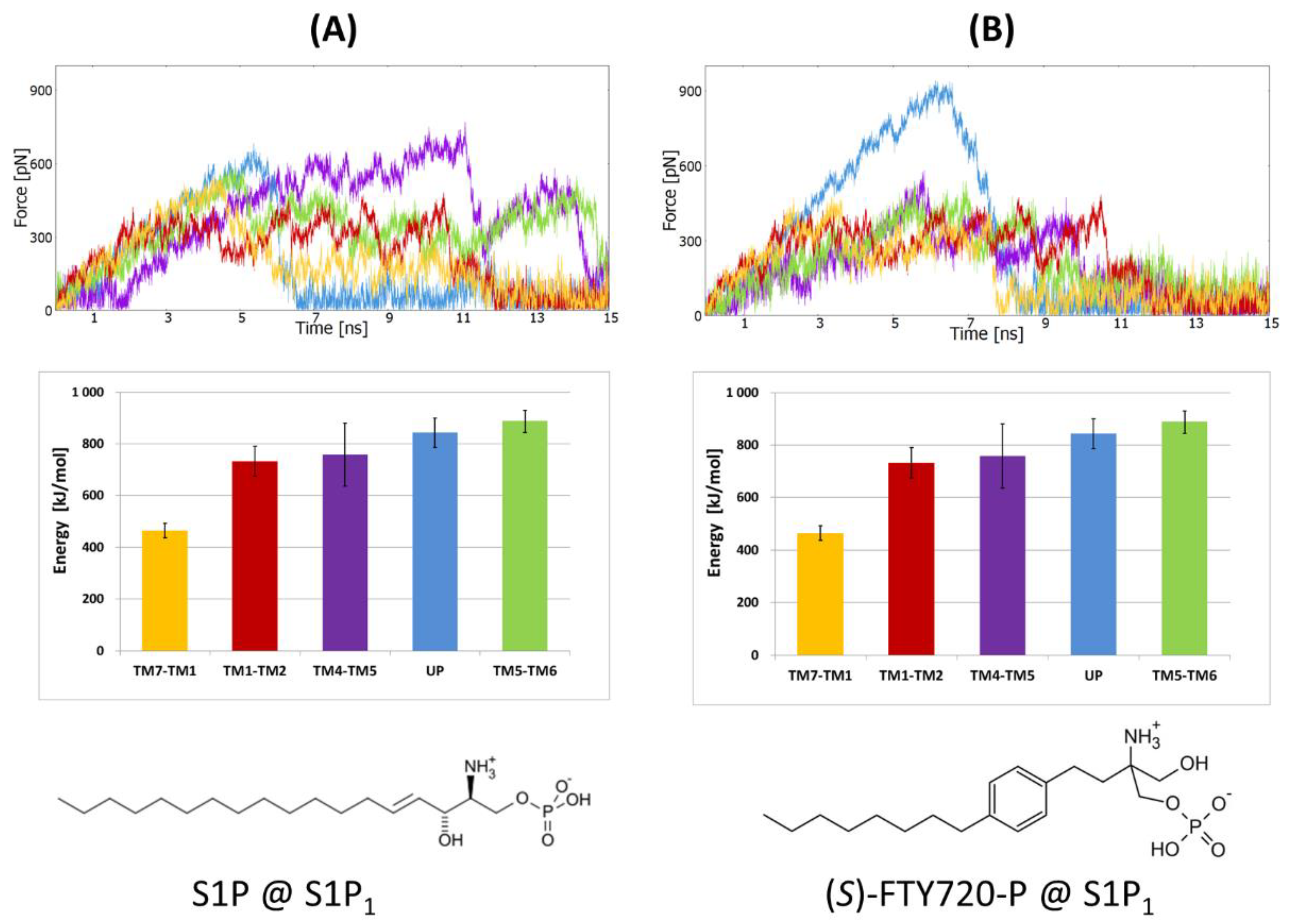
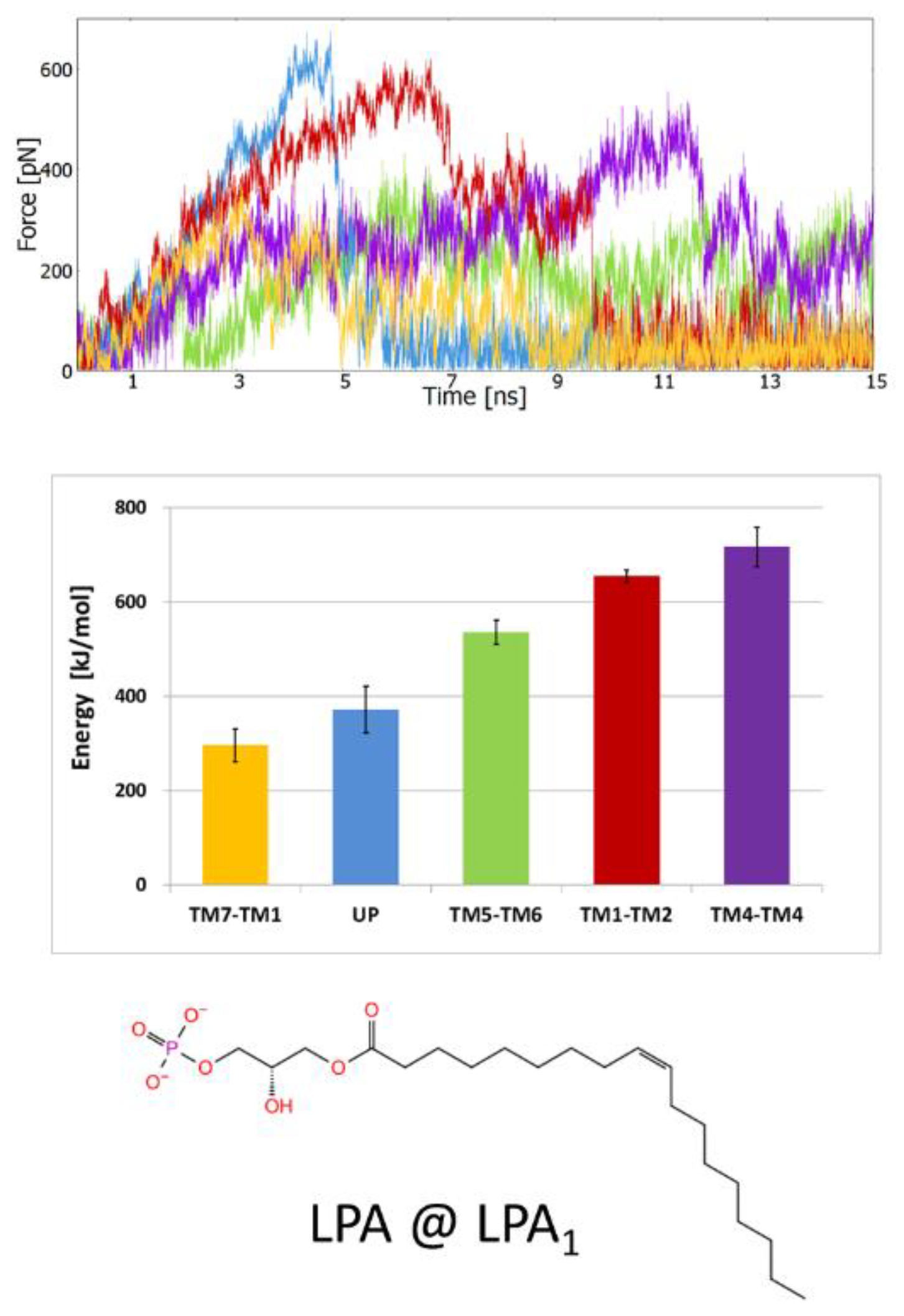
2.1. Steered Molecular Dynamics (SMD) Simulations
2.1.1. SMD Simulations of CB1 Complexes with Two Antagonists and One Agonist
2.1.2. SMD Simulations of S1P1 Complexes with Two Agonists
2.1.3. SMD Simulations of LPA1 Complex with Endogenous Agonist
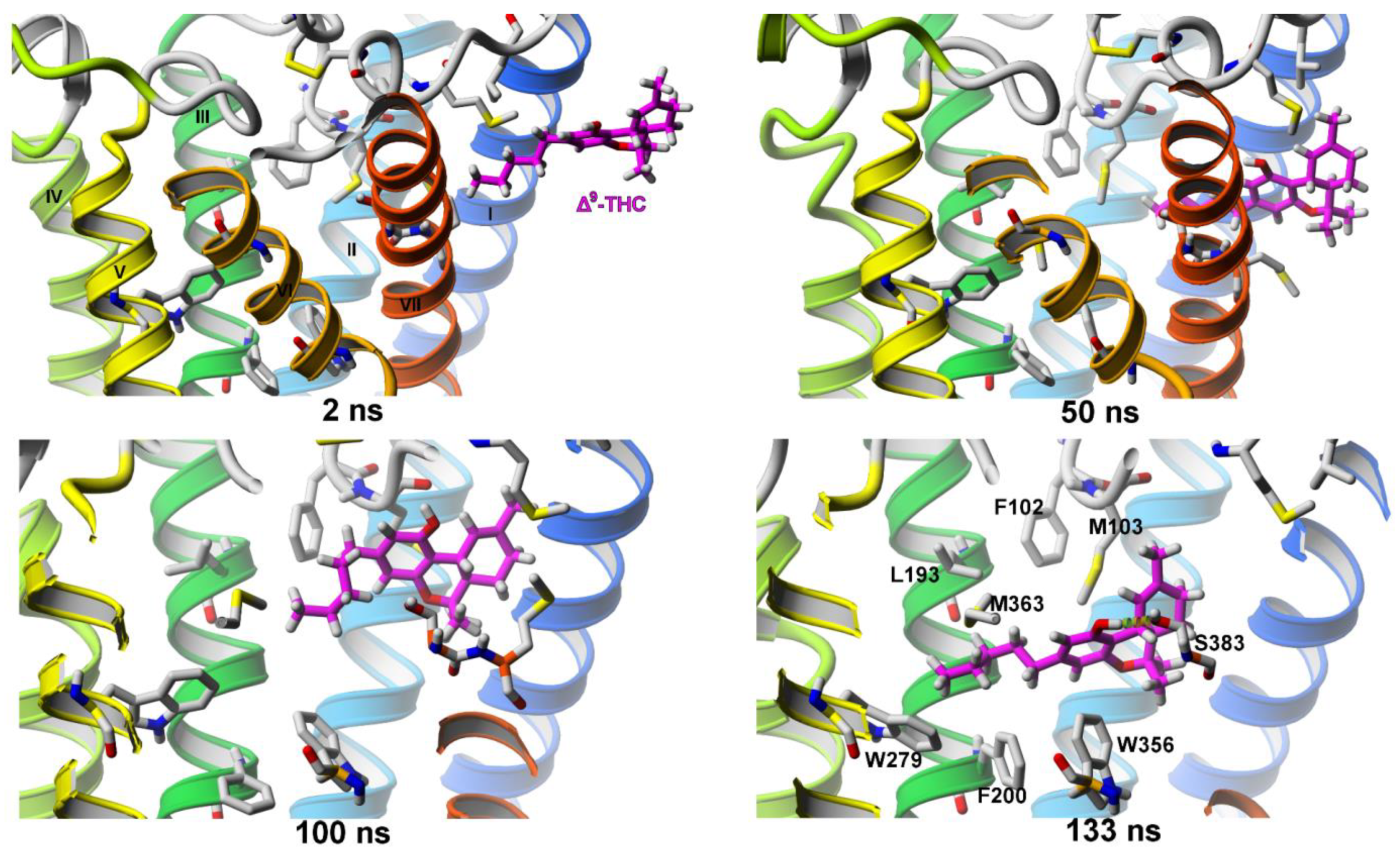
2.2. The Supervised Molecular Dynamics (SuMD) Simulations of Ligand Entrance
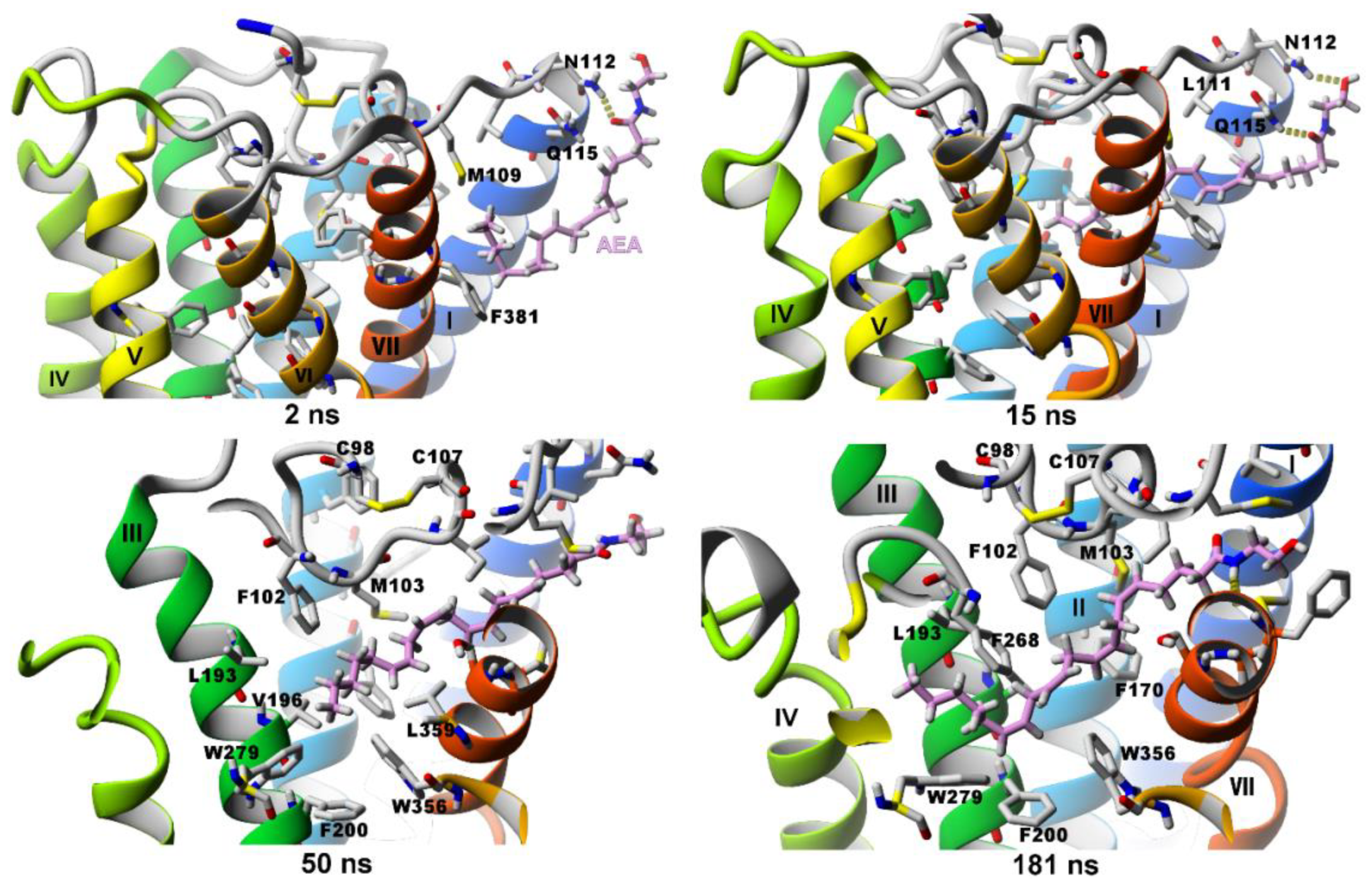
2.2.1. SuMD Simulations of Two Agonists Entering Inactive CB1 Receptor Structure
Δ9-THC Entrance to CB1 Receptor
Anandamide (AEA) Entrance to the CB1 Receptor
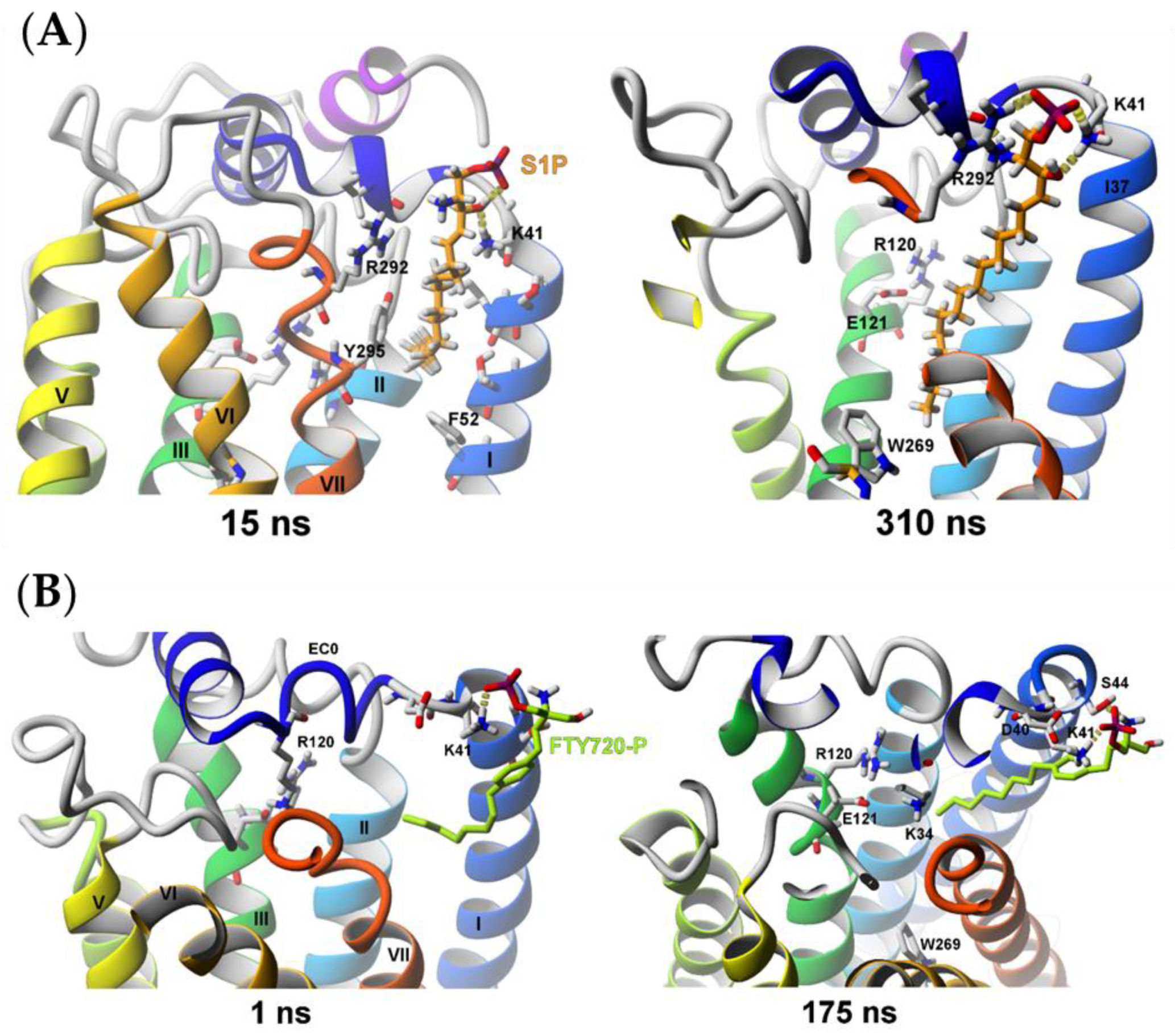
2.2.2. Sphingosine-1-phosphate (S1P) and (S)-FTY720-P Entrance into the S1P1 Receptor
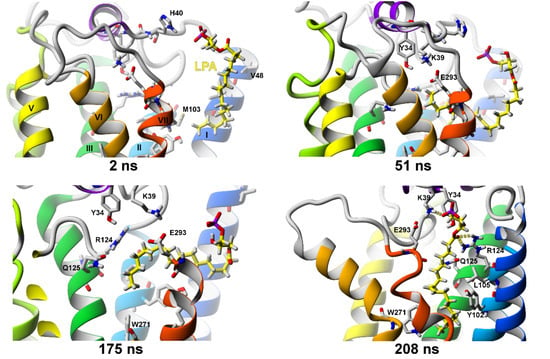
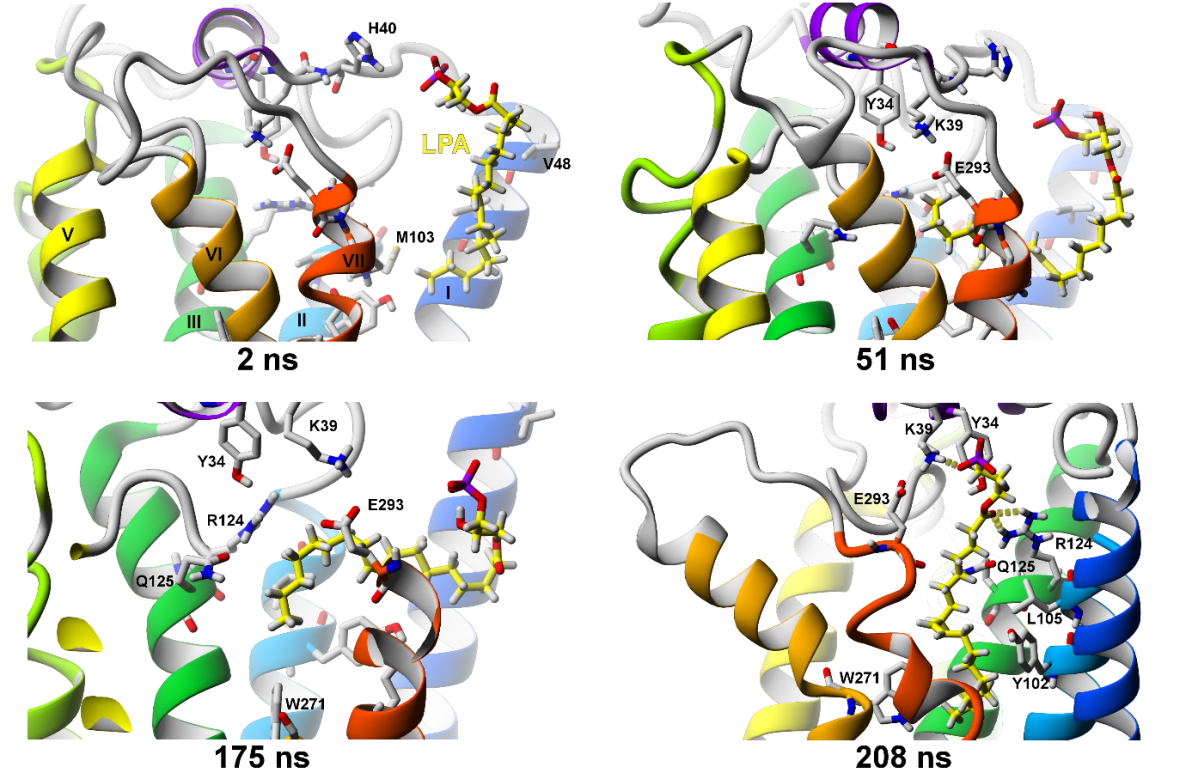
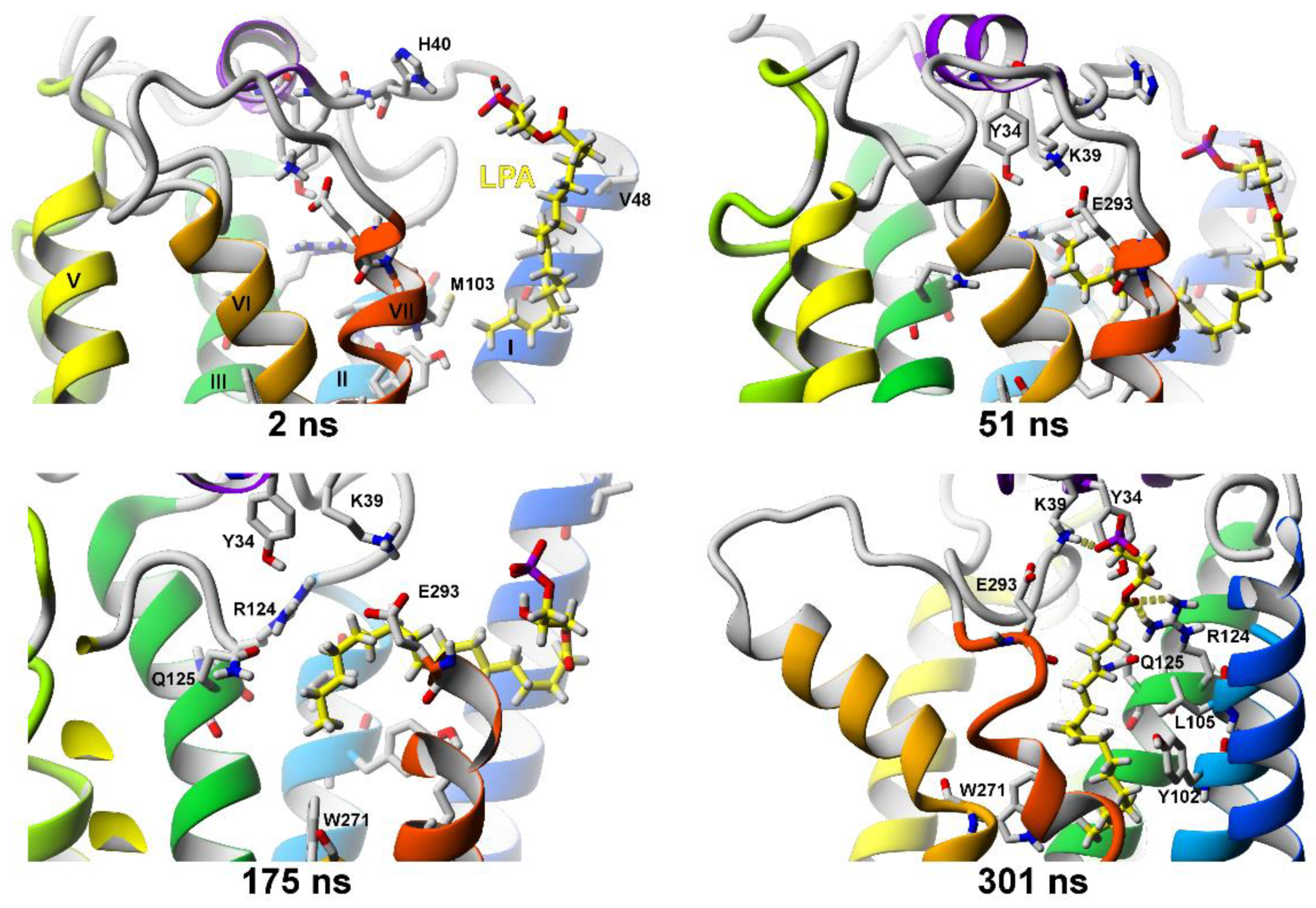
2.2.3. SuMD Simulations of LPA Ligand Entrance into the LPA1 Receptor
2.2.4. Comparison of SuMD Simulations to Earlier Data
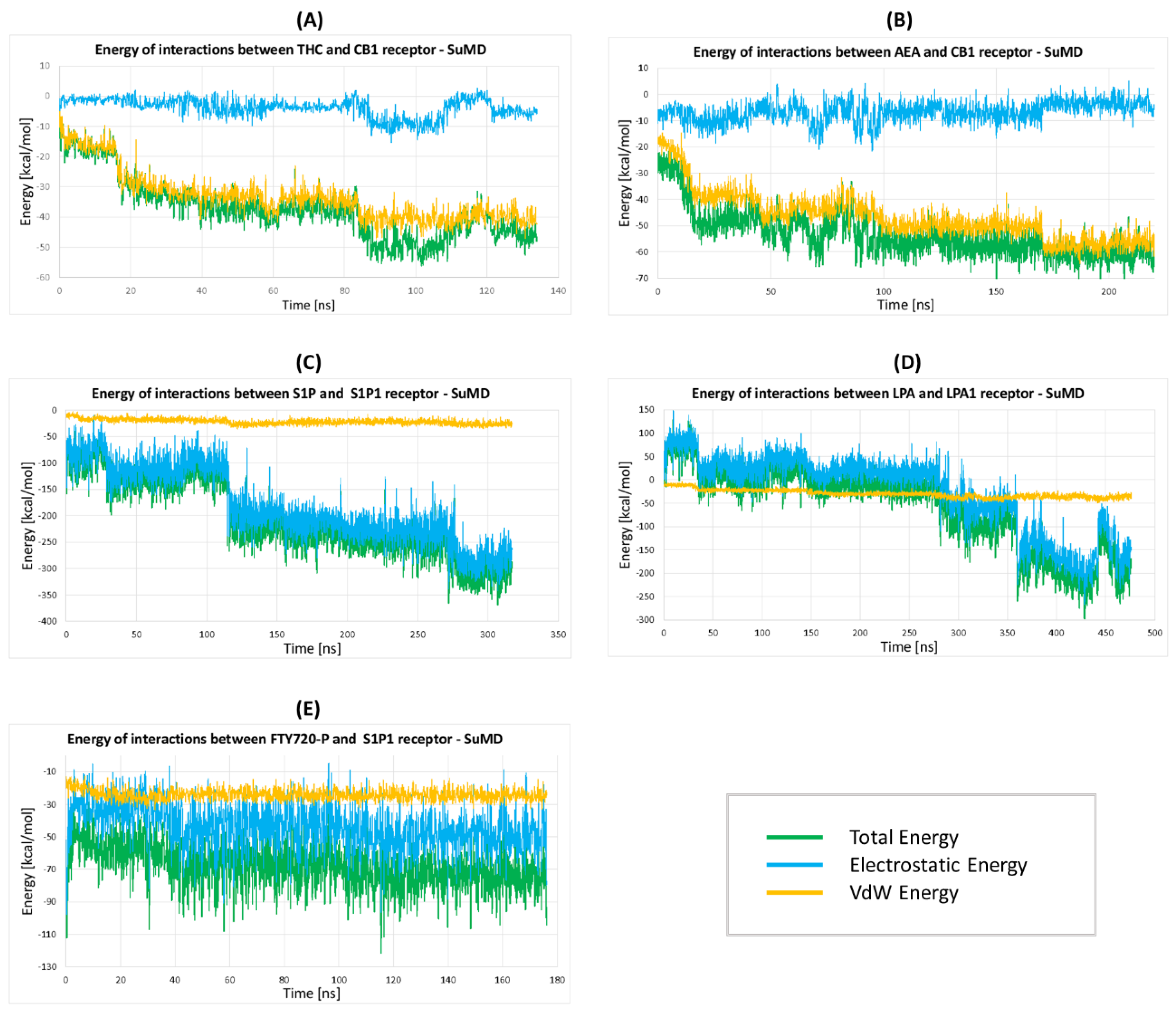
