
Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa

 , , ,
, , ,
Abstract
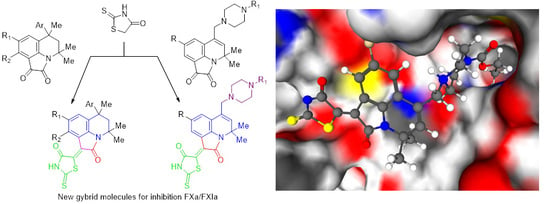
1. Introduction
2. Results and Discussion
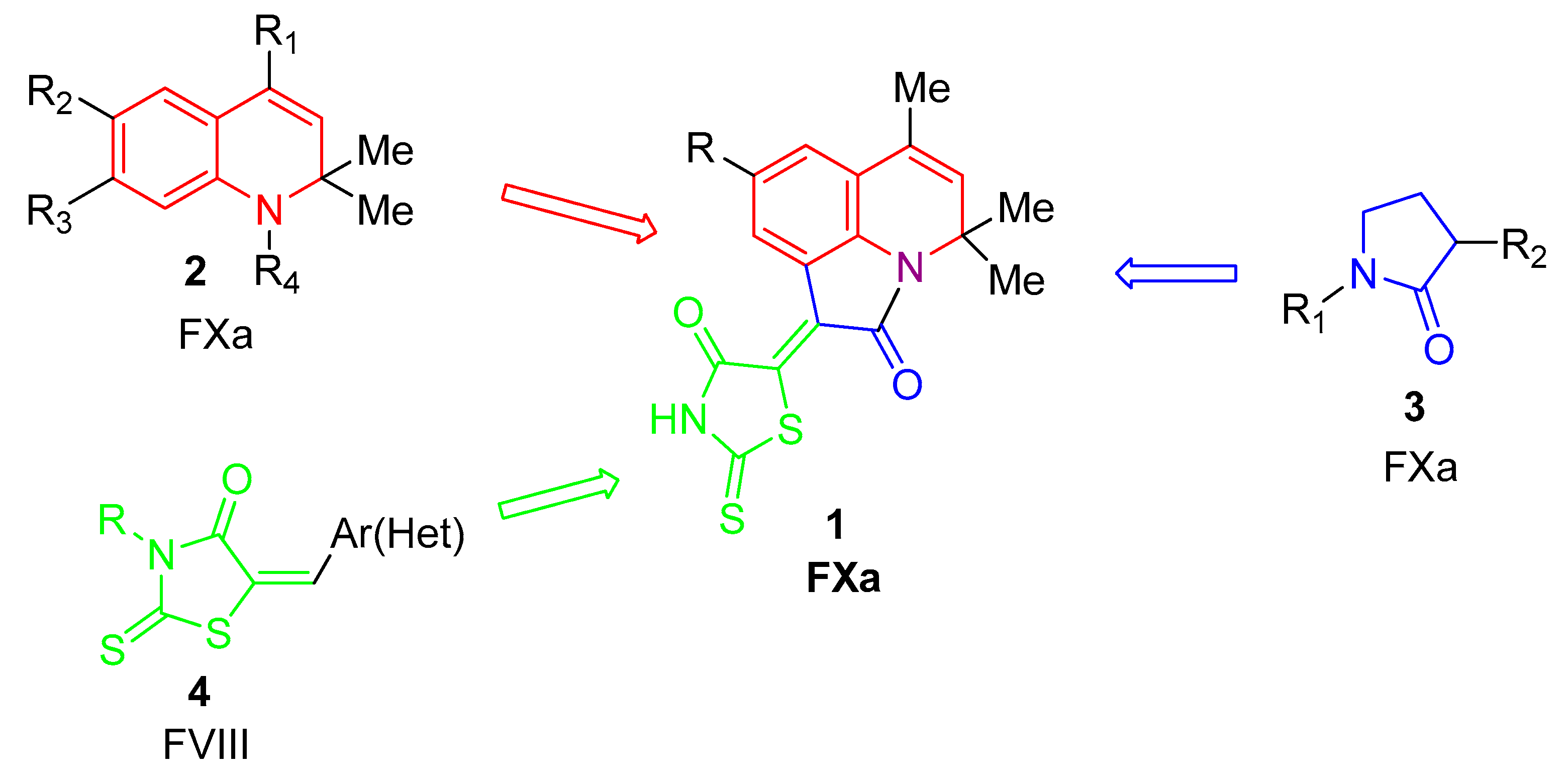
2.1. Design of Pyrrolo[3,2,1-Ij]quinolin-2(1H)-One-Based Derivatives
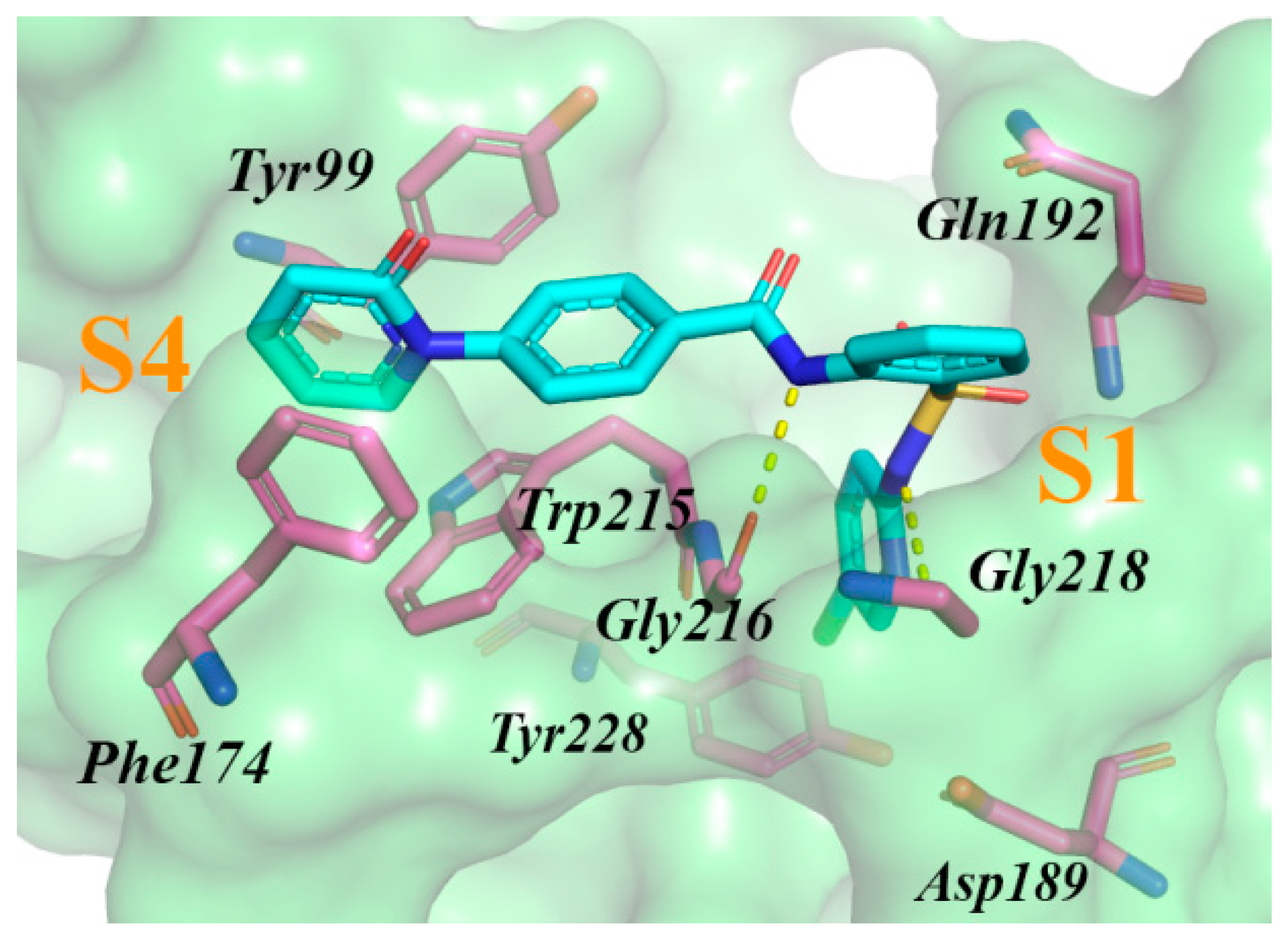
2.2. Results of Virtual Screening and In Vitro Testing
2.3. Determining IC50 Values and Mechanism of Inhibition
3. Material and Methods
3.1. Molecular Docking Studies with Semiempirical Postprocessing
3.2. Virtual Screening of Focused Library
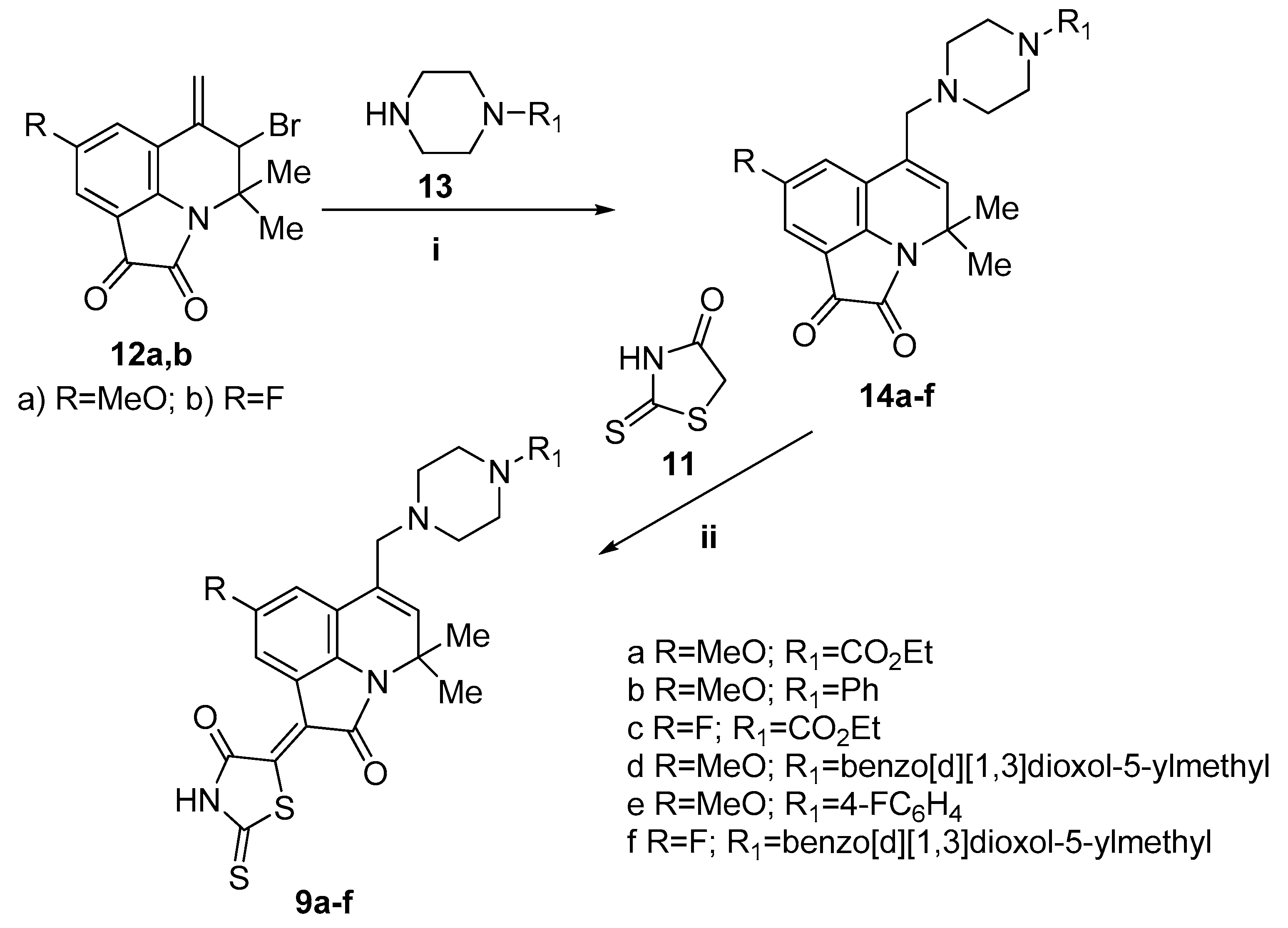
3.3. Synthesis
3.3.1. Instrumentation
3.3.2. Chemicals
General procedure for the synthesis of (2,4,5,6-tetrahydro-1H-pyrrolo[3,2,1-ij]quinolin-1-ylidene)-2-thioxothiazolidin-4-ones 7a–c
General procedure for the synthesis of 5-(8-R-4,4-Dimethyl-2-oxo-6-((4-R1-piperazin-1-yl)methyl)-2,4-dihydro-1H-pyrrolo[3,2,1-ij]quinolin-1-ylidene)-2-thioxothiazolidin-4-ones 9a–f
3.4. In Vitro Assays
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References and Notes
- Wendelboe, A.M.; Raskob, G.E. Global Burden of Thrombosis. Circ. Res. 2016, 118, 1340–1347. [Google Scholar] [CrossRef] [PubMed]
- De Candia, M.; Lopopolo, G.; Altomare, C. Novel factor Xa inhibitors: A patent review. Expert Opin. Ther. Pat. 2009, 19, 1535–1580. [Google Scholar] [CrossRef] [PubMed]
- Gailani, D.; Broze, G.J.J. Factor XI activation in a revised model of blood coagulation. Science 1991, 253, 909–912. [Google Scholar] [CrossRef] [PubMed]
