
Molecular Crystallization Inhibitors for Salt Damage Control in Porous Materials: An Overview

 , ,
, ,  and
and 
Abstract
:1. Introduction
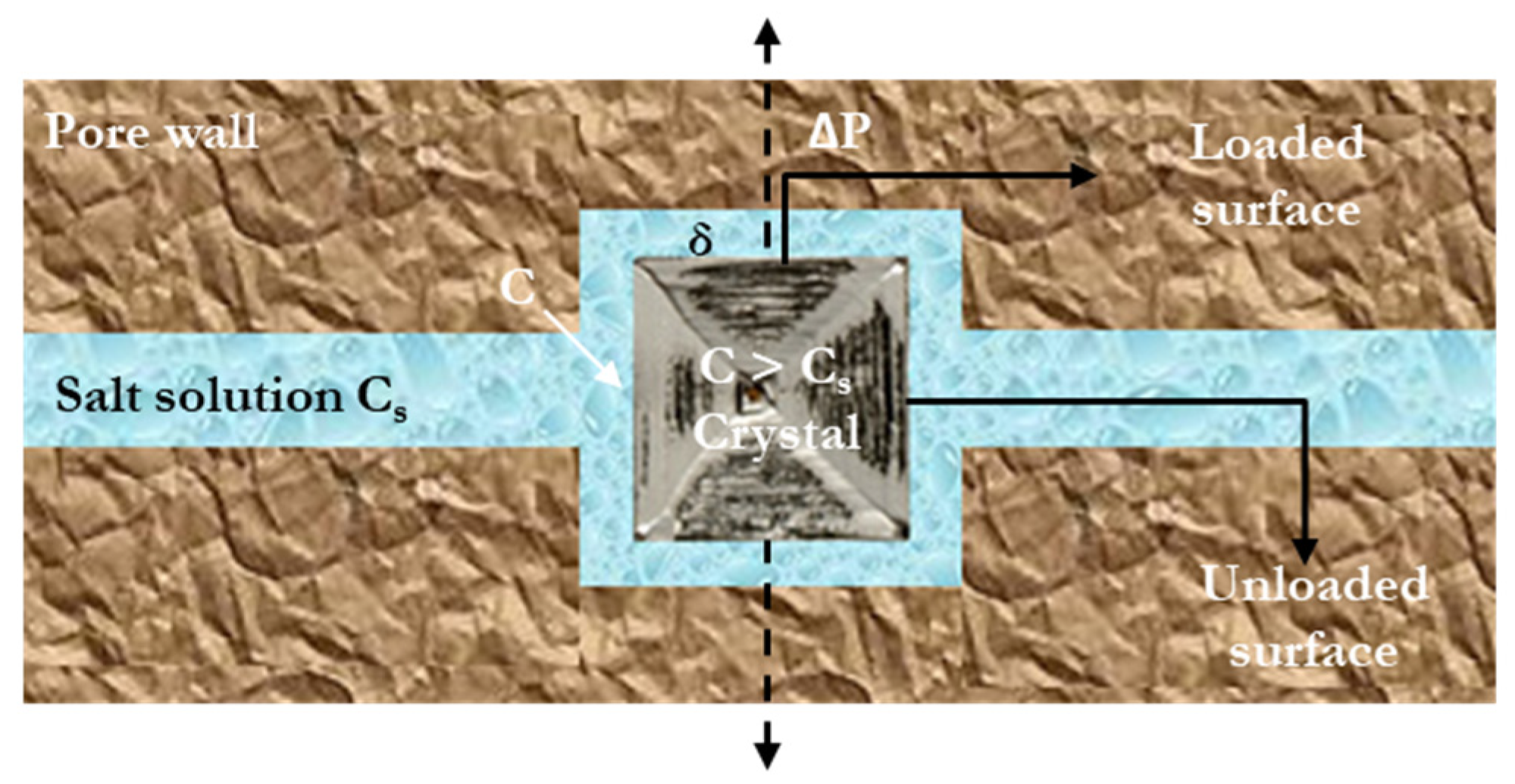
2. Fundamental Mechanisms and Influencing Factors for Salt Crystallization Damage
3. Crystal Growth Inhibitors
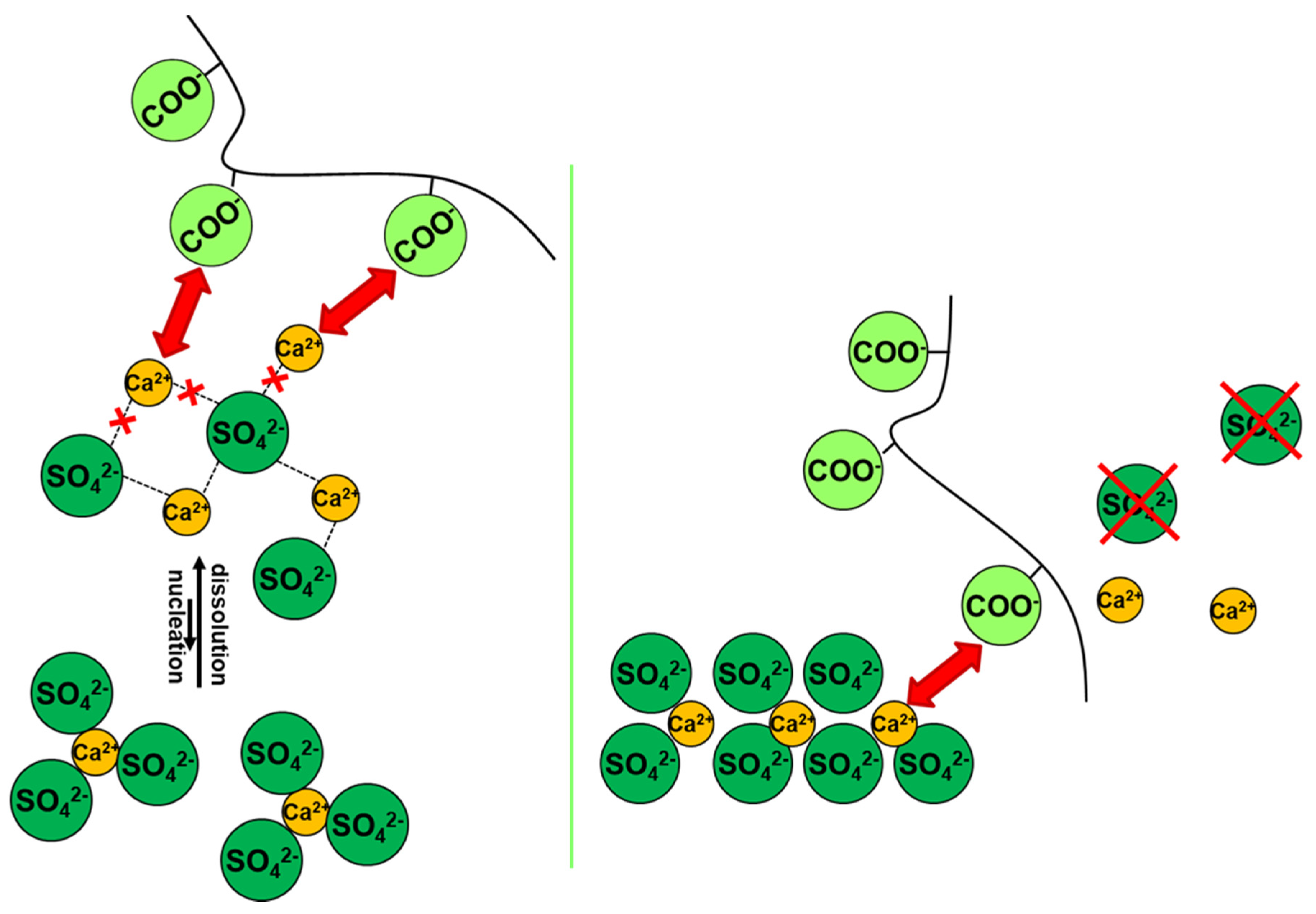
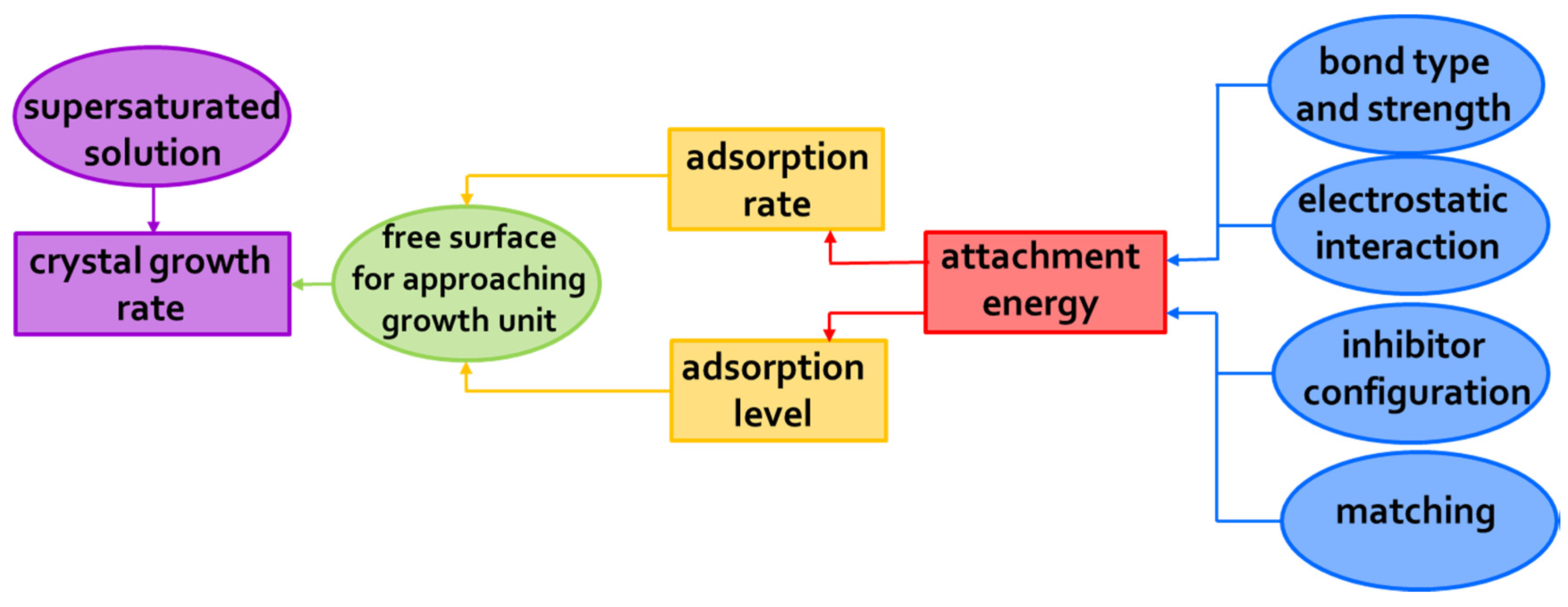
3.1. Mechanisms of Action
3.2. Inhibition Chemicals for Porous Media Applications
3.2.1. Biomass-Derived Species
3.2.2. Phosphonates
3.3. Alkali Ferrocyanides
3.4. Surfactants

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor Type (Concentration) | Salt Type (Concentration) | Effects | Ref. | ||
|---|---|---|---|---|---|
| Biomass-derived | CA[a] | 10−6 M to 10−4 M | Na2SO4 (0.7 M or 0.35 M) |
| [49,75,77,78] |
| NaCl (1.54 M) |
| [76,77] | |||
| NaCl+Na2SO4 (0.68+0.28 M; 1.03+0.14 M; 0.34+0.42 M) |
| [76] | |||
PC[b]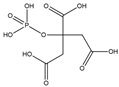 | 10−6 M to 10−4 M | Na2SO4 (0.7 M or 0.35 M) |
| [49,75,77] [78,80] | |
| NaCl (1.54 M) |
| [76,77] | |||
| NaCl+Na2SO4 (0.68+0.28 M; 1.03+0.14 M; 0.34+0.42 M) |
| [76] | |||
| Phosphonates | DTPMP[c]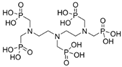 | 10−3 M | Na2SO4 (1 wt%) * |
| [97] |
| 10−4 M to 10−2 M | Na2SO4 (3.1 M) |
| [101] | ||
| MgSO4 (2.1 M) |
| ||||
| Alkali ferrocyanides | 10−3 M To 10−2 M | Na2SO4 (5 wt%; 20 wt%) |
| [102] | |
KFe(CN)[d]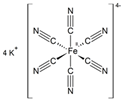 | NaCl (5 wt%; 20 wt%) |
| [102] | ||
| 10−4 M to 10−3 M | NaCl (3 M); sea water |
| [105] | ||
| 10−4 M to 10−2 M | NaCl+KCl (2+2 M; 3+1 M); NaCl+LiCl (3+1 M; 2+2M; 1+3 M) |
| [109] | ||
| 10−3 M To 10−2 M | NaCl (3 M) |
| [106] [108] | ||
| 10−4 M To 10−3 M |
| [103] | |||
NaFe(CN)[e] | NaCl (3 M); sea water |
| [105] | ||
| 10−3 M | NaCl (2 wt%) * |
| [97] | ||
| 0% To 1%† | NaCl (3 M; 1.5 M) |
| [104] | ||
| Surfactants | SDS[f] 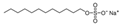 | 10−3 M | Na2SO4 (1.37 M) |
| [110] |
CDBAC[g] 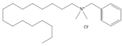 |
| ||||
4. Experimental and Instrumental Techniques, Modelling
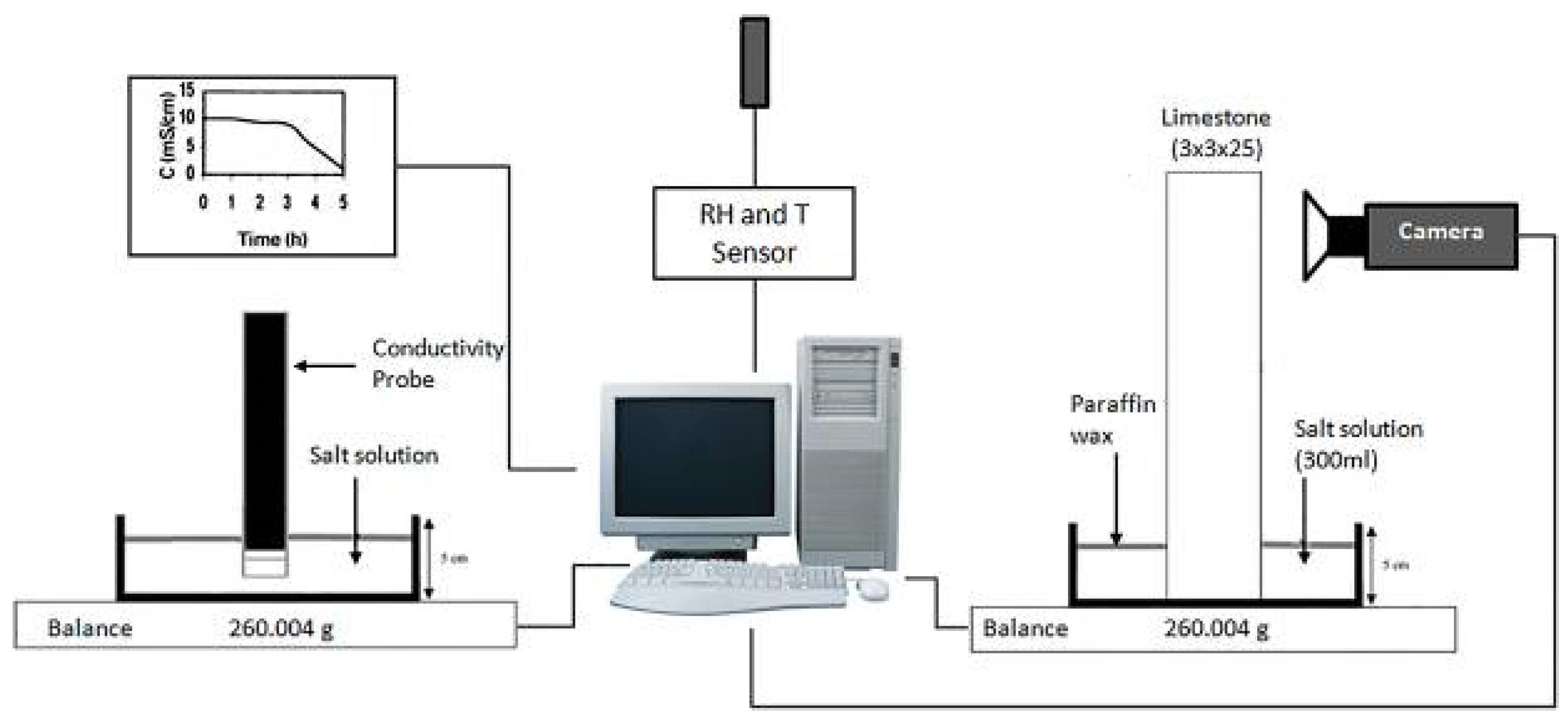
4.1. Macroscale Experiments
4.1.1. Capillary-Rise Experiments
4.1.2. Crystal Growth Experiments
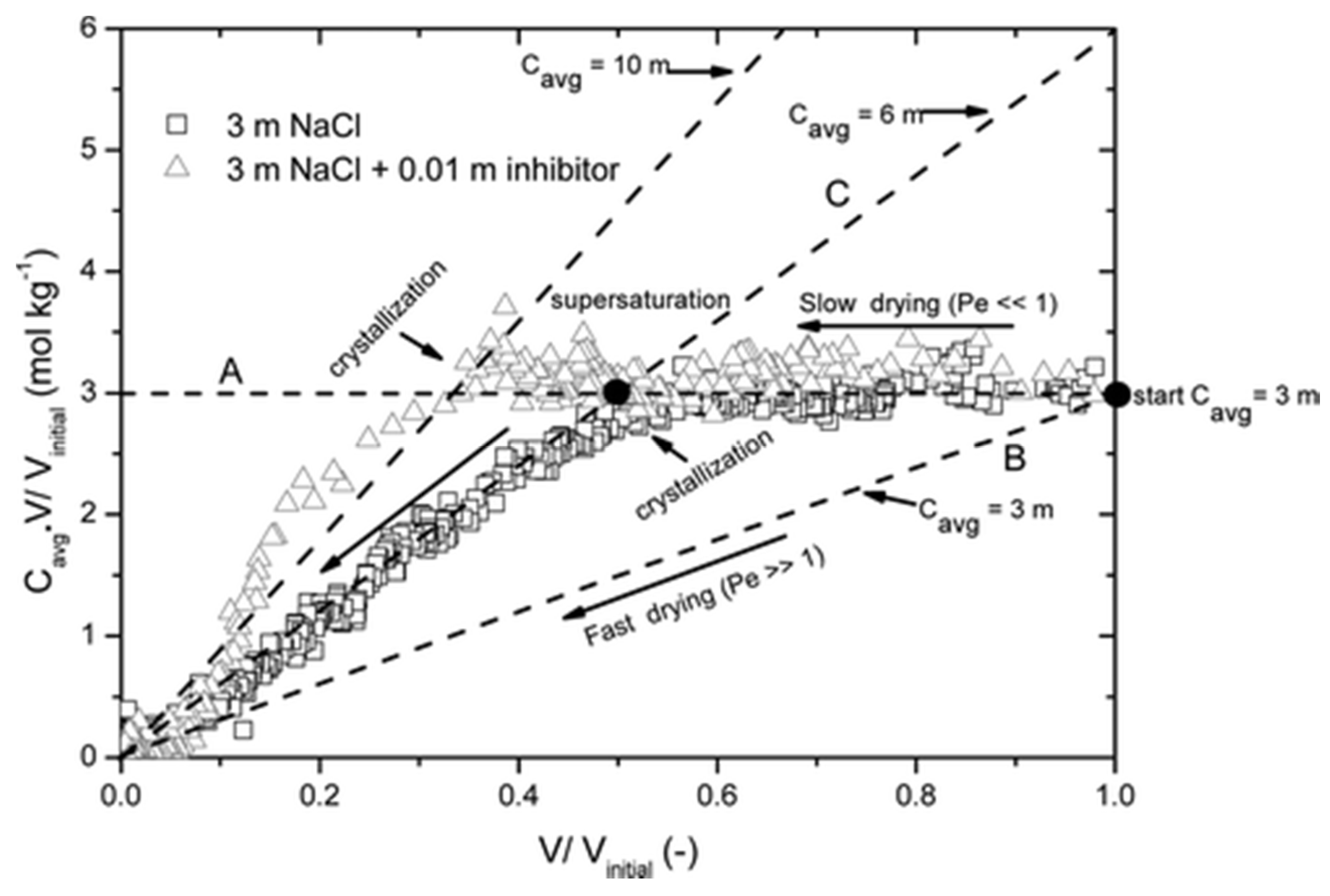
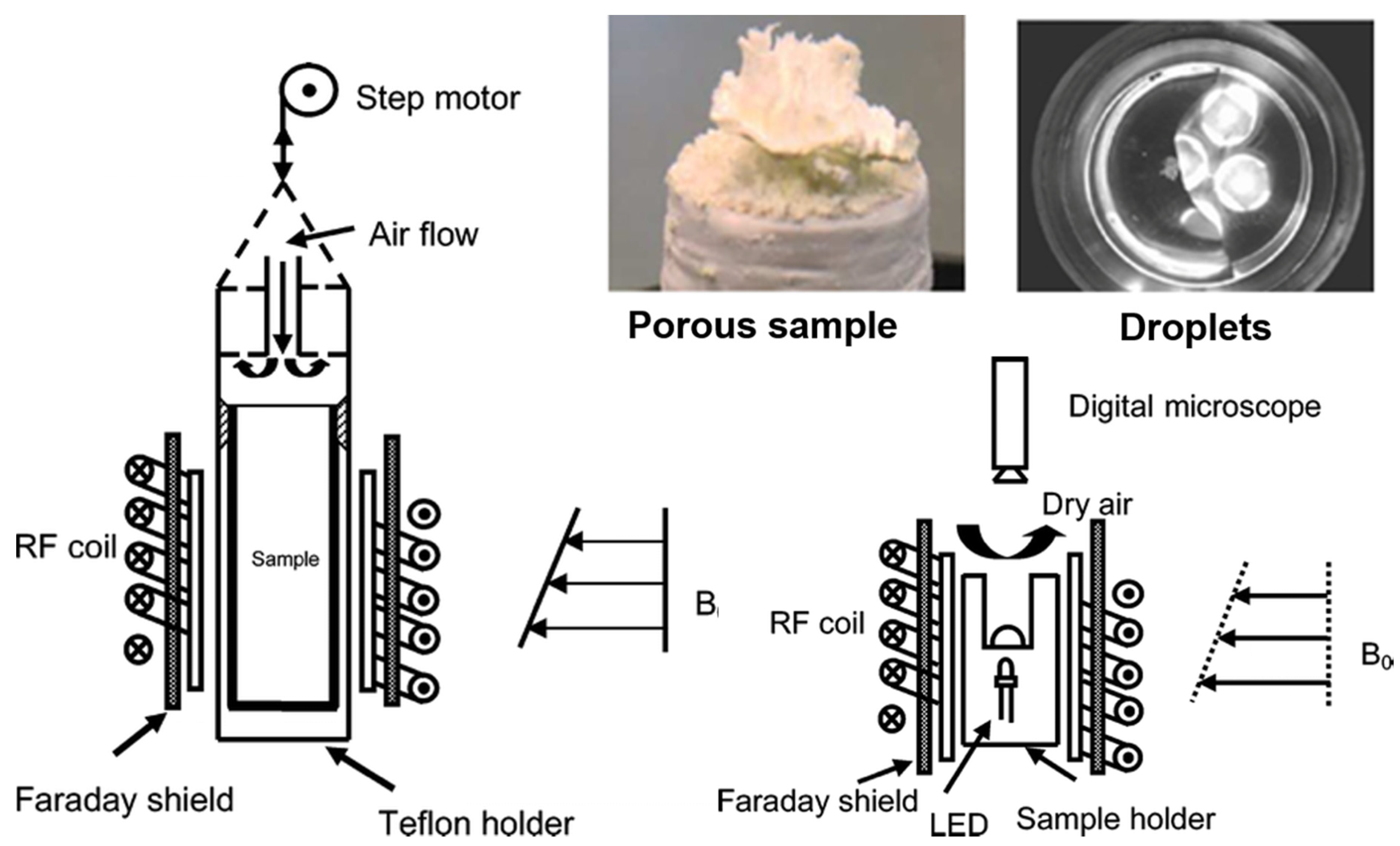
4.2. Microscale Experiments
4.3. Molecular and Numerical Modeling
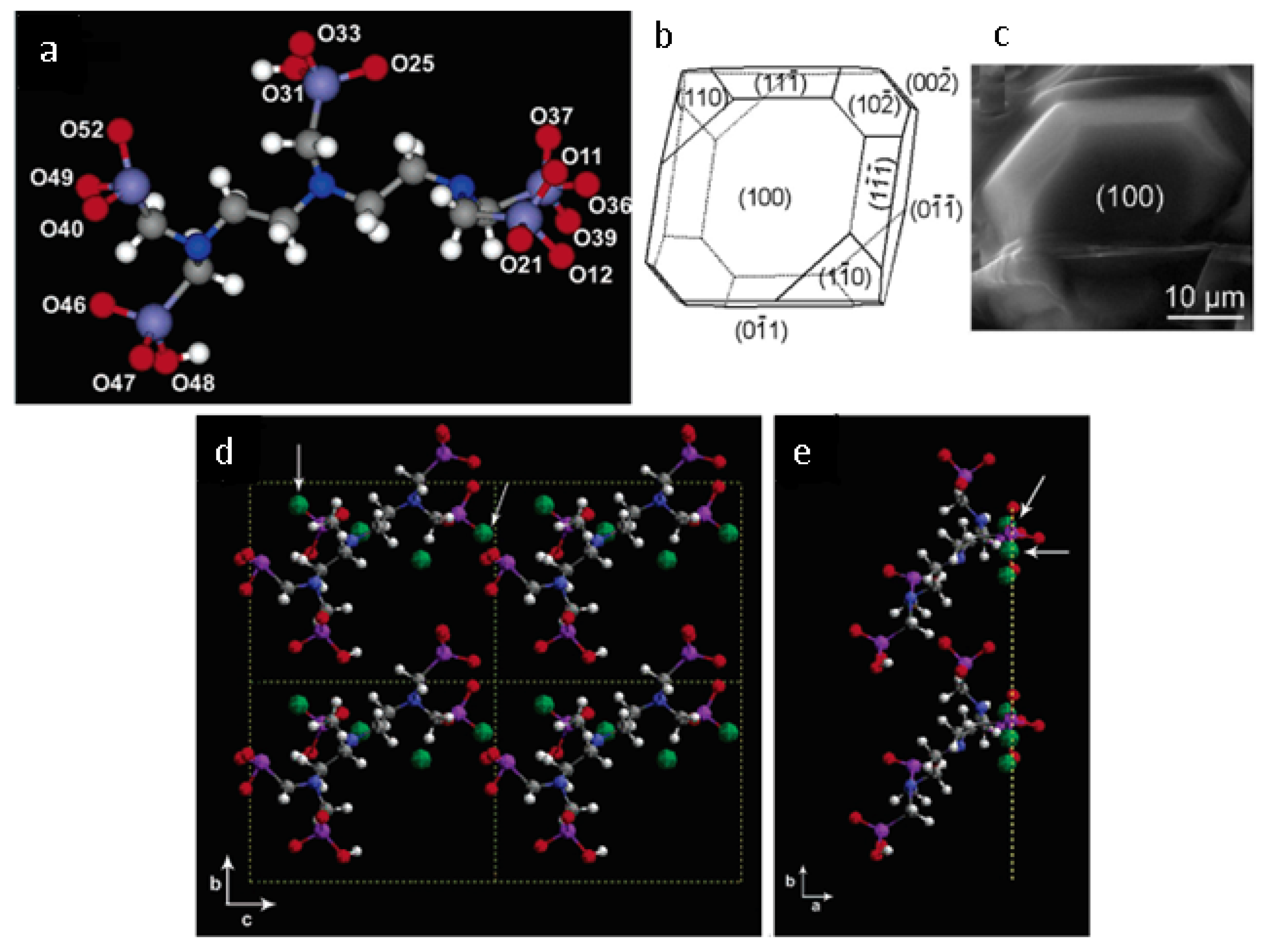