2.2.5. The Energy Profiles and Distances for SuMD Simulations
3. Materials and Methods
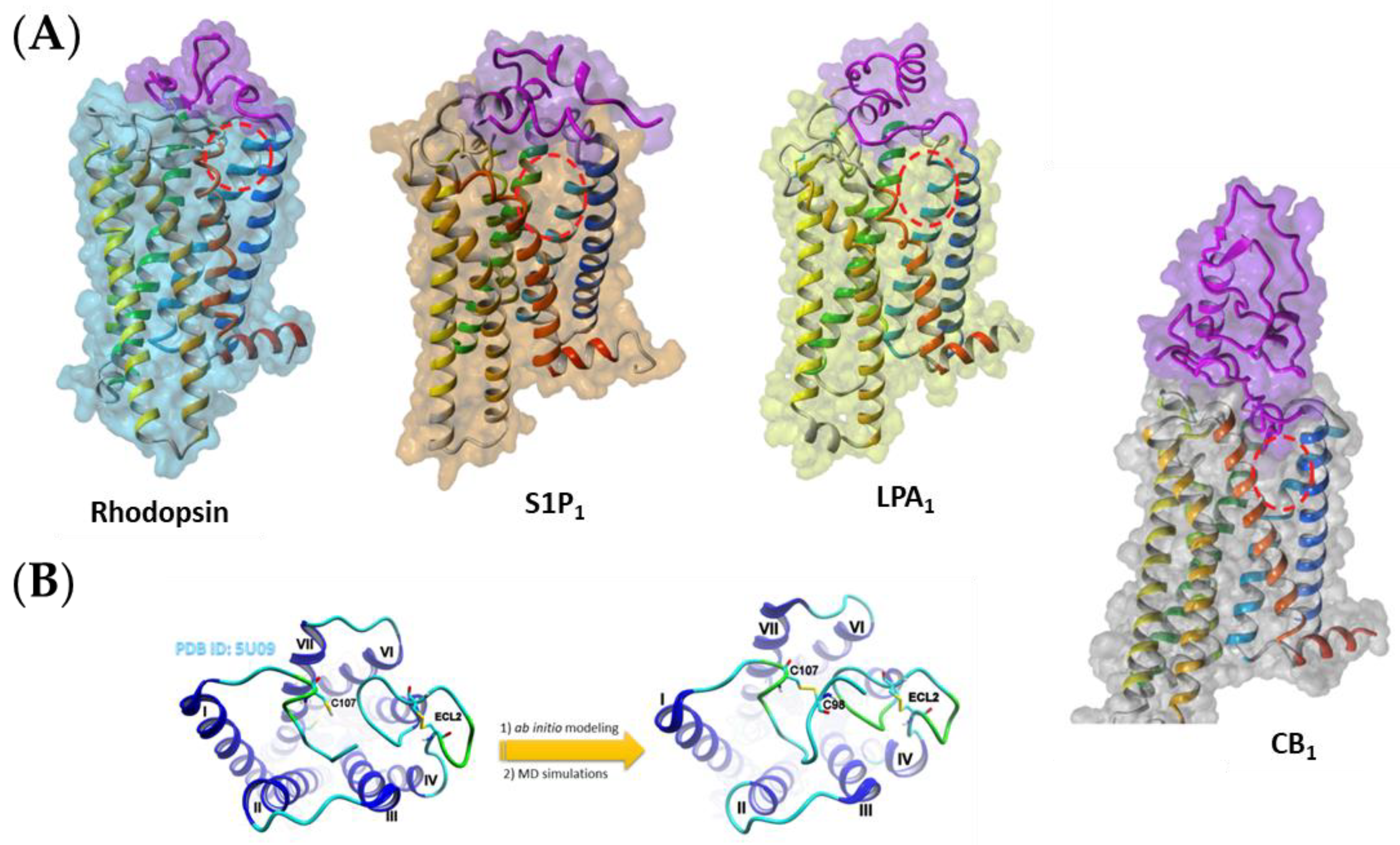
3.1. Obtaining the Receptor Structures
3.2. Ligand Docking and Preparations for SMD and SuMD Simulations
3.3. The SMD and SuMD Simulations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pandy-Szekeres, G.; Munk, C.; Tsonkov, T.M.; Mordalski, S.; Harpsoe, K.; Hauser, A.S.; Bojarski, A.J.; Gloriam, D.E. GPCRdb in 2018: Adding GPCR structure models and ligands. Nucleic Acids Res. 2018, 46, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D. The molecular logic of endocannabinoid signalling. Nat. Rev. Neurosci. 2003, 4, 873–884. [Google Scholar] [CrossRef] [PubMed]
- Yatomi, Y.; Ozaki, Y.; Ohmori, T.; Igarashi, Y. Sphingosine 1-phosphate: Synthesis and release. Prostaglandins Other Lipid Mediat. 2001, 64, 107–122. [Google Scholar] [CrossRef]