- Yeh, C.H.; Hogg, K.; Weitz, J.I. Overview of the new oral anticoagulants: Opportunities and challenges. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1056–1065. [Google Scholar] [CrossRef] [PubMed]
- Ansell, J.E. Reversing the Effect of Oral Anticoagulant Drugs: Established and Newer Options. Am. J. Cardiovasc. Drugs 2016, 16, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Patel, M.R.; Hellkamp, A.S.; Lokhnygina, Y.; Piccini, J.P.; Zhang, Z.; Mohanty, S.; Singer, D.E.; Hacke, W.; Breithardt, G.; Halperin, J.L.; et al. Outcomes of discontinuing rivaroxaban compared with warfarin in patients with nonvalvular atrial fibrillation: Analysis from the ROCKET AF trial (Rivaroxaban Once-Daily, Oral, Direct Factor Xa Inhibition Compared With Vitamin K Antagonism for Preventio). J. Am. Coll. Cardiol. 2013, 61, 651–658. [Google Scholar] [CrossRef]
- Polzin, A.; Dannenberg, L.; Wolff, G.; Helten, C.; Achilles, A.; Hohlfeld, T.; Zeus, T.; Kelm, M.; Massberg, S.; Petzold, T. Non-vitamin K oral anticoagulants (NOAC) and the risk of myocardial infarction: Differences between factor IIa and factor Xa inhibition? Pharmacol. Ther. 2019, 195, 1–4. [Google Scholar] [CrossRef]
- Patel, N.R.; Patel, D.V.; Murumkar, P.R.; Yadav, M.R. Contemporary developments in the discovery of selective factor Xa inhibitors: A review. Eur. J. Med. Chem. 2016, 121, 671–698. [Google Scholar] [CrossRef]
- Quan, M.L.; Pinto, D.J.P.; Smallheer, J.M.; Ewing, W.R.; Rossi, K.A.; Luettgen, J.M.; Seiffert, D.A.; Wexler, R.R. Factor XIa Inhibitors as New Anticoagulants. J. Med. Chem. 2018, 61, 7425–7447. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Kutov, D.C.; Sulimov, A. V Advances in docking. Curr. Med. Chem. 2019, 26, 7555–7580. [Google Scholar] [CrossRef]