4.3.1. Atomic-Scale Molecular Models
4.3.2. Numerical Modeling
5. Conclusions
- A careful design of targets, aided by “in silico” tools; tuning the inhibitor architecture in terms of, e.g., the molecular weight, nature and relative orientation of the functional groups, ionization degree, and charge distribution over the molecules, may enable an accurate control of the adsorption level/adsorption rate of the inhibitors to the crystal surface. On the other hand, it is mandatory to design architectures exhibiting low toxicity for humans and the environment, which will abide by relevant legislation applicable to the chemical substances. Additionally, it is desirable to take into account the stability and persistence in the environment of the inhibitors, as well as the toxicity of their environmental metabolites on soil microbial communities and on organisms of different levels of the trophic aquatic chain.
- The development of simple synthetic protocols (ideally employing biomass as a raw material), in which the use of expensive and toxic reagents as well as the waste associated with the chemical processes are minimized; these aspects will ultimately lead to efficient synthesis of the inhibitors at a scale much greater than that used in the laboratories.
Author Contributions
Funding
Conflicts of Interest
References
- Goudie, A.; Viles, H.A. Salt Weathering Hazard; Wiley: Chichester, UK, 1997; ISBN 9780471958420. [Google Scholar]
- Lewin, S.Z. The Mechanism of Masonry Decay Through Crystallization. In Conservation of Historic Stone Buildings and Monuments; Committee on Conservation of Historic Stone Buildings and Monuments; National Materials Advisory Board; Commission on Engineering and Technical Systems; Division on Engineering and Physical Sciences; National Research Council, Ed.; National Academies Press: Washington, DC, USA, 1982; pp. 120–144. ISBN 978-0-309-03275-9. [Google Scholar]
- Rodriguez-Navarro, C.; Doehne, E. Salt weathering: Influence of evaporation rate, supersaturation and crystallization pattern. Earth Surf. Process. Landforms 1999, 24, 191–209. [Google Scholar] [CrossRef]
- Charola, A.E. Salts in the Deterioration of Porous Materials: An Overview. J. Am. Inst. Conserv. 2000, 39, 327–343. [Google Scholar] [CrossRef]
- Harris, S.Y. Building Pathology: Deterioration, Diagnostics, and Intervention; Wiley: New York, NY, USA, 2001; ISBN 9780471331728. [Google Scholar]