- Bracey, M.H.; Hanson, M.A.; Masuda, K.R.; Stevens, R.C.; Cravatt, B.F. Structural adaptations in a membrane enzyme that terminates endocannabinoid signaling. Science 2002, 298, 1793–1796. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, P.W.; Scheerer, P.; Park, J.H.; Choe, H.W.; Piechnick, R.; Ernst, O.P.; Hofmann, K.P.; Heck, M. A ligand channel through the G protein coupled receptor opsin. PLoS ONE 2009, 4, e4382. [Google Scholar] [CrossRef] [PubMed]
- Jakowiecki, J.; Filipek, S. Hydrophobic Ligand Entry and Exit Pathways of the CB1 Cannabinoid Receptor. J. Chem. Inf. Model. 2016, 56, 2457–2466. [Google Scholar] [CrossRef]
- Albert, R.; Hinterding, K.; Brinkmann, V.; Guerini, D.; Muller-Hartwieg, C.; Knecht, H.; Simeon, C.; Streiff, M.; Wagner, T.; Welzenbach, K.; et al. Novel immunomodulator FTY720 is phosphorylated in rats and humans to form a single stereoisomer. Identification, chemical proof, and biological characterization of the biologically active species and its enantiomer. J. Med. Chem. 2005, 48, 5373–5377. [Google Scholar] [CrossRef]
