
Simultaneous Measurement of Urinary Trimethylamine (TMA) and Trimethylamine N-Oxide (TMAO) by Liquid Chromatography–Mass Spectrometry



Abstract

1. Introduction
2. Results
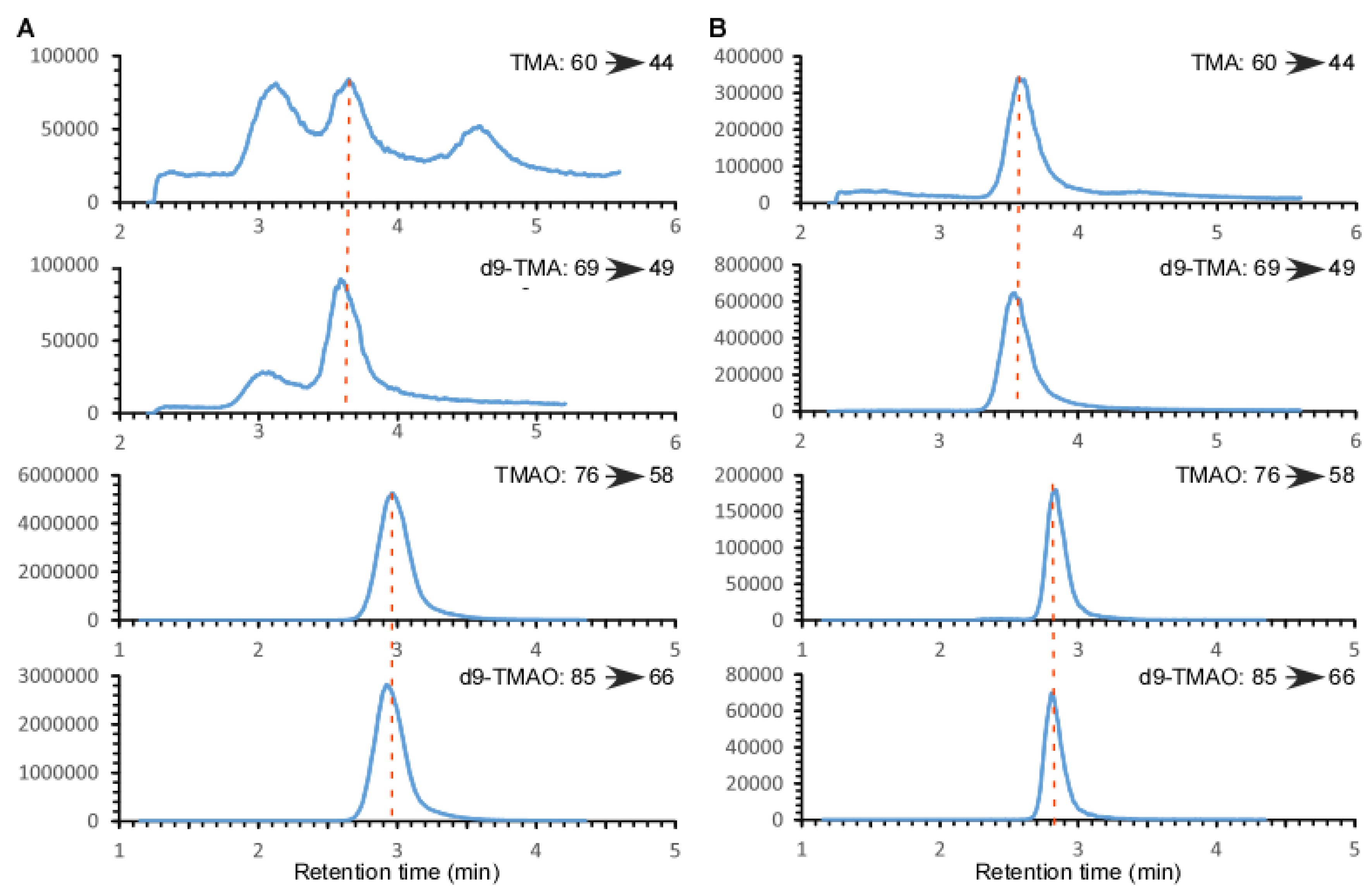
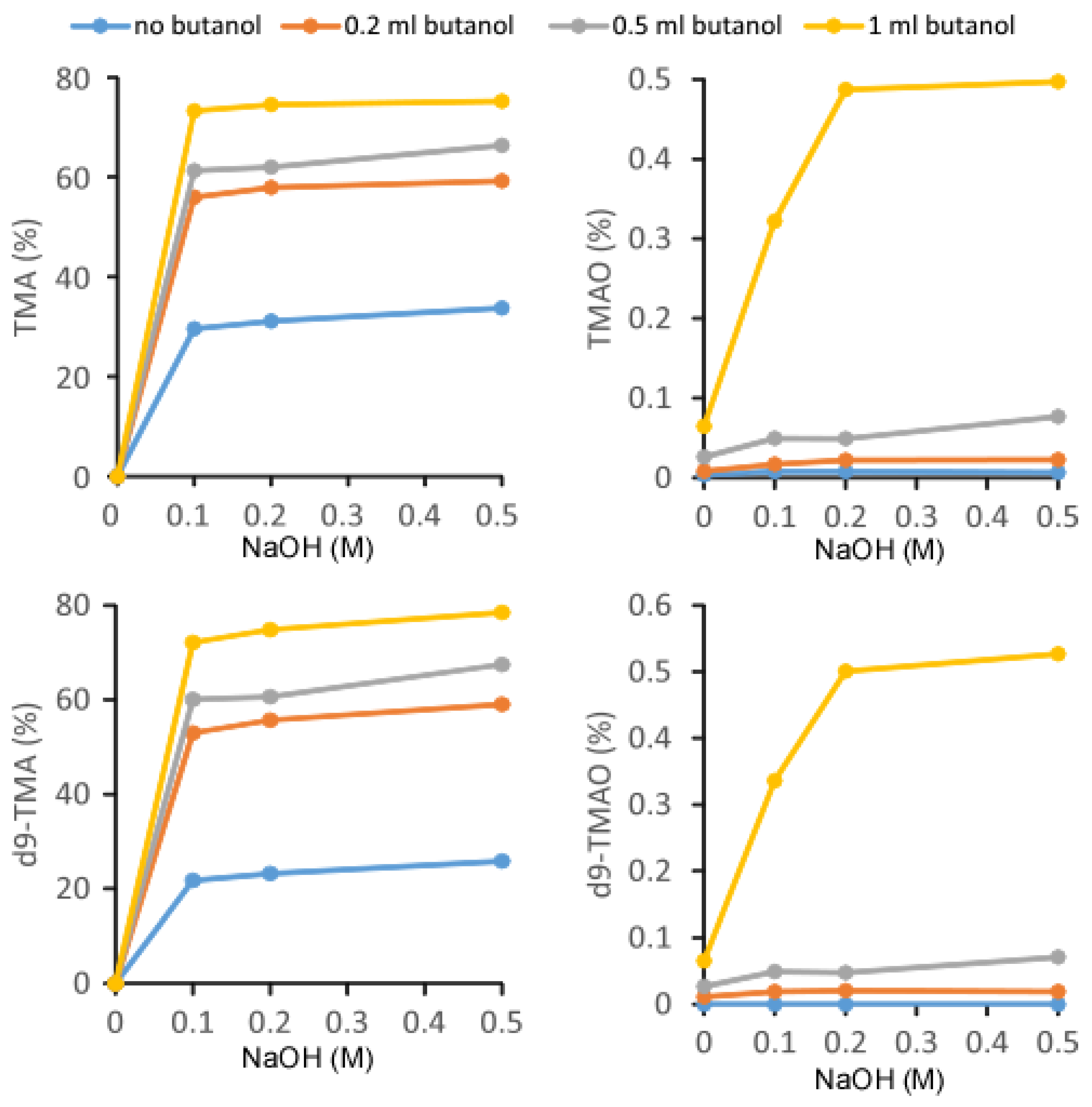
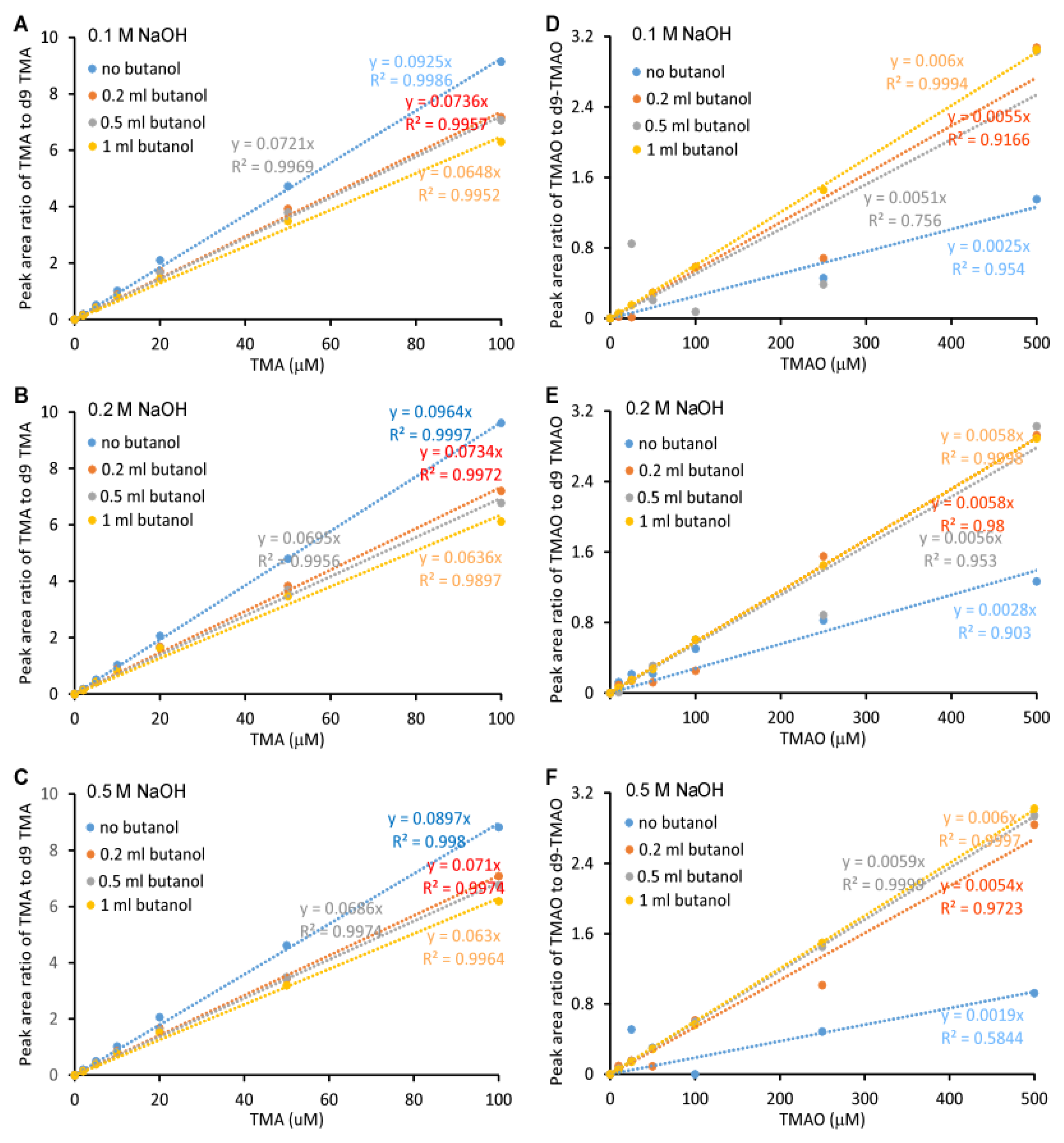
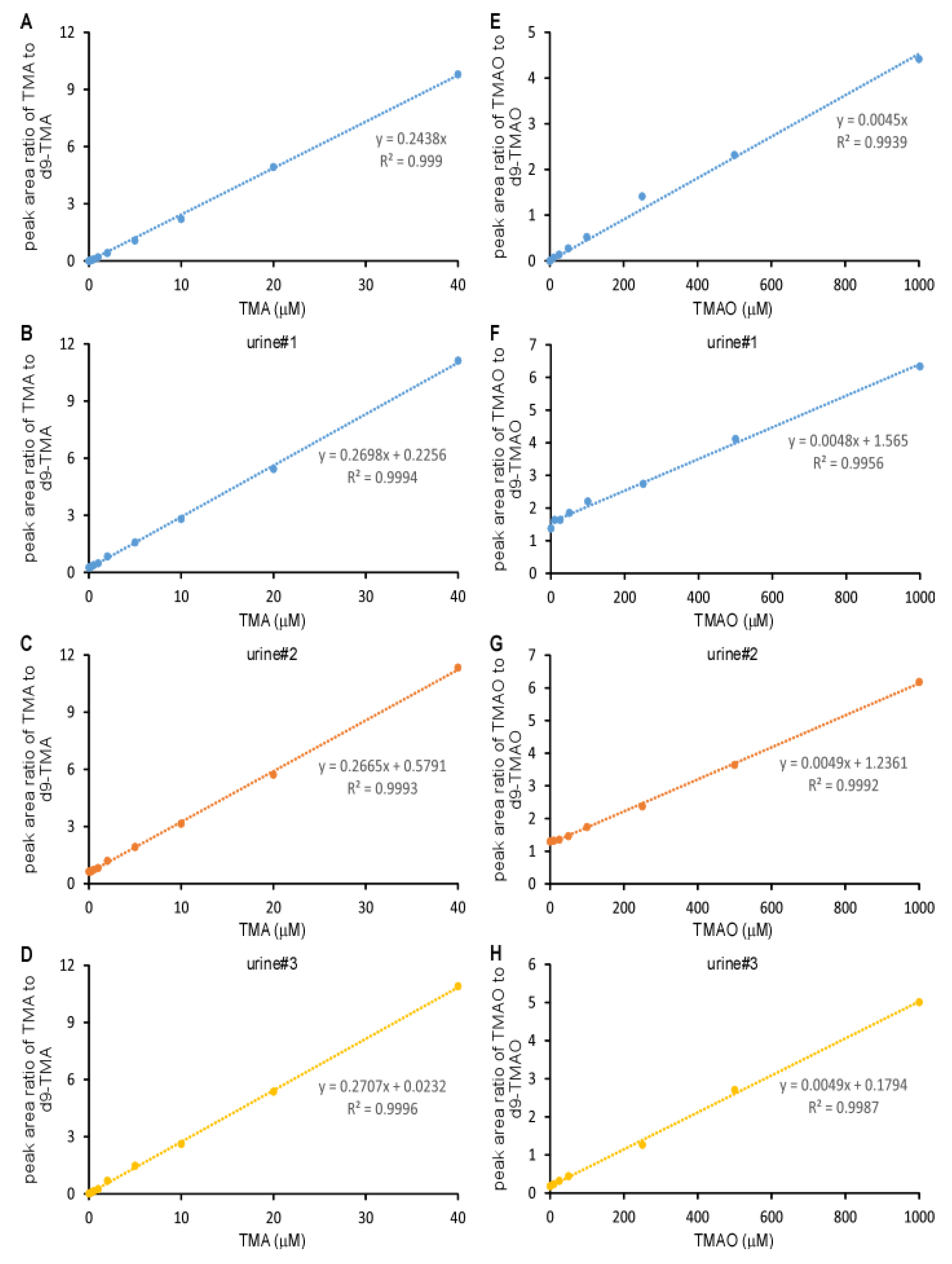
2.1. Validation of the LC/MS/MS Quantitation of TMA and TMAO in Urine
2.2. Normal Range of Urinary TMA and TMAO in Healthy Subjects
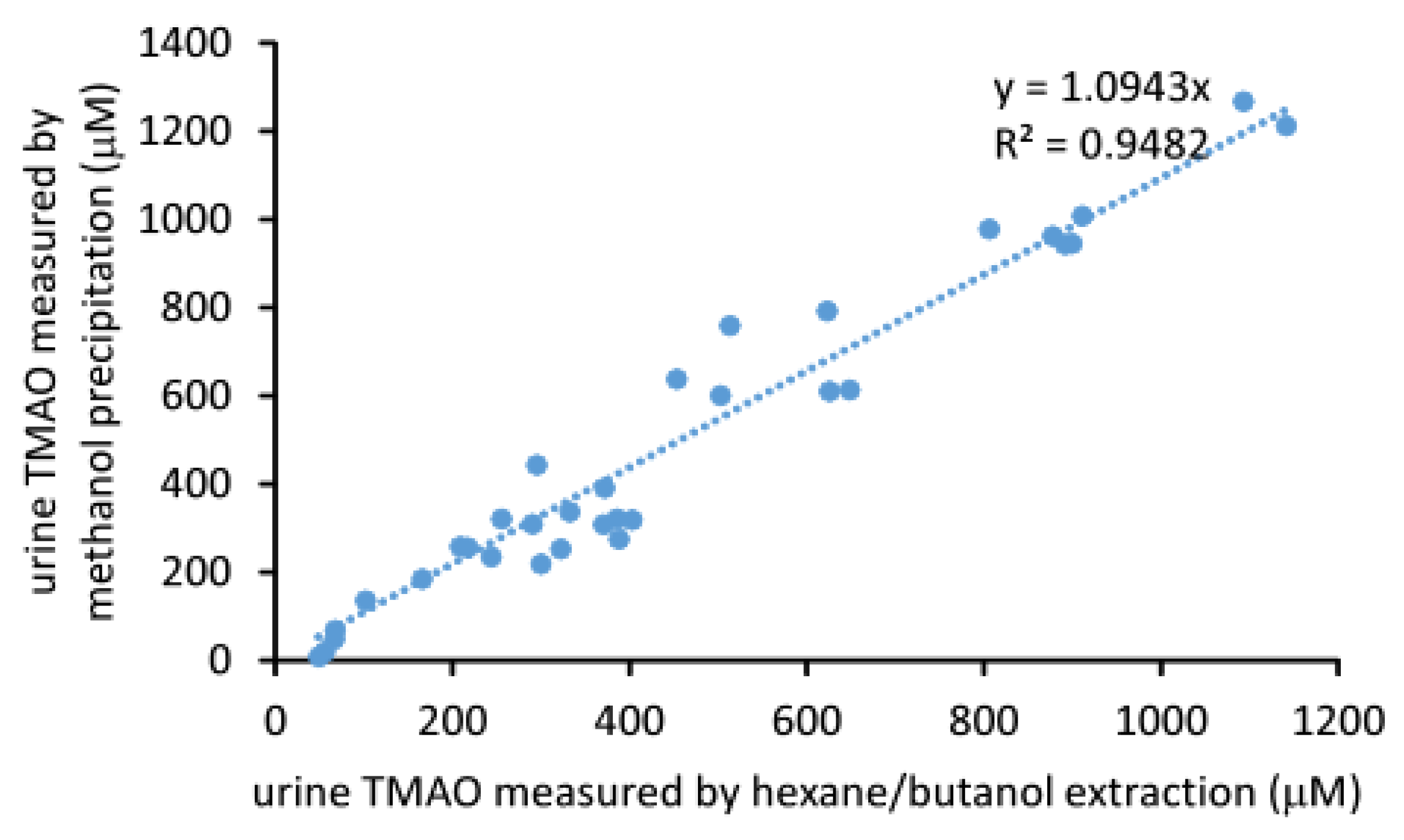
2.3. Comparison of the Measured TMAO Results by Two Different Methods