- Doehne, E. Salt weathering: A selective review. In Natural Stone, Weathering Phenomena, Conservation Strategies and Case Studies; Siegesmund, S., Weiss, T., Vollbrecht, A., Eds.; Geological Society of London: London, UK, 2002; Volume 205, pp. 51–64. ISBN 9781862394537. [Google Scholar]
- Lubelli, B.; van Hees, R.P.J.; Groot, C.J.W.P. The role of sea salts in the occurrence of different damage mechanisms and decay patterns on brick masonry. Constr. Build. Mater. 2004, 18, 119–124. [Google Scholar] [CrossRef]
- Bersani, D.; Madariaga, J.M. Applications of Raman spectroscopy in art and archaeology. J. Raman Spectrosc. 2012, 43, 1523–1528. [Google Scholar] [CrossRef]
- Verges-Belmin, V.; Siedel, H. Desalination of Masonries and Monumental Sculptures by Poulticing: A Review/Entsalzen von Mauerwerk und Steinfiguren mit Hilfe von Kompressen: Ein Überblick. Restor. Build. Monum. 2005, 11, 391–408. [Google Scholar] [CrossRef]
- Sawdy, A.; Heritage, A.; Pel, L. A review of salt transport in porous media: Assessment methods and salt reduction treatments. In Salt Weathering on Buildings and Stone Sculptures, 22–24 October 2008, The National Museum Copenhagen, Denmark; Technical University of Denmark, Department of Civil Engineering: Copenhagen, Denmark, 2008; pp. 1–27. [Google Scholar]
- Lubelli, B.; van Hees, R.P.J. Desalination of masonry structures: Fine tuning of pore size distribution of poultices to substrate properties. J. Cult. Herit. 2010, 11, 10–18. [Google Scholar] [CrossRef]
- Pel, L.; Sawdy, A.; Voronina, V. Physical principles and efficiency of salt extraction by poulticing. J. Cult. Herit. 2010, 11, 59–67. [Google Scholar] [CrossRef]
- Normandin, K.C.; Slaton, D.; Weiss, N.; Pearce, J. Cleaning Techniques in Conservation Practice; Donhead: Dorset, UK, 2005; ISBN 9781873394748. [Google Scholar]
- Sammut, S. Limiting Salt Damage–A Study on the Effectiveness of Salt Inhibitors on Globigerina Limestone. Ph.D. Thesis, University of Malta, Faculty for the Built Environment, Msida, Malta, 2017. [Google Scholar]
- Granneman, S.J.C.; Lubelli, B.; van Hees, R.P.J. Mitigating salt damage in building materials by the use of crystallization modifiers–a review and outlook. J. Cult. Herit. 2019, 40, 183–194. [Google Scholar] [CrossRef]
- EUR-Lex. Available online: https://eur-lex.europa.eu/legal-content/EN/TXT/PDF/?uri=CELEX:02006R1907-20180509&from=EN (accessed on 28 March 2020).