- Nilsson, U.K.; Andersson, R.G.G.; Ekeroth, J.; Hallin, E.C.; Konradsson, P.; Lindberg, J.; Svensson, S.P.S. Lack of stereospecificity in lysophosphatidic acid enantiomer-induced calcium mobilization in human erythroleukemia cells. Lipids 2003, 38, 1057–1064. [Google Scholar] [CrossRef]
- Latek, D.; Kolinski, M.; Ghoshdastider, U.; Debinski, A.; Bombolewski, R.; Plazinska, A.; Jozwiak, K.; Filipek, S. Modeling of ligand binding to G protein coupled receptors: Cannabinoid CB(1), CB(2) and adrenergic beta(2)AR. J. Mol. Model. 2011, 17, 2353–2366. [Google Scholar] [CrossRef]
- Latek, D.; Modzelewska, A.; Trzaskowski, B.; Palczewski, K.; Filipek, S. G protein-coupled receptors-recent advances. Acta Biochim. Pol. 2012, 59, 515–529. [Google Scholar] [CrossRef]
- Filipek, S. Molecular switches in GPCRs. Curr. Opin. Struct. Biol. 2019, 55, 114–120. [Google Scholar] [CrossRef] [PubMed]
- Hanson, M.A.; Roth, C.B.; Jo, E.; Griffith, M.T.; Scott, F.L.; Reinhart, G.; Desale, H.; Clemons, B.; Cahalan, S.M.; Schuerer, S.C.; et al. Crystal structure of a lipid G protein-coupled receptor. Science 2012, 335, 851–855. [Google Scholar] [CrossRef] [PubMed]