- Sulimov, A.V.; Kutov, D.C.; Sulimov, V.B. Supercomputer docking. Supercomput. Front. Innov. 2019, 6, 26–50. [Google Scholar] [CrossRef]
- Meng, X.Y.; Zhang, H.X.; Mezei, M.; Cui, M. Molecular docking: A powerful approach for structure-based drug discovery. Curr. Comput. Aided Drug Des. 2011, 7, 146–157. [Google Scholar] [CrossRef] [PubMed]
- Fan, J.; Fu, A.; Zhang, L. Progress in molecular docking. Quant. Biol. 2019, 7, 83–89. [Google Scholar] [CrossRef]
- Lyu, J.; Wang, S.; Balius, T.E.; Singh, I.; Levit, A.; Moroz, Y.S.; O’Meara, M.J.; Che, T.; Algaa, E.; Tolmachova, K.; et al. Ultra-large library docking for discovering new chemotypes. Nature 2019, 566, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C. Beware of docking! Trends Pharmacol. Sci. 2015, 36, 78–95. [Google Scholar] [CrossRef] [PubMed]
- Fraga, C.A.M. Drug hybridization strategies: Before or after lead identification? Expert Opin. Drug Discov. 2009, 4, 605–609. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Gribkova, I.V.; Kochugaeva, M.P.; Katkova, E.V.; Sulimov, A.V.; Kutov, D.C.; Shikhaliev, K.S.; Medvedeva, S.M.; Krysin, M.Y.; Sinauridze, E.I.; et al. Application of molecular modeling to development of new factor Xa inhibitors. Biomed Res. Int. 2015, 2015. [Google Scholar] [CrossRef]