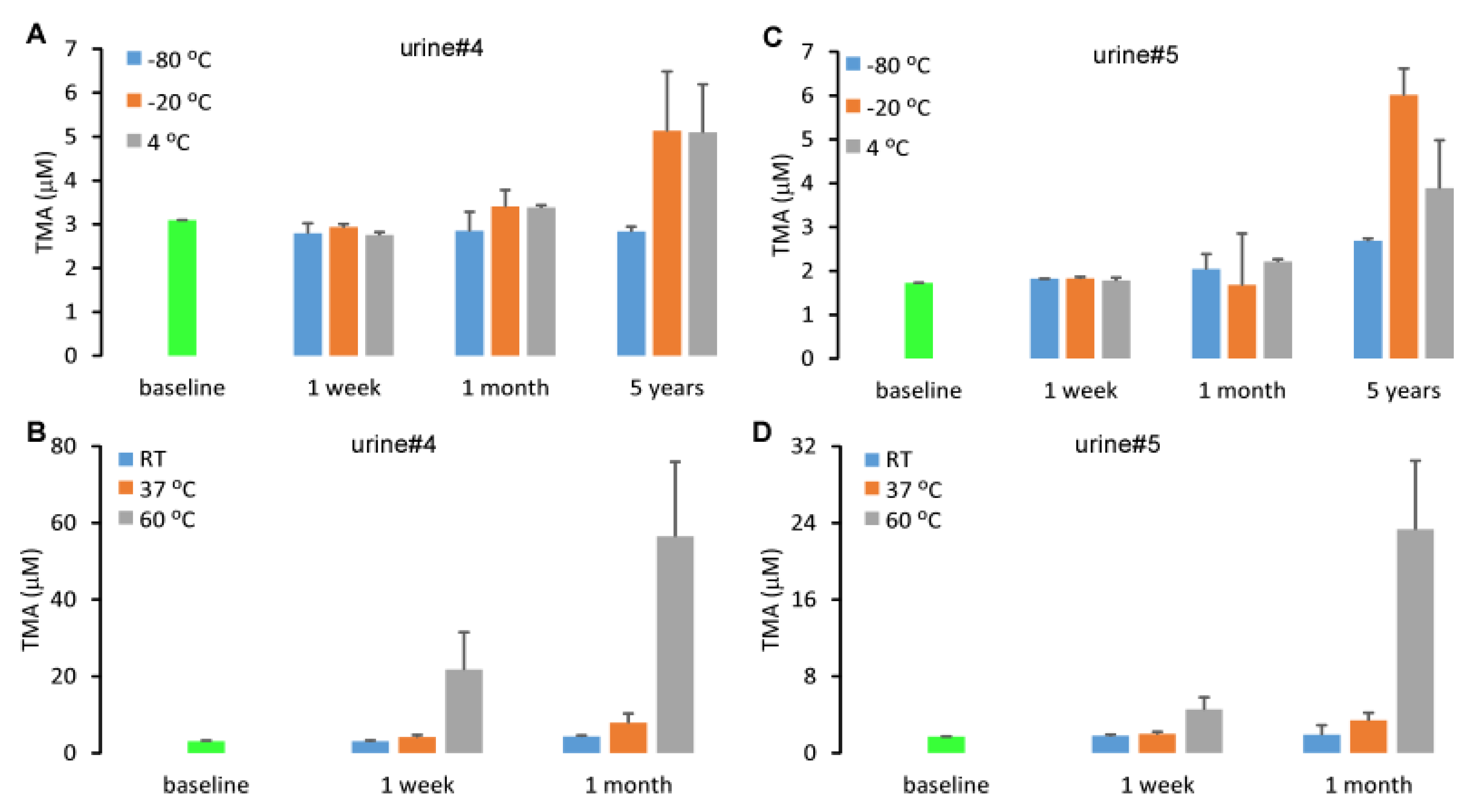
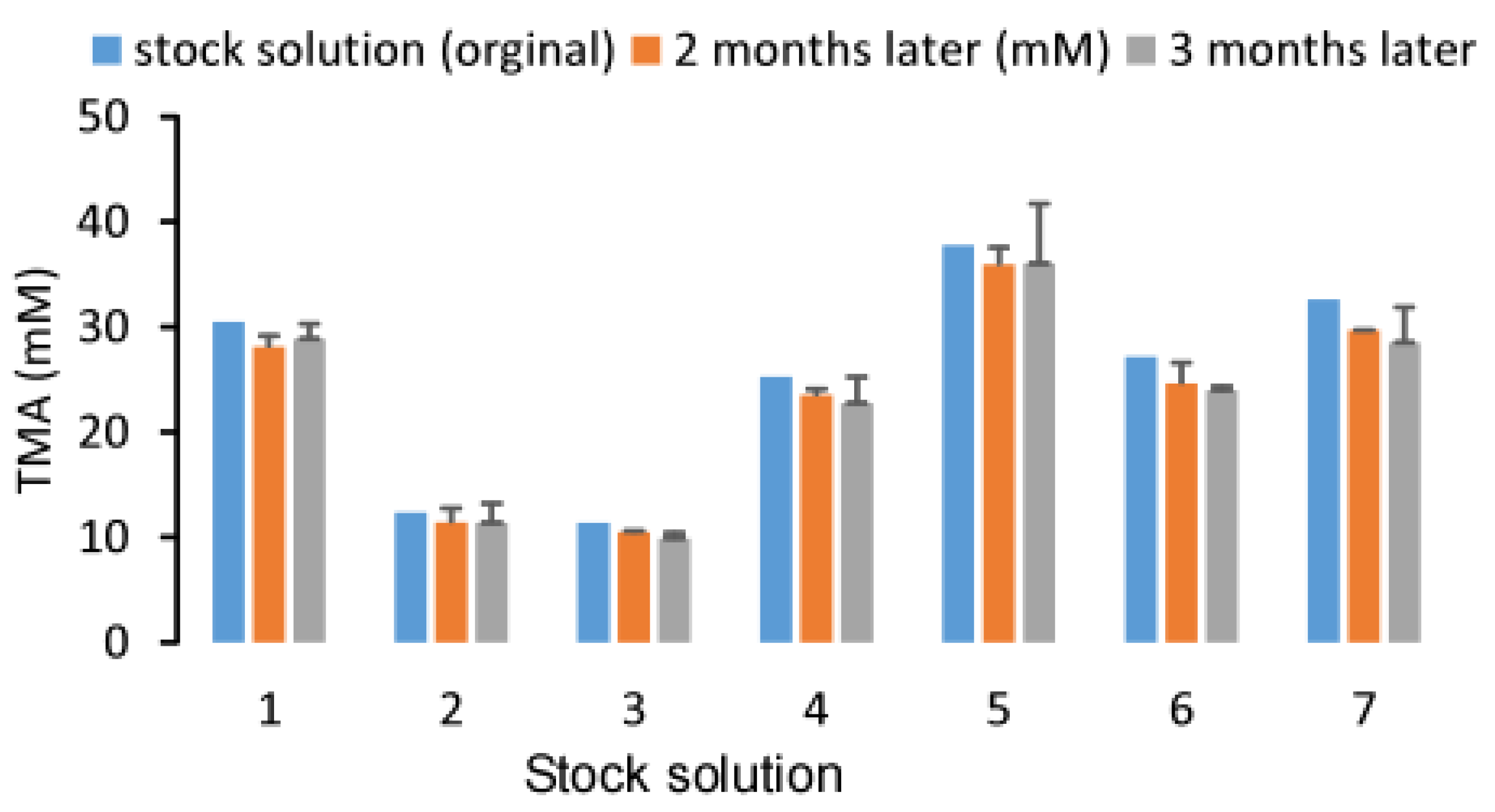
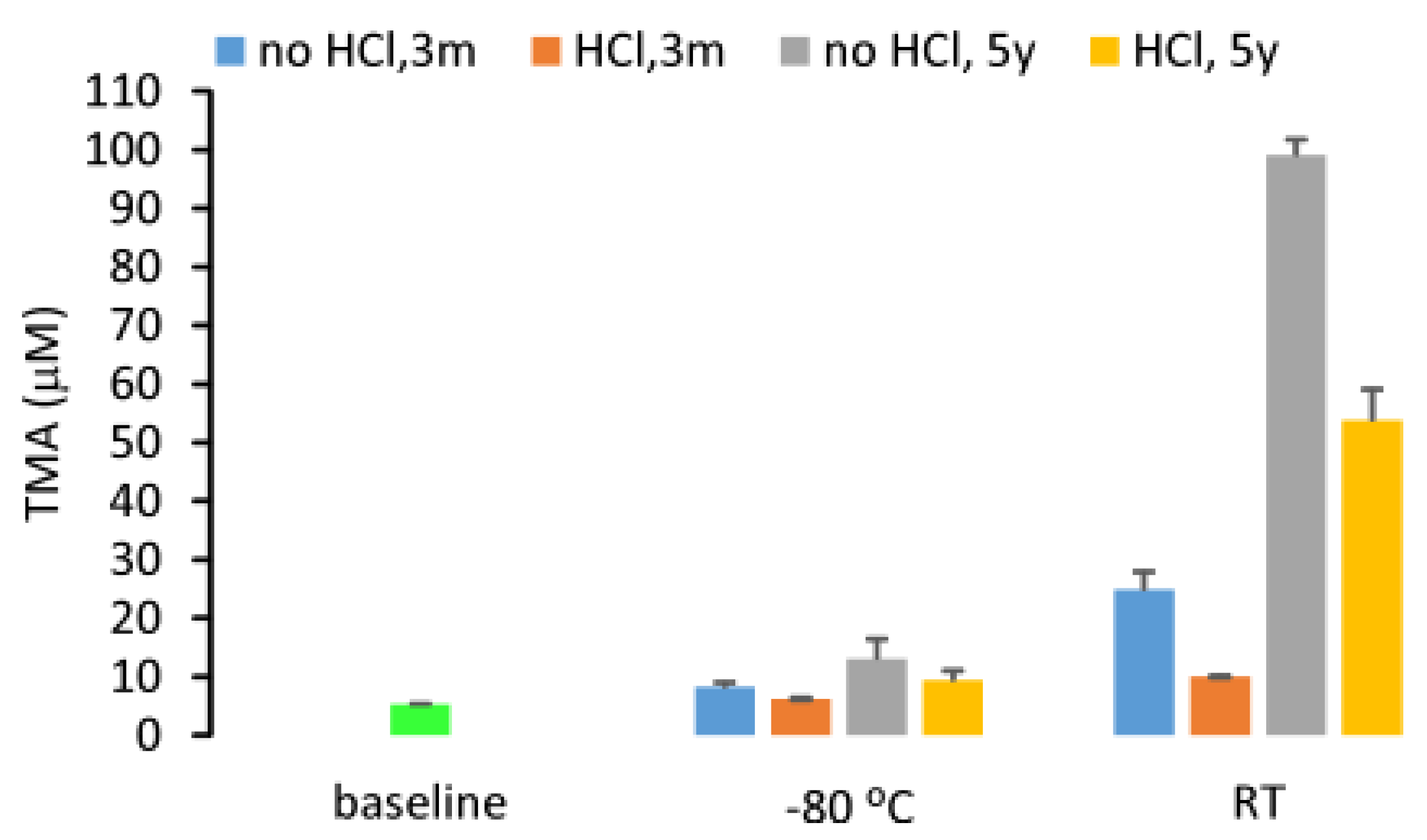
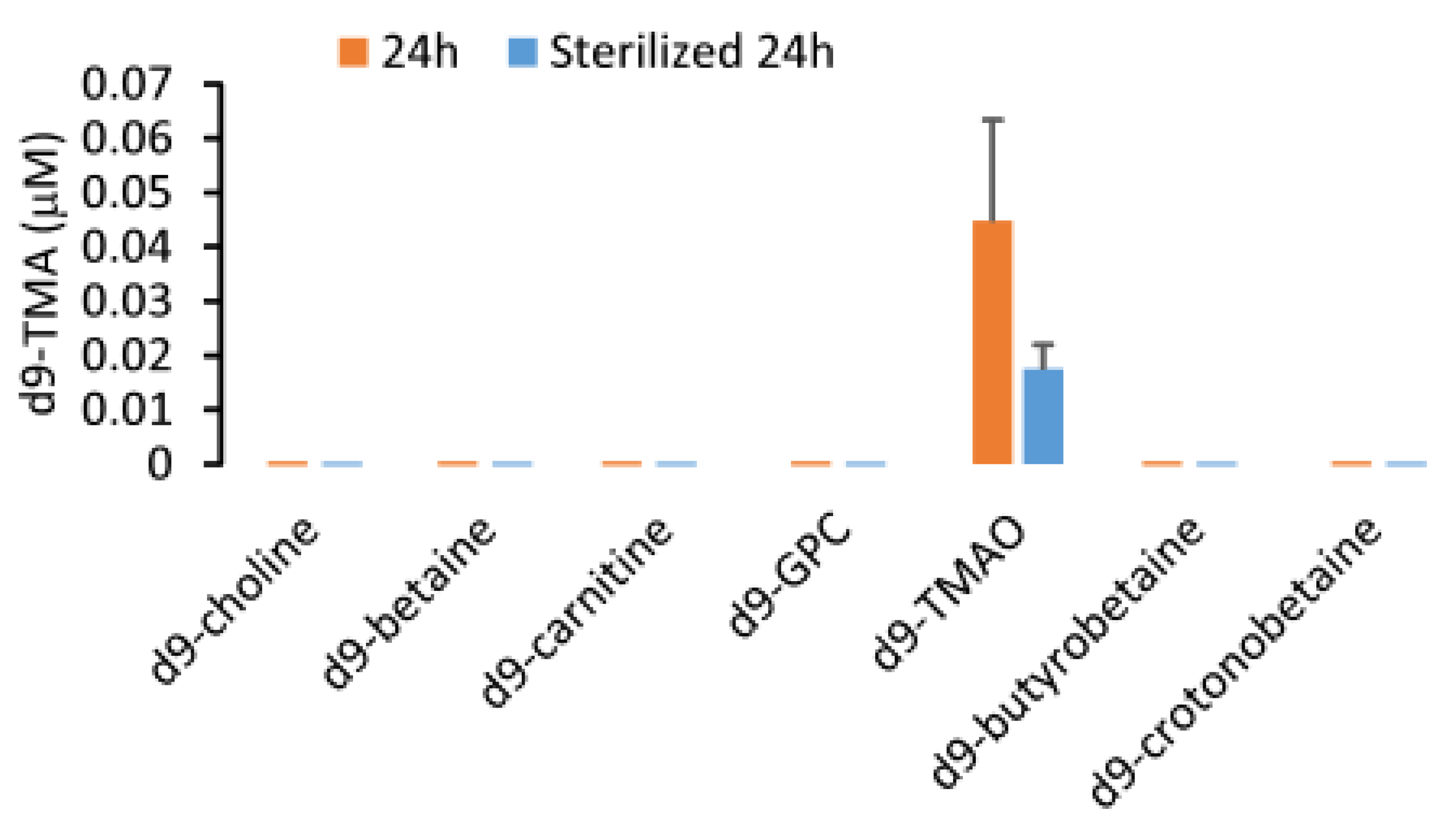
2.4. Stability of Urine TMA
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Research Subjects
4.3. Sample Processing
4.4. LC/MS/MS
4.5. Precision, Accuracy, Limit of Quantitation and Linearity
Author Contributions
Funding
Conflicts of Interest
References
- Wang, Z.; Klipfell, E.; Bennett, B.J.; Koeth, R.; Levison, B.S.; DuGar, B.; Feldstein, A.E.; Britt, E.B.; Fu, X.; Chung, Y.M.; et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011, 472, 57–63. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Wang, Z.; Levison, B.S.; Buffa, J.A.; Org, E.; Sheehy, B.T.; Britt, E.B.; Fu, X.; Wu, Y.; Li, L.; et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat. Med. 2013, 19, 576–585. [Google Scholar] [CrossRef] [PubMed]
- Zhu, W.; Gregory, J.C.; Org, E.; Buffa, J.A.; Gupta, N.; Wang, Z.; Li, L.; Fu, X.; Wu, Y.; Mehrabian, M.; et al. Gut Microbial Metabolite TMAO Enhances Platelet Hyperreactivity and Thrombosis Risk. Cell 2016, 165, 111–124. [Google Scholar] [CrossRef] [PubMed]
- Seldin, M.M.; Meng, Y.; Qi, H.; Zhu, W.; Wang, Z.; Hazen, S.L.; Lusis, A.J.; Shih, D.M. Trimethylamine N-Oxide Promotes Vascular Inflammation Through Signaling of Mitogen-Activated Protein Kinase and Nuclear Factor-kappa B. J. Am. Heart Assoc. 2016, 5, e002767. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Levison, B.S.; Koeth, R.A.; Britt, E.B.; Fu, X.; Wu, Y.; Hazen, S.L. Intestinal Microbial Metabolism of Phosphatidylcholine and Cardiovascular Risk. N. Engl. J. Med. 2013, 368, 1575–1584. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Kennedy, D.J.; Wu, Y.; Buffa, J.A.; Agatisa-Boyle, B.; Li, X.S.; Levison, B.S.; Hazen, S.L. Gut Microbiota-Dependent Trimethylamine N-Oxide (TMAO) Pathway Contributes to Both Development of Renal Insufficiency and Mortality Risk in Chronic Kidney Disease. Circ. Res. 2015, 116, 448–455. [Google Scholar] [CrossRef]
- Shan, Z.; Sun, T.; Huang, H.; Chen, S.; Chen, L.; Luo, C.; Yang, W.; Yang, X.; Yao, P.; Cheng, J.; et al. Association between microbiota-dependent metabolite trimethylamine-N-oxide and type 2 diabetes. Am. J. Clin. Nutr. 2017, 106, 888–894. [Google Scholar] [CrossRef]
- Lever, M.; George, P.M.; Slow, S.; Bellamy, D.; Young, J.M.; Ho, M.; McEntyre, C.J.; Elmslie, J.L.; Atkinson, W.; Molyneux, S.L.; et al. Betaine and Trimethylamine-N-Oxide as Predictors of Cardiovascular Outcomes Show Different Patterns in Diabetes Mellitus: An Observational Study. PLoS ONE 2014, 9, e114969. [Google Scholar] [CrossRef]
- Barrea, L.; Annunziata, G.; Muscogiuri, G.; Di Somma, C.; Laudisio, D.; Maisto, M.; De Alteriis, G.; Tenore, G.C.; Colao, A.; Savastano, S. Trimethylamine-N-oxide (TMAO) as Novel Potential Biomarker of Early Predictors of Metabolic Syndrome. Nutrients 2018, 10, 1971. [Google Scholar] [CrossRef]