- Hurst, D.P.; Schmeisser, M.; Reggio, P.H. Endogenous lipid activated G protein-coupled receptors: Emerging structural features from crystallography and molecular dynamics simulations. Chem. Phys. Lipids 2013, 169, 46–56. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Chrencik, J.E.; Roth, C.B.; Terakado, M.; Kurata, H.; Omi, R.; Kihara, Y.; Warshaviak, D.; Nakade, S.; Asmar-Rovira, G.; Mileni, M.; et al. Crystal Structure of Antagonist Bound Human Lysophosphatidic Acid Receptor 1. Cell 2015, 161, 1633–1643. [Google Scholar] [CrossRef] [PubMed]
- Audet, M.; Stevens, R.C. Emerging structural biology of lipid G protein-coupled receptors. Protein Sci. 2019, 28, 292–304. [Google Scholar] [CrossRef] [PubMed]
- Hua, T.; Vemuri, K.; Pu, M.; Qu, L.; Han, G.W.; Wu, Y.; Zhao, S.; Shui, W.; Li, S.; Korde, A.; et al. Crystal Structure of the Human Cannabinoid Receptor CB1. Cell 2016, 167, 750–762. [Google Scholar] [CrossRef]
- Shao, Z.; Yin, J.; Chapman, K.; Grzemska, M.; Clark, L.; Wang, J.; Rosenbaum, D.M. High-resolution crystal structure of the human CB1 cannabinoid receptor. Nature 2016, 540, 602–606. [Google Scholar] [CrossRef]