- Ilin, I.S.; Lipets, E.N.; Sulimov, A.V.; Kutov, D.C.; Shikhaliev, K.S.; Potapov, A.Y.; Krysin, M.Y.; Zubkov, F.I.; Sapronova, L.V.; Ataullakhanov, F.I.; et al. New factor Xa inhibitors based on 1,2,3,4-tetrahydroquinoline developed by molecular modelling. J. Mol. Graph. Model. 2019, 89, 215–224. [Google Scholar] [CrossRef]
- Young, R.J.; Borthwick, A.D.; Brown, D.; Burns-Kurtis, C.L.; Campbell, M.; Chan, C.; Charbaut, M.; Chung, C.; Convery, M.A.; Kelly, H.A.; et al. Structure and property based design of factor Xa inhibitors: Pyrrolidin-2-ones with biaryl P4 motifs. Bioorg. Med. Chem. Lett. 2008, 18, 23–27. [Google Scholar] [CrossRef]
- Nicolaes, G.A.F.; Kulharia, M.; Voorberg, J.; Kaijen, P.H.; Wroblewska, A.; Wielders, S.; Schrijver, R.; Sperandio, O.; Villoutreix, B.O. Rational design of small molecules targeting the C2 domain of coagulation factor VIII. Blood 2014, 123, 113–120. [Google Scholar] [CrossRef]
- Quan, M.L.; Wong, P.C.; Wang, C.; Woerner, F.; Smallheer, J.M.; Barbera, F.A.; Bozarth, J.M.; Brown, R.L.; Harpel, M.R.; Luettgen, J.M.; et al. Tetrahydroquinoline derivatives as potent and selective factor XIa inhibitors. J. Med. Chem. 2014, 57, 955–969. [Google Scholar] [CrossRef]
- Fjellstrom, O.; Akkaya, S.; Beisel, H.-G.; Eriksson, P.-O.; Erixon, K.; Gustafsson, D.; Jurva, U.; Kang, D.; Karis, D.; Knecht, W.; et al. Creating novel activated factor XI inhibitors through fragment based lead generation and structure aided drug design. PLoS ONE 2015, 10, e0113705. [Google Scholar] [CrossRef] [PubMed]