- Buckley, H.E. Crystal growth. Q. J. R. Meteorol. Soc. 1951, 691. [Google Scholar] [CrossRef]
- Sangwal, K. Additives and Crystallization Processes: From Fundamentals to Applications; John Wiley and Sons: Hoboken, NJ, USA, 2007; ISBN 9780470061534. [Google Scholar]
- Hasson, D.; Shemer, H.; Sher, A. State of the Art of Friendly “Green” Scale Control Inhibitors: A Review Article. Ind. Eng. Chem. Res. 2011, 50, 7601–7607. [Google Scholar] [CrossRef]
- Amjad, Z. (Ed.) Mineral Scale Formation and Inhibition; Springer US: Boston, MA, USA, 1995; ISBN 978-1-4899-1402-6. [Google Scholar]
- Amjad, Z. (Ed.) Advances in Crystal Growth Inhibition Technologies; Kluwer Academic Publishers: Boston, MA, USA, 2002; ISBN 0-306-46499-3. [Google Scholar]
- Xyla, A.G.; Mikroyannidis, J.; Koutsoukos, P.G. The inhibition of calcium carbonate precipitation in aqueous media by organophosphorus compounds. J. Colloid Interface Sci. 1992, 153, 537–551. [Google Scholar] [CrossRef]
- Van Rosmalen, G.M. Scale Prevention with Special Reference to Threshold Treatment. Chem. Eng. Commun. 1983, 20, 209–233. [Google Scholar] [CrossRef]
- Butt, F.H.; Rahman, F.; Baduruthamal, U. Pilot plant evaluation of advanced vs. conventional scale inhibitors for RO desalination. Desalination 1995, 103, 189–198. [Google Scholar] [CrossRef]
- Klepetsanis, P.G.; Koutsoukos, P.G. Kinetics of calcium sulfate formation in aqueous media: Effect of organophosphorus compounds. J. Cryst. Growth 1998, 193, 156–163. [Google Scholar] [CrossRef]
- Garcia, C.; Courbin, G.; Ropital, F.; Fiaud, C. Study of the scale inhibition by HEDP in a channel flow cell using a quartz crystal microbalance. Electrochim. Acta 2001, 46, 973–985. [Google Scholar] [CrossRef]
- Tadros, M.E.; Mayes, I. Linear growth rates of calcium sulfate dihydrate crystals in the presence of additives. J. Colloid Interface Sci. 1979, 72, 245–254. [Google Scholar] [CrossRef]
- Badens, E.; Veesler, S.; Boistelle, R. Crystallization of gypsum from hemihydrate in presence of additives. J. Cryst. Growth 1999, 198, 704–709. [Google Scholar] [CrossRef]
- Zafiropoulou, A.; Dalas, E. The effect of benzotriazoles on calcium carbonate scale formation. J. Cryst. Growth 2000, 219, 477–480. [Google Scholar] [CrossRef]
- Black, S.N.; Bromley, L.A.; Cottier, D.; Davey, R.J.; Dobbs, B.; Rout, J.E. Interactions at the organic/inorganic interface: Binding motifs for phosphonates at the surface of barite crystals. J. Chem. Soc. Faraday Trans. 1991, 87, 3409–3414. [Google Scholar] [CrossRef]
- Espinosa-Marzal, R.M.; Scherer, G.W. Advances in Understanding Damage by Salt Crystallization. Acc. Chem. Res. 2010, 43, 897–905. [Google Scholar] [CrossRef]
- Correns, C.W. Growth and dissolution of crystals under linear pressure. Discuss. Faraday Soc. 1949, 5, 267. [Google Scholar] [CrossRef]
- Scherer, G.W. Crystallization in pores. Cem. Concr. Res. 1999, 29, 1347–1358. [Google Scholar] [CrossRef]
- Steiger, M. Crystal growth in porous materials—I: The crystallization pressure of large crystals. J. Cryst. Growth 2005, 282, 455–469. [Google Scholar] [CrossRef]
- Houck, J.; Scherer, G.W. Controlling Stress from Salt Crystallization. In Fracture and Failure of Natural Building Stones; Kourkoulis, S.K., Ed.; Springer Netherlands: Dordrecht, The Netherlands, 2006; pp. 299–312. [Google Scholar]
- Désarnaud, J.; Grauby, O.; Bromblet, P.; Vallet, J.-M.; Baronnet, A. Growth and Dissolution of Crystal under Load: New Experimental Results on KCl. Cryst. Growth Des. 2013, 13, 1067–1074. [Google Scholar] [CrossRef]
- Espinoza-Marzal, R.M.; Scherer, G.W. Mechanisms of damage by salt. In Limestone in the Built Environment: Present-Day Challenges for the Preservation of the Past; Smith, B.J., Gomez-Heras, M., Viles, H.A., Cassar, J., Eds.; GSL: London, UK, 2010; pp. 61–77. ISBN 978-1-86239-294-6. [Google Scholar]
- Benavente, D.; García Del Cura, M.A.; Fort, R.; Ordóñez, S. Thermodynamic modelling of changes induced by salt pressure crystallization in porous media of stone. J. Cryst. Growth 1999, 204, 168–178. [Google Scholar] [CrossRef]
- Min, D.; Mingshu, T. Formation and expansion of ettringite crystals. Cem. Concr. Res. 1994, 24, 119–126. [Google Scholar] [CrossRef]
- Desarnaud, J.; Bonn, D.; Shahidzadeh, N. The Pressure induced by salt crystallization in confinement. Sci. Rep. 2016, 6, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sekine, K.; Hayashi, K. In situ observation of the crystallization pressure induced by halite crystal growth in a microfluidic channel. Am. Mineral. 2011, 96, 1012–1019. [Google Scholar] [CrossRef]
- Hamilton, A.; Koutsos, V.; Hall, C. Direct measurement of salt-mineral repulsion using atomic force microscopy. Chem. Commun. 2010, 46, 5235–5237. [Google Scholar] [CrossRef] [Green Version]
- Pitzer, K.S.; Kenneth, S. Activity Coefficients in Electrolyte Solutions; CRC: Boca Raton, FL, USA, 1991; ISBN 9781351069472. [Google Scholar]
- Neugebauer, J. The diagenetic problem of chalk. Diagenetic Probl. Chalk 1973, 143, 223–245. [Google Scholar]
- Everett, D.H. The thermodynamics of frost damage to porous solids. Trans. Faraday Soc. 1961, 57, 1541–1551. [Google Scholar] [CrossRef]
- Shahidzadeh-Bonn, N.; Desarnaud, J.; Bertrand, F.; Chateau, X.; Bonn, D. Damage in porous media due to salt crystallization. Phys. Rev. E 2010, 81, 066110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espinosa-Marzal, R.M.; Scherer, G.W. Impact of in-pore salt crystallization on transport properties. Environ. Earth Sci. 2013, 69, 2657–2669. [Google Scholar] [CrossRef] [Green Version]
- Steiger, M.; Asmussen, S. Crystallization of sodium sulfate phases in porous materials: The phase diagram Na2SO4–H2O and the generation of stress. Geochim. Cosmochim. Acta 2008, 72, 4291–4306. [Google Scholar] [CrossRef]
- Marrocchi, A.; Taticchi, A.; Minuti, L. Inhibitori della crescita di cristalli di solfato di sodio in materiali lapidei 1. Sci. Technol. Cult. Herit. 2006, 15, 101–108. [Google Scholar]
- Rothert, E.; Eggers, T.; Cassar, J.; Ruedrich, J.; Fitzner, B.; Siegesmund, S. Stone properties and weathering induced by salt crystallization of Maltese Globigerina Limestone; Přikryl, R., Smith, B.J., Eds.; GSL: London, UK; Volume SP 271, pp. 189–198.
- Ruiz-Agudo, E.; Rodriguez-Navarro, C.; Sebastián-Pardo, E. Sodium sulfate crystallization in the presence of phosphonates: Implications in ornamental stone conservation. Cryst. Growth Des. 2006, 6, 1575–1583. [Google Scholar] [CrossRef]