- Hua, T.; Vemuri, K.; Nikas, S.P.; Laprairie, R.B.; Wu, Y.; Qu, L.; Pu, M.; Korde, A.; Jiang, S.; Ho, J.H.; et al. Crystal structures of agonist-bound human cannabinoid receptor CB1. Nature 2017, 547, 468–471. [Google Scholar] [CrossRef]
- Yarov-Yarovoy, V.; Schonbrun, J.; Baker, D. Multipass membrane protein structure prediction using Rosetta. Proteins 2006, 62, 1010–1025. [Google Scholar] [CrossRef]
- Krieger, E.; Vriend, G. New ways to boost molecular dynamics simulations. J. Comput. Chem. 2015, 36, 996–1007. [Google Scholar] [CrossRef]
- Ballesteros, J.A.; Weinstein, H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995, 25, 366–428. [Google Scholar]
- Vanommeslaeghe, K.; Raman, E.P.; MacKerell, A.D., Jr. Automation of the CHARMM General Force Field (CGenFF) II: Assignment of bonded parameters and partial atomic charges. J. Chem. Inf. Model. 2012, 52, 3155–3168. [Google Scholar] [CrossRef] [PubMed]
- Brooks, B.R.; Brooks, C.L., 3rd; Mackerell, A.D., Jr.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Jo, S.; Kim, T.; Iyer, V.G.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI Input Generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM Simulations Using the CHARMM36 Additive Force Field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Sabbadin, D.; Moro, S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR-ligand recognition pathway in a nanosecond time scale. J. Chem. Inf. Model. 2014, 54, 372–376. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| SuMD Ligand | Crystal Ligand | Distance [Å] |
|---|---|---|
| CB1 receptor ligands | ||
| THC | 7DY (PDB id: 5u09) | 3.780 |
| THC | 8D0 (PDB id: 5xr8) | 3.596 |
| THC | 8D3 (PDB id: 5xra) | 3.595 |
| THC | ZDG (PDB id: 5tgz) | 2.745 |
| AEA | 7DY (PDB id: 5u09) | 3.020 |
| AEA | 8D0 (PDB id: 5xr8) | 3.211 |
| AEA | 8D3 (PDB id: 5xra) | 3.463 |
| AEA | ZDG (PDB id: 5tgz) | 2.759 |
| S1P1 receptor ligands | ||
| S1P | ML5 (PDB id: 3V2Y) | 7.920 |
| S1P | ML5 (PDB id: 3V2W) | 8.069 |
| LPA1 receptor ligands | ||
| (S)-LPA | ON3 (PDB id: 4z36) | 3.346 |
| (S)-LPA | ON7 (PDB id: 4z34) | 3.541 |
| (S)-LPA | ON9 (PDB id: 4z35) | 3.556 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jakowiecki, J.; Orzeł, U.; Chawananon, S.; Miszta, P.; Filipek, S. The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules 2020, 25, 1930. https://doi.org/10.3390/molecules25081930
Jakowiecki J, Orzeł U, Chawananon S, Miszta P, Filipek S. The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules. 2020; 25(8):1930. https://doi.org/10.3390/molecules25081930
Chicago/Turabian StyleJakowiecki, Jakub, Urszula Orzeł, Sathapana Chawananon, Przemysław Miszta, and Sławomir Filipek. 2020. "The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations" Molecules 25, no. 8: 1930. https://doi.org/10.3390/molecules25081930
APA StyleJakowiecki, J., Orzeł, U., Chawananon, S., Miszta, P., & Filipek, S. (2020). The Hydrophobic Ligands Entry and Exit from the GPCR Binding Site-SMD and SuMD Simulations. Molecules, 25(8), 1930. https://doi.org/10.3390/molecules25081930