- Santana-Romo, F.; Lagos, C.F.; Duarte, Y.; Castillo, F.; Moglie, Y.; Maestro, M.A.; Charbe, N.; Zacconi, F.C. Innovative Three-Step Microwave-Promoted Synthesis of N-Propargyltetrahydroquinoline and 1,2,3-Triazole Derivatives as a Potential Factor Xa (FXa) Inhibitors: Drug Design, Synthesis, and Biological Evaluation. Molecules 2020, 25, 491. [Google Scholar] [CrossRef] [PubMed]
- Shaquiquzzaman, M.; Verma, G.; Marella, A.; Akhter, M.; Akhtar, W.; Khan, M.F.; Tasneem, S.; Alam, M.M. Piperazine scaffold: A remarkable tool in generation of diverse pharmacological agents. Eur. J. Med. Chem. 2015, 102, 487–529. [Google Scholar] [CrossRef] [PubMed]
- Rathi, A.K.; Syed, R.; Shin, H.-S.; Patel, R. V Piperazine derivatives for therapeutic use: A patent review (2010-present). Expert Opin. Ther. Pat. 2016, 26, 777–797. [Google Scholar] [CrossRef] [PubMed]
- Brito, A.F.; Moreira, L.K.S.; Menegatti, R.; Costa, E.A. Piperazine derivatives with central pharmacological activity used as therapeutic tools. Fundam. Clin. Pharmacol. 2019, 33, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Novichikhina, N.P.; Shestakov, A.S.; Potapov, A.Y.; Kosheleva, E.A.; Shatalov, G.V.; Verezhnikov, V.N.; Vandyshev, D.Y.; Ledeneva, I.V.; Shikhaliev, K.S. Synthesis of 4H-pyrrolo[3,2,1-ij]quinoline-1,2-diones containing a piperazine fragment and study of their inhibitory properties against protein kinases. Russ. Chem. Bull. 2020, 4, 787–792. [Google Scholar]
- Sulimov, A.V.; Kutov, D.C.; Oferkin, I.V.; Katkova, E.V.; Sulimov, V.B. Application of the docking program SOL for CSAR benchmark. J. Chem. Inf. Model 2013, 53, 1946–1956. [Google Scholar] [CrossRef]