- Rijniers, L.A.; Huinink, H.P.; Pel, L.; Kopinga, K. Experimental Evidence of Crystallization Pressure inside Porous Media. Phys. Rev. Lett. 2005, 94, 075503. [Google Scholar] [CrossRef] [Green Version]
- Espinosa Marzal, R.M.; Scherer, G.W. Crystallization of sodium sulfate salts in limestone. Environ. Geol. 2008, 56, 605–621. [Google Scholar] [CrossRef]
- Angeli, M.; Hébert, R.; Menéndez, B.; David, C.; Bigas, J.-P. Influence of temperature and salt concentration on the salt weathering of a sedimentary stone with sodium sulfate. Eng. Geol. 2010, 115, 193–199. [Google Scholar] [CrossRef]
- Broggi, A.; Petrucci, E.; Bracciale, M.P.; Santarelli, M.L. FT-Raman spectroscopy for quantitative analysis of salt efflorescences. J. Raman Spectrosc. 2012, 43, 1560–1566. [Google Scholar] [CrossRef]
- Schaffer, R.J. The Weathering of Natural Building Stones; Donhead: London, UK, 2004; ISBN 9781873394694. [Google Scholar]
- Scherer, G.W. Stress from crystallization of salt. Cem. Concr. Res. 2004, 34, 1613–1624. [Google Scholar] [CrossRef]
- Arnold, A.; Zehnder, K. Salt weathering on monuments. In 1st International Symposium on the Conservation of Monuments in the Mediterranean Basin; Zezza, F., Ed.; Springer: Bari, Italy, 1989; pp. 31–58. [Google Scholar]
- Rousset-Tournier, B. Transferts par Capillarité et évaporation dans des Roches: Rôle des Structures de Porosité. Ph.D. Thesis, Université Louis Pasteur, Strasbourg, France, 2001. [Google Scholar]
- Diaz-Gonçalves, T. Salt Crystallization in Plastered or Rendered Walls. Ph.D. Thesis, UNIVERSIDADE TÉCNICA DE LISBOA INSTITUTO SUPERIOR TÉCNICO, Lisboa, Portugal, 2007. [Google Scholar]
- Ruiz-Agudo, E.; Mees, F.; Jacobs, P.; Rodriguez-Navarro, C. The role of saline solution properties on porous limestone salt weathering by magnesium and sodium sulfates. Environ. Geol. 2007, 52, 269–281. [Google Scholar] [CrossRef]
- Cardell, C.; Benavente, D.; Rodríguez-Gordillo, J. Weathering of limestone building material by mixed sulfate solutions. Characterization of stone microstructure, reaction products and decay forms. Mater. Charact. 2008, 59, 1371–1385. [Google Scholar] [CrossRef]
- Bläuer Böhm, C. Restoration of Buildings and Monuments Quantitative Salt Analysis in Conservation of Buildings. Restor. Build. Monum. 2005, 11, 409–418. [Google Scholar] [CrossRef]
- Arnold, A.; Küng, A. Crystallization and habits of salt efflorescences on walls. Part I: Methods of investigation and hahits. In 5th International Congress on the Deterioration and Conservation of Stone; Félix, G., Ed.; Presses Polytechniques Romandes: Lausanne, Switzerland, 1985; pp. 255–267. [Google Scholar]
- Arnold, A.; Zehnder, K. Crystallization and habits of salt efflorescences on walls. II. Conditions of crystallization. In 5th International Congress on the Deterioration and Conservation of Stone; Félix, G., Ed.; Presses Polytechniques Romandes: Lausanne, Switzerland, 1985; pp. 269–277. [Google Scholar]
- Bloss, F.D.; Fred, D. Crystallography and crystal chemistry: An introduction; Mineralogical Society of America: Chantilly, VA, USA, 1994; ISBN 0939950375. [Google Scholar]
- Chatterji, S. Aspects of generation of destructive crystal growth pressure. J. Cryst. Growth 2005, 277, 566–577. [Google Scholar] [CrossRef]
- Van der Leeden, M.C.; van Rosmalen, G.M. Adsorption Behaviour of Polyelectrolytes on Barium Sulfate Crystals. J. Colloid Interface Sci. 1995, 171, 142–149. [Google Scholar] [CrossRef]
- Van der Leeden, M.C.; van Rosmalen, G.M. Adsorption Behaviour of Polyelectrolytes in Relation to the Crystal Growth Kinetics of Barium Sulfate. In Mineral scale formation and inhibition; Amjad, Z., Ed.; Springer Science+Business Media, LLC: New York, NY, USA, 1995; pp. 99–110. ISBN 978-1-4899-1402-6. [Google Scholar]
- Öner, M.; Doğan, Ö.; Öner, G. The influence of polyelectrolytes architecture on calcium sulfate dihydrate growth retardation. J. Cryst. Growth 1998, 186, 427–437. [Google Scholar] [CrossRef]
- Weijnen, M.P.C.; van Rosmalen, G.M.; Bennema, P.; Rijpkema, J.J.M. The adsorption of additives at the gypsum crystal surface: A theoretical approach: I. Determination of the interfacial bond energies. J. Cryst. Growth 1987, 82, 509–527. [Google Scholar] [CrossRef]
- Weijnen, M.P.C.; van Rosmalen, G.M.; Bennema, P. The adsorption of additives at the gypsum crystal surface: A theoretical approach: II. Determination of the surface coverage required for growth inhibition. J. Cryst. Growth 1987, 82, 528–542. [Google Scholar] [CrossRef]
- Christoffersen, J.; Christoffersen, M.R.; Christensen, S.B.; Nancollas, G.H. Kinetics of dissolution of calcium hydroxyapatite: VI. The effects of adsorption of methylene diphosphonate, stannous ions and partly-peptized collagen. J. Cryst. Growth 1983, 62, 254–264. [Google Scholar] [CrossRef]
- Gill, J.S.; Varsanik, R.G. Computer modeling of the specific matching between scale inhibitors and crystal structure of scale forming minerals. J. Cryst. Growth 1986, 76, 57–62. [Google Scholar] [CrossRef]
- Marrocchi, A.; Taticchi, A.; Santarelli, M.L.; Minuti, L.; Broggi, A.; Garibaldi, V. Acidi organici quail inhibitori della cristallizzazione di sali nei materiali lapidei. 2. Sci. Technol. Cult. Herit. 2006, 15, 109–114. [Google Scholar]
- Marrocchi, A.; Taticchi, A.; Santarelli, M.L.; Broggi, A.; Minuti, L.; Librando, V. Acidi organici quali inibitori della cristalizzazione del cloruro di sodio e di miscele cloruro di sodio-solfato di sodio nei materiali lapidei porosi 3. Sci. Technol. Cult. Herit. 2006, 15, 115–122. [Google Scholar]
- Marrocchi, A.; Taticchi, A.; Orrú, M.; Minuti, L.; Santarelli, M.L.; Librando, V. Inibitori organici della cristallizzazione salina nei materiali lapidei porosi 4. Sci. Technol. Cult. Herit. 2007, 16, 143–151. [Google Scholar]
- Cassar, J.; Marrocchi, A.; Santarelli, M.L.; Muscat, M. Control de los daños por cristalización en la caliza de Malta mediante inhibidores de sales. Mater. Construcción 2008, 58, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Franceschini, M.; Broggi, A.; Bracciale, M.P.; Sommei, L.; Santarelli, M.L.; Marrocchi, A. Effectiveness of Phosphocitrate as Salt Crystallization Inhibitor in Porous Materials: Case Study of the Roman Mosaic of Orpheus and the Beasts (Perugia, Italy). Int. J. Archit. Herit. 2015, 9, 195–200. [Google Scholar] [CrossRef]