- Katkova, E. V Investigation of influence of genetic algorithm parameters on the docking effectivness with the SOL program (in Russian). Numer. methods Program. 2012, 13, 536–550. [Google Scholar]
- Sinauridze, E.I.; Romanov, A.N.; Gribkova, I.V.; Kondakova, O.A.; Surov, S.S.; Gorbatenko, A.S.; Butylin, A.A.; Monakov, M.Y.; Bogolyubov, A.A.; Kuznetsov, Y.V.; et al. New synthetic thrombin inhibitors: Molecular design and experimental verification. PLoS ONE 2011, 6, e19969. [Google Scholar] [CrossRef]
- Sulimov, V.B.; Katkova, E.V.; Oferkin, I.V.; Sulimov, A.V.; Romanov, A.N.; Roschin, A.I.; Beloglazova, I.B.; Plekhanova, O.S.; Tkachuk, V.A.; Sadovnichiy, V.A. Application of molecular modeling to urokinase inhibitors development. Biomed Res. Int. 2014, 2014, 625176. [Google Scholar] [CrossRef] [PubMed]
- Hanwell, M.D.; Curtis, D.E.; Lonie, D.C.; Vandermeersch, T.; Zurek, E.; Hutchison, G.R. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012, 4, 17. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J. Optimization of parameters for semiempirical methods VI: More modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Klamt, A.; Schuurmann, G. COSMO: A new approach to dielectric screening in solvents with explicit expressions for the screening energy and its gradient. J. Chem. Soc. Perkin Trans. 1993, 2, 799–805. [Google Scholar] [CrossRef]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An open chemical toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef] [PubMed]
- Stewart, J.J.P. MOPAC2016, Stewart Computational Chemistry. Available online: http://openmopac.net (accessed on 20 October 2018).