- Sammut, S.; Cassar, J.; Marrocchi, A.; Russo, C.; Sommei, L.; Santarelli, M.L. Investigating a method to limit damage in Globigerina Limestone, a soft porous stone widely used in historic buildings. In Proceedings of the Third International Conference on Salt Weathering of Buildings and Stone Sculptures (SWBSS 2014), 14–16 October 2014, Brussels, Belgium.
- Bracciale, M.P.; Bretti, G.; Broggi, A.; Ceseri, M.; Marrocchi, A.; Natalini, R.; Russo, C. Mathematical modelling of experimental data for crystallization inhibitors. Appl. Math. Model. 2017, 48, 21–38. [Google Scholar] [CrossRef]
- Werpy, T.; Petersen, G. Top Value Added Chemicals from Biomass Volume I-Results of Screening for Potential Candidates from Sugars and Synthesis Gas Pacific Northwest National Laboratory (PNNL), National Renewable Energy Laboratory (NREL), and Office of Biomass Program. 2004. Available online: https://www.nrel.gov/docs/fy04osti/35523.pdf (accessed on 17 April 2020).
- Turhanen, P.A.; Demadis, K.D.; Peräniemi, S.; Vepsäläinen, J.J. A novel strategy for the preparation of naturally occurring phosphocitrate and its partially esterified derivatives. J. Org. Chem. 2007, 72, 1468–1471. [Google Scholar] [CrossRef]
- Tew, W.P.; Malis, C.D.; Howard, J.E.; Lehninger, A.L.T. Phosphocitrate inhibits mitochondrial and cytosolic accumulation of calcium in kidney cells in vivo. Proc. Natl. Acad. Sci. USA 1981, 78, 5528–5532. [Google Scholar] [CrossRef] [Green Version]
- Sikes, C.S.; Wierzbicki, A. Mechanisms of Regulation of Crystal Growth in Selected Biological Systems. In Mineral Scale Formation and Inhibition; Amjad, Z., Ed.; Springer Science + Business Media, LLC: New York, NY, USA, 1995; pp. 183–206. ISBN 978-1-4899-1402-6. [Google Scholar]
- Cheung, H.S.; Sallis, J.D.; Demadis, K.D.; Wierzbicki, A. Phosphocitrate blocks calcification-induced articular joint degeneration in a guinea pig model. Arthritis Rheum. 2006, 54, 2452–2461. [Google Scholar] [CrossRef]
- Demadis, K.D. Structure and in vivo anticalcification properties of a polymeric calcium-sodium-phosphocitrate organic-inorganic hybrid. Inorg. Chem. Commun. 2003, 6, 527–530. [Google Scholar] [CrossRef]
- Demadis, K.D.; Sallis, J.D.; Raptis, R.G.; Baran, P. A crystallographically characterized nine-coordinate calcium -Phosphocitrate complex as calcification inhibitor in vivo [16]. J. Am. Chem. Soc. 2001, 123, 10129–10130. [Google Scholar] [CrossRef] [PubMed]
- Sallis, J.; Demadis, K.; Cheung, H. Phosphocitrate, A Potential Therapeutic Agent for Calcium Crystal Deposition Diseases. Curr. Rheumatol. Rev. 2006, 2, 95–99. [Google Scholar] [CrossRef]
- Sallis, J.D.; Wierzbicki, A.; Cheung, H.S. Calcium Pyrophosphate Crystal Salt forms and the Influence of Phosphocitrate. In Advances in Crystal Growth Inhibition Technologies; Kluwer Academic Publishers: Boston, MA, USA; pp. 57–69.
- Kofina, A.N.; Demadis, K.D.; Koutsoukos, P.G. The Effect of Citrate and Phosphocitrate On Struvite Spontaneous Precipitation. Cryst. Growth Des. 2007, 7, 2705–2712. [Google Scholar] [CrossRef]
- Cassar, J. Deterioration of the Globigerina Limestone of the Maltese Islands. In Natural Stone, Weathering Phenomena, Conservation Strategies and Case Studies; Siegesmund, S., Weiss, T., Vollbrecht, A., Eds.; GSL: London, UK, 2001; Volume SP 205, pp. 33–50. ISBN 978-1-86239-453-7. [Google Scholar]
- Cassar, J.; Vella, A.J. Methodology to identify badly weathering limestone using geochemistry: Case study on the Lower Globigerina Limestone of the Maltese islands. Q. J. Eng. Geol. Hydrogeol. 2003, 36, 85–96. [Google Scholar] [CrossRef]
- Cassar, J. The use of limestone in a historic context-the experience of Malta. In Limestone in the Built Environment: Present-Day Challenges for the Preservation of the Past; Smith, B., Gomez-Heras, M., Viles, H., Cassar, J., Eds.; GSL: London, UK, 2010; Volume SP 331, pp. 13–25. ISBN 978-1-86239-294-6. [Google Scholar]
- Zammit, T.; Cassar, J.; Vella, A.J.; Torpiano, A. STONE: Newsletter on stone decay; Queen University of Belfast: Belfast, Ireland, 2007; pp. 32–33. [Google Scholar]
- Bracciale, M.P.; Broggi, A.; Chandraiahgari, C.R.; De Bellis, G.; Santarelli, M.L.; Sarto, M.S.; Uccelletti, D.; Zanni, E.; Marrocchi, A. Coating Composition with Antimicrobial and Antisaline Activity, and Process for the Preparation Thereof. WO 2017/125388 A1, 27 July 2017. [Google Scholar]
- Lubelli, B.; van Hees, R.P.J. Effectiveness of crystallization inhibitors in preventing salt damage in building materials. J. Cult. Herit. 2007, 8, 223–234. [Google Scholar] [CrossRef]
- Ruiz-Agudo, E.; Lubelli, B.; Sawdy, A.; van Hees, R.; Price, C.; Rodriguez-Navarro, C. An integrated methodology for salt damage assessment and remediation: The case of San Jerónimo Monastery (Granada, Spain). Environ. Earth Sci. 2011, 63, 1475–1486. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Agudo, E.; Rodriguez-Navarro, C.; Sebastian, E. The use of additives (phosphonates) as inhibitors for the crystallization of magnesium sulfate. In Proocedings of the Heritage, Weathering and Conservation HWC International Conference, Madrid, 21-24 June 2006; Fort, R., Alvarez de Buergo, M., Gomez-Heras, M., Vazquez-Calvo, C., Eds.; Consejo Superior de Investigaciones Cientificas: Madrid, Spain, 2006; p. 160. [Google Scholar]
- Ruiz-Agudo, E.; Putnis, C.V.; Rodriguez-Navarro, C. Interaction between Epsomite Crystals and Organic Additives. Cryst. Growth Des. 2008, 8, 2665–2673. [Google Scholar] [CrossRef] [Green Version]
- Ruiz-Agudo, E.; Putnis, C.V.; Pel, L.; Rodriguez-Navarro, C. Template-Assisted Crystallization of Sulfates onto Calcite: Implications for the Prevention of Salt Damage. Cryst. Growth Des. 2013, 13, 40–51. [Google Scholar] [CrossRef] [Green Version]
- Selwitz, C.; Doehne, E. The evaluation of crystallization modifiers for controlling salt damage to limestone. J. Cult. Herit. 2002, 3, 205–216. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Linares-Fernandez, L.; Doehne, E.; Sebastian, E. Effects of ferrocyanide ions on NaCl crystallization in porous stone. J. Cryst. Growth 2002, 243, 503–516. [Google Scholar] [CrossRef] [Green Version]
- Lubelli, B.; Nijland, T.G.; van Hees, R.P.J.; Hacquebord, A. Effect of mixed in crystallization inhibitor on resistance of lime–cement mortar against NaCl crystallization. Constr. Build. Mater. 2010, 24, 2466–2472. [Google Scholar] [CrossRef]
- Rivas, T.; Alvarez, E.; Mosquera, M.J.; Alejano, L.; Taboada, J. Crystallization modifiers applied in granite desalination: The role of the stone pore structure. Constr. Build. Mater. 2010, 24, 766–776. [Google Scholar] [CrossRef]
- Gupta, S.; Terheiden, K.; Pel, L.; Sawdy, A. Influence of Ferrocyanide Inhibitors on the Transport and Crystallization Processes of Sodium Chloride in Porous Building Materials. Cryst. Growth Des. 2012, 12, 3888–3898. [Google Scholar] [CrossRef] [Green Version]
- Pel, L.; Huinink, H.; Kopinga, K. Ion transport and crystallization in inorganic building materials as studied by nuclear magnetic resonance. Appl. Phys. Lett. 2002, 81, 2893–2895. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Huinink, H.P.; Pel, L.; Kopinga, K. How Ferrocyanide Influences NaCl Crystallization under Different Humidity Conditions. Cryst. Growth Des. 2014, 14, 1591–1599. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Pel, L.; Steiger, M.; Kopinga, K. The effect of ferrocyanide ions on sodium chloride crystallization in salt mixtures. J. Cryst. Growth 2015, 410, 7–13. [Google Scholar] [CrossRef]
- Rodriguez-Navarro, C.; Doehne, E.; Sebastian, E. Influencing crystallization damage in porous materials through the use of surfactants: Experimental results using sodium dodecyl sulfate and cetyldimethylbenzylammonium chloride. Langmuir 2000, 16, 947–954. [Google Scholar] [CrossRef]
- Flatt, R.J. Salt damage in porous materials: How high supersaturations are generated. J. Cryst. Growth 2002, 242, 435–454. [Google Scholar] [CrossRef]
- Genkinger, S.; Putnis, A. Crystallisation of sodium sulfate: Supersaturation and metastable phases. Environ. Geol. 2007, 52, 329–337. [Google Scholar] [CrossRef]
- Linnow, K.; Zeunert, A.; Steiger, M. Investigation of sodium sulfate phase transitions in a porous material using humidity- and temperature-controlled x-ray diffraction. Anal. Chem. 2006, 78, 4683–4689. [Google Scholar] [CrossRef] [PubMed]
- Steiger, M.; Linnow, K.; Juling, H.; Gülker, G.; Jarad, A.E.; Brüggerhoff, S.; Kirchner, D. Hydration of MgSO 4 ·H 2 O and Generation of Stress in Porous Materials. Cryst. Growth Des. 2008, 8, 336–343. [Google Scholar] [CrossRef]
- Shahidzadeh-Bonn, N.; Rafaı, S.; Bonn, D.; Wegdam, G. Salt Crystallization during Evaporation: Impact of Interfacial Properties. Langmuir 2008, 24, 8599–8605. [Google Scholar] [CrossRef] [PubMed]
- Derluyn, H.; Saidov, T.A.; Espinosa-Marzal, R.M.; Pel, L.; Scherer, G.W. Sodium sulfate heptahydrate I: The growth of single crystals. J. Cryst. Growth 2011, 329, 44–51. [Google Scholar] [CrossRef]
- Saidov, T.A.; Shahidzadeh, N.; Pel, L. Crystallization of sodium sulfate on hydrophilic/hydrophobic surfaces during drying: An NMR study. EPL 2013, 102, 28003. [Google Scholar] [CrossRef]
- Desarnaud, J.; Bertrand, F.; Shahidzadeh-Bonn, N. Impact of the Kinetics of Salt Crystallization on Stone Damage During Rewetting/Drying and Humidity Cycling. J. Appl. Mech. 2013, 80, 020911–020917. [Google Scholar] [CrossRef]
- Hamilton, A.; Hall, C.; Pel, L. Sodium sulfate heptahydrate: Direct observation of crystallization in a porous material. J. Phys. D. Appl. Phys. 2008, 41, 212002–212006. [Google Scholar] [CrossRef] [Green Version]
- Espinosa Marzal, R.; Hamilton, A.; McNall, M.; Whitaker, K.; Scherer, G. The chemomechanics of sodium sulfate crystallization in thenardite impregnated limestones during re-wetting. J. Mater. Res. 2011, 26, 1472–1481. [Google Scholar] [CrossRef] [Green Version]
- Derluyn, H.; Dewanckele, J.; Boone, M.N.; Derome, D.; Carmeliet, J. Crystallization of hydrated and anhydrous salts in porous limestone resolved by synchrotron X-ray microtomography. Nucl. Instruments Methods Phys. Res. Sect. B Beam Interact. Mater. Atoms 2014, 324, 102–112. [Google Scholar] [CrossRef] [Green Version]
- Kelnar, J.; Maděra, J.; Černý, R. Computational simulation of the effect of crystallization inhibitors on salt transport and crystallization in porous materials. In WIT Transactions on Modelling and Simulation; WIT Transactions on Modelling and Simulation, Vol 46; WIT Press: Southampton, UK, 2007; Volume 46, pp. 367–375. [Google Scholar]
- Nicolai, A.; Grunewald, J.; Zhang, J.S. Salztransport und Phasenumwandlung–Modellierung und numerische Lösung im Simulationsprogramm Delphin 5. Bauphysik 2007, 29, 231–239. [Google Scholar] [CrossRef]
- Koniorczyk, M. Salt transport and crystallization in non-isothermal, partially saturated porous materials considering ions interaction model. Int. J. Heat Mass Transf. 2012, 55, 665–679. [Google Scholar] [CrossRef]
- Derluyn, H.; Moonen, P.; Carmeliet, J. Deformation and damage due to drying-induced salt crystallization in porous limestone. J. Mech. Phys. Solids 2014, 63, 242–255. [Google Scholar] [CrossRef] [Green Version]
- Espinosa, R.M.; Franke, L.; Deckelmann, G.; Gunstmann, C. Gekoppelter Wärme- und Stofftransport einschließlich der Korrosionsprozesse in porösen Baustoffen mit dem Simulationsprogramm AStra. Bauphysik 2007, 29, 187–193. [Google Scholar] [CrossRef]
- Espinosa, R.M.; Franke, L.; Deckelmann, G. Model for the mechanical stress due to the salt crystallization in porous materials. Constr. Build. Mater. 2008, 22, 1350–1367. [Google Scholar] [CrossRef]
- Espinosa, R.M.; Franke, L.; Deckelmann, G. Phase changes of salts in porous materials: Crystallization, hydration and deliquescence. Constr. Build. Mater. 2008, 22, 1758–1773. [Google Scholar] [CrossRef]
- Koniorczyk, M.; Gawin, D. Numerical modelling of coupled heat, moisture and salt transport in porous materials. Comput. Assist. Mech. Eng. Sci. 2006, 565–574. [Google Scholar]
- Koniorczyk, M.; Gawin, D. Heat and Moisture Transport in Porous Building Materials Containing Salt. J. Build. Phys. 2008, 31, 279–300. [Google Scholar] [CrossRef]
- Koniorczyk, M.; Wojciechowski, M. Influence of salt on desorption isotherm and hygral state of cement mortar–Modelling using neural networks. Constr. Build. Mater. 2009, 23, 2988–2996. [Google Scholar] [CrossRef]
- Koniorczyk, M. Modelling the phase change of salt dissolved in pore water–Equilibrium and non-equilibrium approach. Constr. Build. Mater. 2010, 24, 1119–1128. [Google Scholar] [CrossRef]
- Nguyen, T.Q.; Petković, J.; Dangla, P.; Baroghel-Bouny, V. Modelling of coupled ion and moisture transport in porous building materials. Constr. Build. Mater. 2008, 22, 2185–2195. [Google Scholar] [CrossRef]