- Stewart, J.J.P. Application of localized molecular orbitals to the solution of semiempirical self-consistent field equations. Int. J. Quantum Chem. 1996, 58, 133–146. [Google Scholar] [CrossRef]
- Marvin A Full Featured Chemical Editor for Making Science. Available online: http://www.chemaxon.com (accessed on 20 October 2018).
- DeLano, W.L. The PyMOL Molecular Graphics System, Version 1.4.1; Schrodinger, LLC: Portland, OR, USA, 2010. [Google Scholar]
Sample Availability: Samples of the compounds 7, 9, 14 are available from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| # | Compound | Structure | Factor Xa | Factor XIa | ||||
|---|---|---|---|---|---|---|---|---|
| Scoring Kunction in SOL, kcal/mol | Binding Enthalpy by PM7, kcal/mol | Percent Inhibition at 30 μM | Scoring Function in SOL, kcal/mol | Binding Enthalpy by PM7, kcal/mol | Percent Inhibition at 30 μM | |||
| 1 | 7a |  | −6.22 | −4.6 | 74 ± 8.8 | −4.40 | −26.20 | 100 ± 0.3 |
| 2 | 7b |  | −5.77 | −4.9 | 33 ± 1.6 | −4.61 | −17.74 | 7 ± 3.1 |
| 3 | 7c |  | −6.12 | −4.52 | 47 ± 1.7 | −5.64 | −33.20 | 87 ± 1.4 |
| 4 | 9a |  | −5.70 | −42.69 | 0 | −4.12 | −20.07 | 0 |
| 5 | 9b |  | −5.74 | −43.62 | 9 ± 4 | −5.67 | −32.29 | 54 ± 1.6 |
| 6 | 9c |  | −5.53 | −78.71 | 20 ± 7 | −4.82 | −26.69 | 96 ± 2.2 |
| 7 | 9d |  | −6.17 | −50.13 | 56 ± 5 | −5.35 | −20.05 | 49 ± 0.6 |
| 8 | 9e |  | −5.89 | −43.47 | 69 ± 1 | −6.57 | −22.50 | 71 ± 2.1 |
| 9 | 9f |  | −6.21 | −45.44 | 37 ± 2 | −5.84 | −25.71 | 64 ± 2 |
| Compound | Inhibition of Hydrolysis Rate Specific to the Factor XIa Substrate in the Buffer System IC50, µM |
|---|---|
| 7a | 4.30 ± 0.346 |
| 7c | 9.42 ± 0.986 |
| 9c | 5.31 ± 0.355 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Novichikhina, N.; Ilin, I.; Tashchilova, A.; Sulimov, A.; Kutov, D.; Ledenyova, I.; Krysin, M.; Shikhaliev, K.; Gantseva, A.; Gantseva, E.; et al. Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa. Molecules 2020, 25, 1889. https://doi.org/10.3390/molecules25081889
Novichikhina N, Ilin I, Tashchilova A, Sulimov A, Kutov D, Ledenyova I, Krysin M, Shikhaliev K, Gantseva A, Gantseva E, et al. Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa. Molecules. 2020; 25(8):1889. https://doi.org/10.3390/molecules25081889
Chicago/Turabian StyleNovichikhina, Nadezhda, Ivan Ilin, Anna Tashchilova, Alexey Sulimov, Danil Kutov, Irina Ledenyova, Mikhail Krysin, Khidmet Shikhaliev, Anna Gantseva, Ekaterina Gantseva, and et al. 2020. "Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa" Molecules 25, no. 8: 1889. https://doi.org/10.3390/molecules25081889
APA StyleNovichikhina, N., Ilin, I., Tashchilova, A., Sulimov, A., Kutov, D., Ledenyova, I., Krysin, M., Shikhaliev, K., Gantseva, A., Gantseva, E., Podoplelova, N., & Sulimov, V. (2020). Synthesis, Docking, and In Vitro Anticoagulant Activity Assay of Hybrid Derivatives of Pyrrolo[3,2,1-ij]Quinolin-2(1H)-one as New Inhibitors of Factor Xa and Factor XIa. Molecules, 25(8), 1889. https://doi.org/10.3390/molecules25081889