- Paz-García, J.M.; Johannesson, B.; Ottosen, L.M.; Ribeiro, A.B.; Rodríguez-Maroto, J.M. Modeling of electrokinetic processes by finite element integration of the Nernst–Planck–Poisson system of equations. Sep. Purif. Technol. 2011, 79, 183–192. [Google Scholar] [CrossRef]
- Poupeleer, A.S. Transport and Crystallization of Dissolved Salts in Cracked Porous Building Materials. Ph.D. Thesis, Katholieke Universiteit Leuven, Leuven, Belgium, 2001. [Google Scholar]
- Espinosa-Marzal, R.M.; Scherer, G.W. Crystallization Pressure Exerted by in-Pore Confined Crystals. In Poromechanics IV, Proc. Fourth Biot Conf. on Poromechanics, New York, 2009; Ling, H.I., Smyth, A., Betti, R., Eds.; DEStech Publications: Lancaster, UK, 2009; pp. 1013–1018. [Google Scholar]
- Schiro, M.; Ruiz-Agudo, E.; Rodriguez-Navarro, C. Damage Mechanisms of Porous Materials due to In-Pore Salt Crystallization. Phys. Rev. Lett. 2012, 109, 265503. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Agudo, E.; Rodriguez-Navarro, C. Suppression of salt weathering of porous limestone by borax-induced promotion of sodium and magnesium sulfate crystallization. Geol. Soc. London, Spec. Publ. 2010, 331, 93–102. [Google Scholar] [CrossRef]
- Washburn, E.R. The Creeping of Solutions. J. Phys. Chem. 1926, 31, 1246–1248. [Google Scholar] [CrossRef]
- Docherty, R.; Clydesdale, G.; Roberts, K.J.; Bennema, P. Application of bravais-friedel-donnay-harker, attachment energy and ising models to predicting and understanding the morphology of molecular crystals. J. Phys. D. Appl. Phys. 1991, 24, 89–99. [Google Scholar] [CrossRef]
- Wulff, G. Zur frage der geschwindigkeit des wachstums und der auflösung der kristallflächen. Z. Krist. Miner. 1901, 34, 449–530. [Google Scholar]
- Gibbs, J.W. The Collected Works of J. Willard Gibbs.; Yale University Press: New Heaven, CT, USA, 1948. [Google Scholar]
- Sikirić, M.; Babić-Ivančić, V.; Milat, O.; Sarig, S.; Füredi-Milhofer, H. Factors influencing additive interactions with calcium hydrogenphosphate dihydrate crystals. Langmuir 2000, 16, 9261–9266. [Google Scholar] [CrossRef]
- Rohl, A.L.; Gay, D.H.; Davey, R.J.; Catlow, C.R.A. Interactions at the Organic/Inorganic Interface: Molecular Modeling of the Interaction between Diphosphonates and the Surfaces of Barite Crystals. J. Am. Chem. Soc. 1996, 118, 642–648. [Google Scholar] [CrossRef]

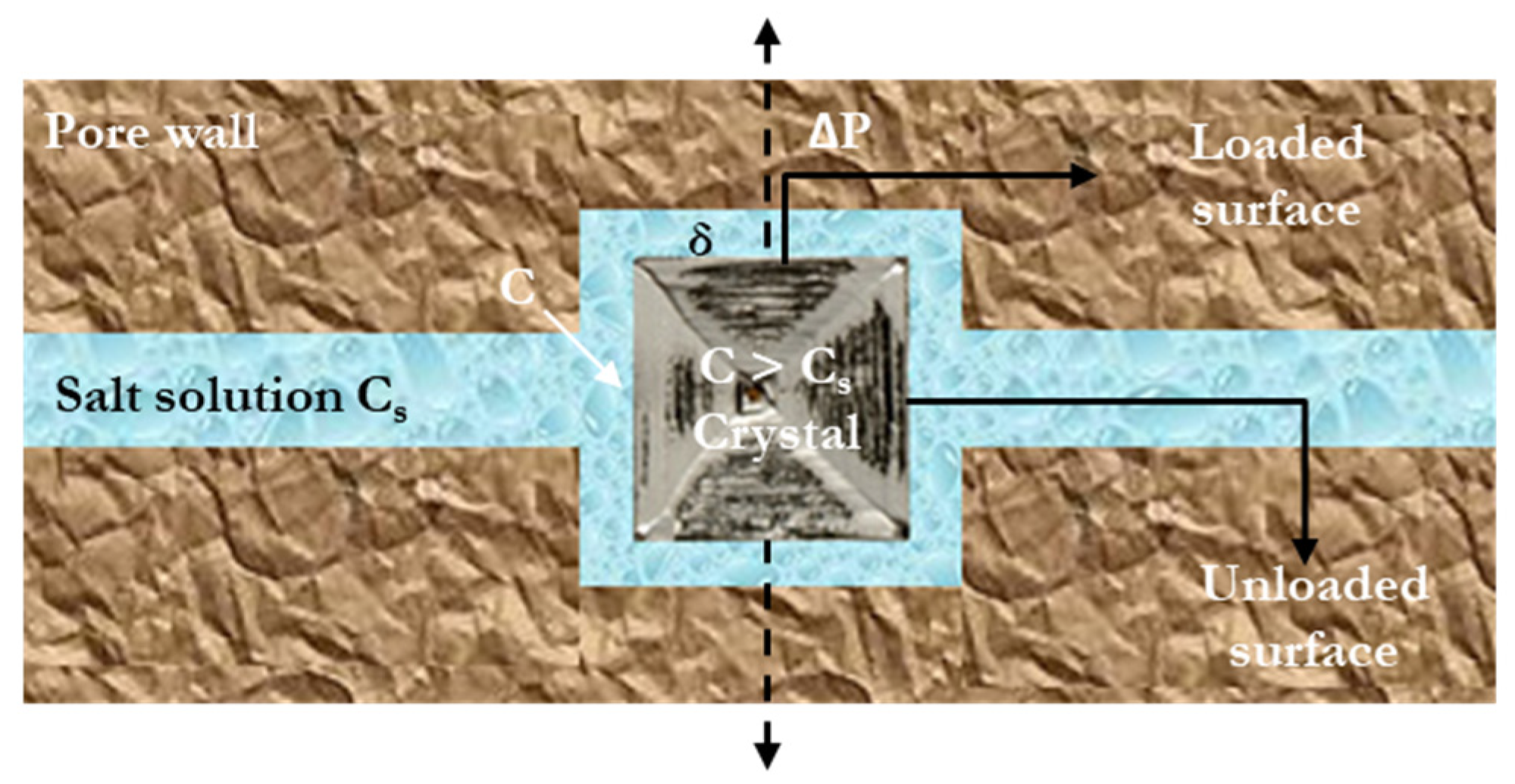

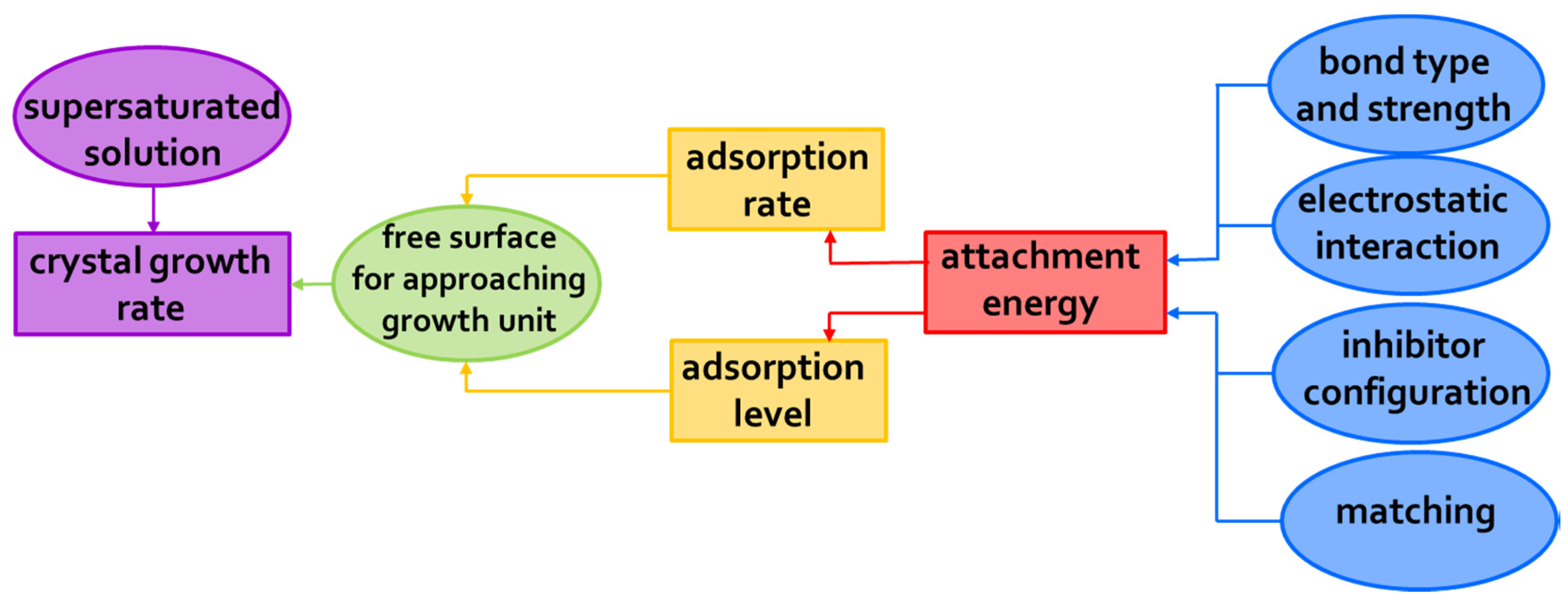


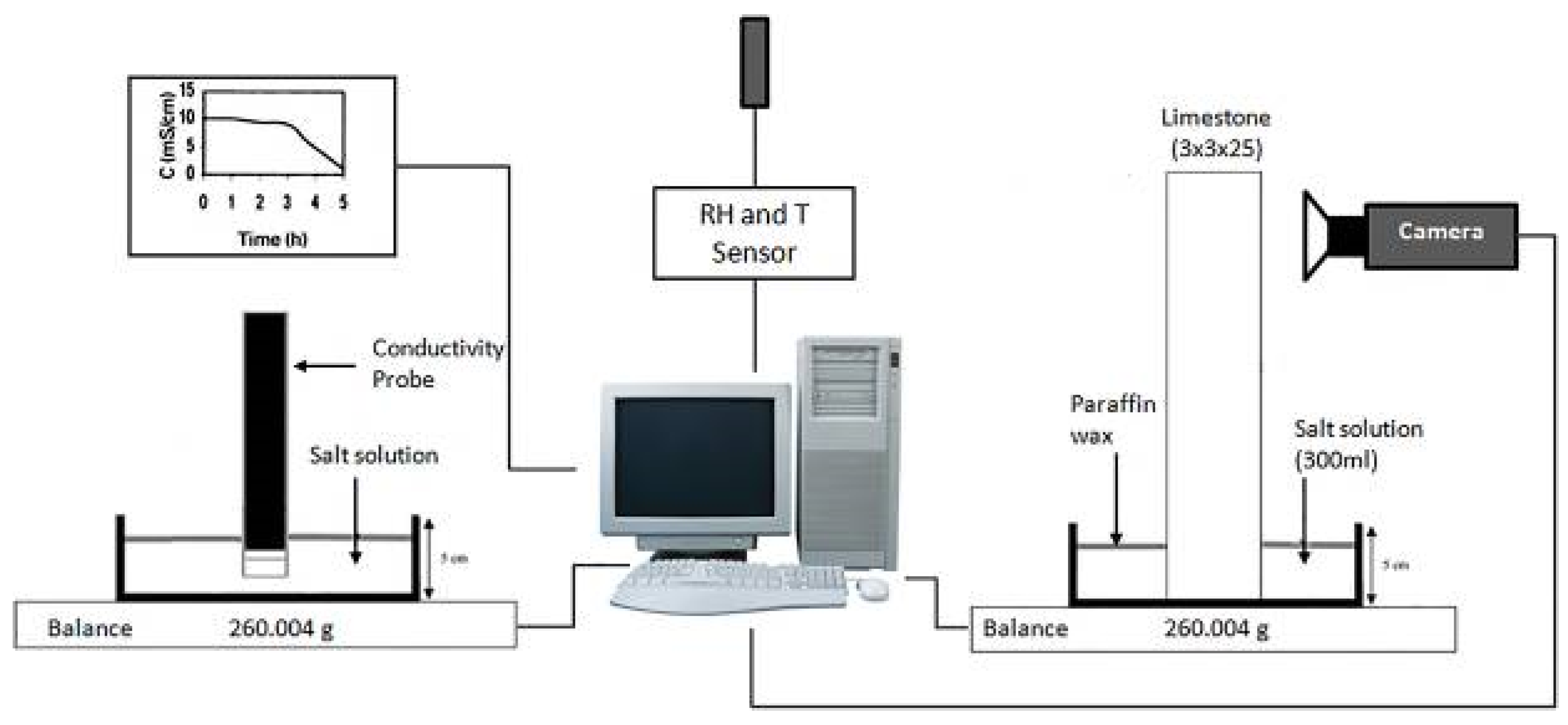
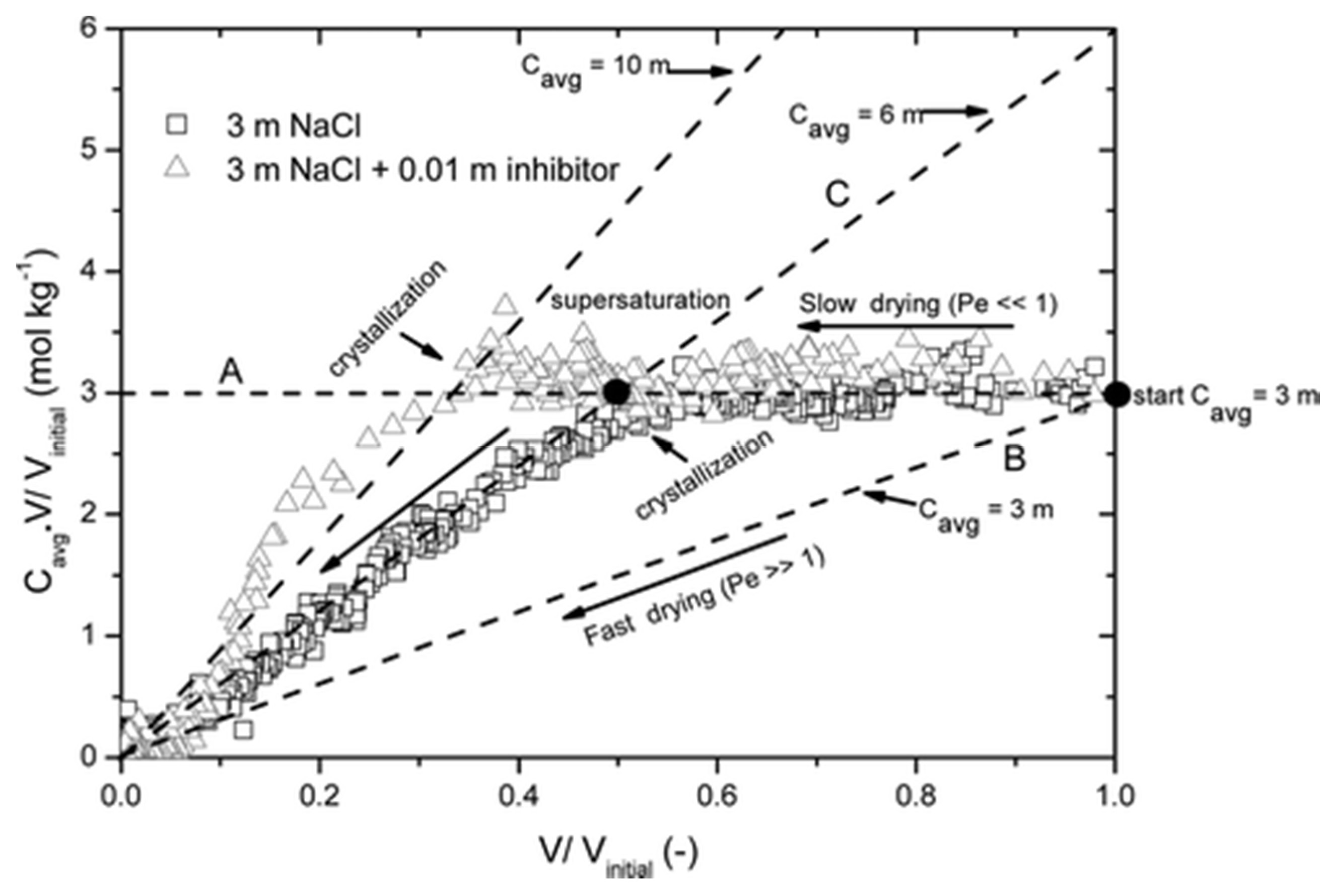
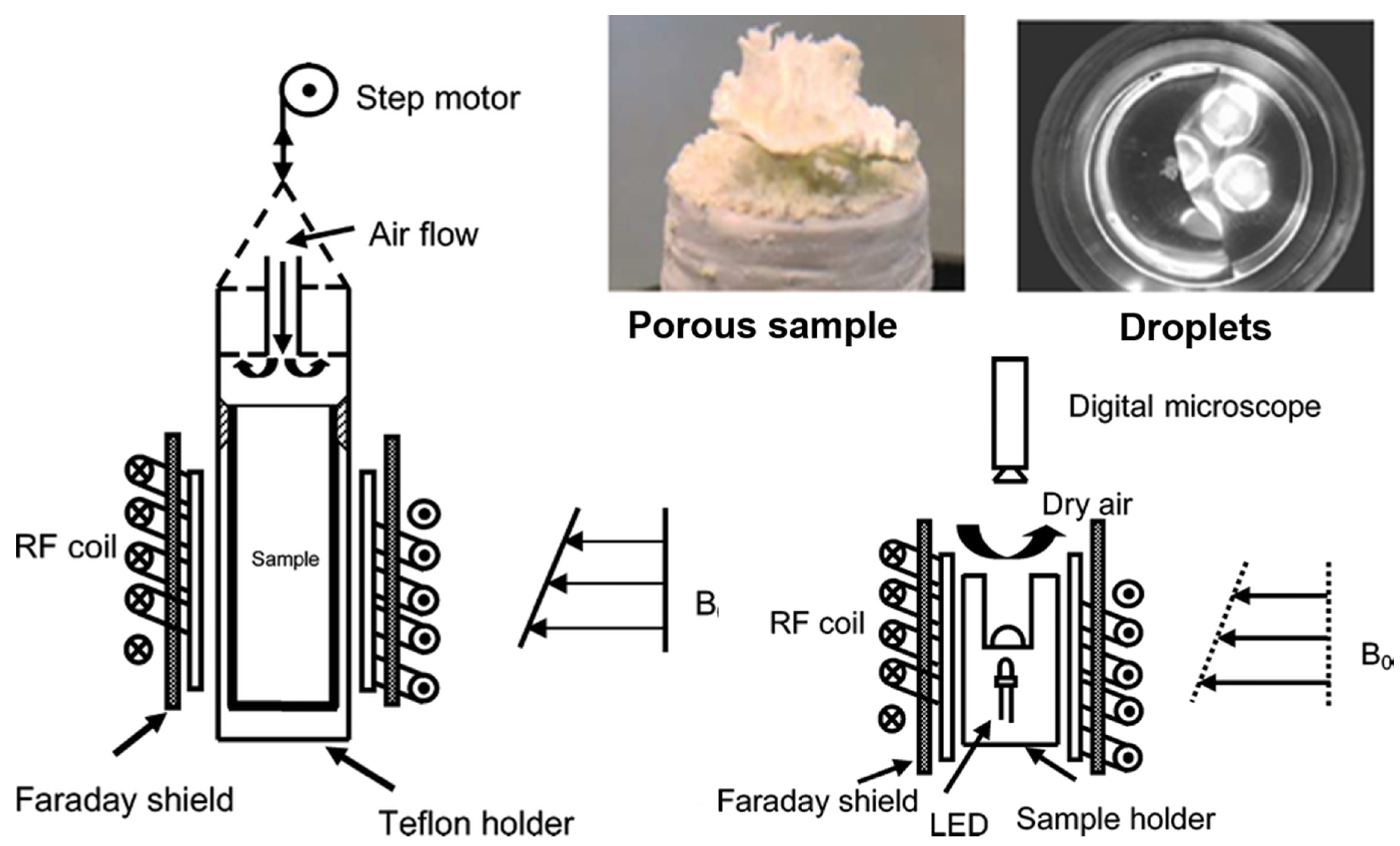
| Salt Type | Experiment Type | Material | Measurements | Ref. |
|---|---|---|---|---|
| CaSO4 | Batch Crystallization Test | Salt Solution | Conductimetry; SEM | [70] |
| Na2SO4 | Macroscale Crystallization Test | Monks Park Oolitic Limestone | ESEM; Evaporation Rate; MIP | [110] |
| Batch Crystallization Test | Salt Solution | Conductimetry; Surface Tension; Contact Angle; Viscosity; Evaporation Rate; XRD; SEM-EDX | ||
| Conductimetry; Evaporation Rate; XRD; ESEM; Molecular Modelling | [51] | |||
| Macroscale Crystallization Test | Brick; Tuff | Evaporation Rate; SEM | [49,75,78,80] | |
| Globigerina Limestone (franka (bajda and safra) and soll) | ||||
| Microscale Crystallization Test | Glass frits | ESEM; 2D-XRD; TG-DSC | [137] | |
| NaCl | Macroscale Crystallization Test | Limestone (Calcarenite from Spain) | SEM-EDX-CTL; XRD; AAS; MIP; Evaporation Rate | [103] |
| Inhibitor addition Lime-Cement mortars | ESEM-EDX; efflorescence and Debris Content | [104] | ||
| Batch Crystallization Test | Salt Solution | Conductimetry; Surface Tension; Evaporation Rate | [103] | |
| Drying Test | Salt Solution Droplets; Granada Limestone; Brick | NMR+Microscopy | [106] | |
| NaCl+KCl; NaCl+LiCl | Salt Solution Droplets | [109] | ||
| NaCl; sea water | Macroscale Crystallization Test | Monçao and Rodas Granites | Evaporation rate; efflorescence/subflorescence Ratio; XRD; SEM | [105] |
| Desalination Test | Conductimetry | |||
| NaCl;Na2SO4 | Macroscale Crystallization Test | Monks Park Limestone; Texas Cream Limestone | efflorescence/subflorescence Ratio | [102] |
| Calcarenites from Noto and Palazzolo | Evaporation Rate; SEM-EDX | [77] | ||
| Absorption-Drying Test; Macroscale Crystallization Test | Spanish Limestone; Czech Sandstone; Dutch Brick | ESEM-EDX; Evaporation Rate; HMC Content | [97] | |
| NaCl; NaCl+Na2SO4 | Macroscale Crystallization Test | Brick; Tuff | Evaporation Rate; SEM | [76] |
| MgSO4 | Microscale Crystallization Test | {110} Optically Clear Epsomite Single Crystals | AFM | [100] |
| Batch Crystallization Test | Salt Solution | Conductimetry; Evaporation Rate; XRD; ESEM; FTIR; Molecular Modeling | ||
| Na2SO4; MgSO4 | Macroscale Crystallization Test | Limestone (biocalcarenite from Granada) | ESEM; MIP | [138] |
| Microscale Crystallization Test | {101̅4} Cleaved Iceland Spar Single Crystals; Salt Solution Droplets | ESEM; XRD; Morphology Simulation | ||
| Batch Crystallization Test | Salt Solution | XRD; Surface Tension; ESEM | ||
| Na2SO4; MgSO4 | Macroscale Crystallization Test | Granada Limestone | XRD; NMR; MIP | [102] |
| Microscale Crystallization Test | {101̅4} Cleaved Iceland Spar Single Crystals | ESEM; AFM; RHXRD; Molecular Modelling |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bracciale, M.P.; Sammut, S.; Cassar, J.; Santarelli, M.L.; Marrocchi, A. Molecular Crystallization Inhibitors for Salt Damage Control in Porous Materials: An Overview. Molecules 2020, 25, 1873. https://doi.org/10.3390/molecules25081873
Bracciale MP, Sammut S, Cassar J, Santarelli ML, Marrocchi A. Molecular Crystallization Inhibitors for Salt Damage Control in Porous Materials: An Overview. Molecules. 2020; 25(8):1873. https://doi.org/10.3390/molecules25081873
Chicago/Turabian StyleBracciale, Maria Paola, Svetlana Sammut, JoAnn Cassar, Maria Laura Santarelli, and Assunta Marrocchi. 2020. "Molecular Crystallization Inhibitors for Salt Damage Control in Porous Materials: An Overview" Molecules 25, no. 8: 1873. https://doi.org/10.3390/molecules25081873
APA StyleBracciale, M. P., Sammut, S., Cassar, J., Santarelli, M. L., & Marrocchi, A. (2020). Molecular Crystallization Inhibitors for Salt Damage Control in Porous Materials: An Overview. Molecules, 25(8), 1873. https://doi.org/10.3390/molecules25081873