- Wang, Z.; Bergeron, N.; Levison, B.S.; Li, X.S.; Chiu, S.; Jia, X.; Koeth, R.A.; Li, L.; Wu, Y.; Tang, W.W.; et al. Impact of chronic dietary red meat, white meat, or non-meat protein on trimethylamine N-oxide metabolism and renal excretion in healthy men and women. Eur. Heart J. 2019, 40, 583–594. [Google Scholar] [CrossRef]
- Dolphin, C.T.; Riley, J.H.; Smith, R.L.; Shephard, E.A.; Phillips, I.R. Structural organization of the human flavin-containing monooxygenase 3 gene (FMO3), the favored candidate for fish-odor syndrome, determined directly from genomic DNA. Genomics 1997, 46, 260–267. [Google Scholar] [CrossRef] [PubMed]
- Al-Waiz, M.; Ayesh, R.; Mitchell, S.C.; Idle, J.R.; Smith, R.L. Trimethylaminuria (‘fish-odour syndrome’): A study of an affected family. Clin. Sci. 1988, 74, 231–236. [Google Scholar] [CrossRef]
- al-Waiz, M.; Ayesh, R.; Mitchell, S.C.; Idle, J.R.; Smith, R.L. Trimethylaminuria: The detection of carriers using a trimethylamine load test. J. Inherit. Metab. Dis. 1989, 12, 80–85. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W. A flow injection electrospray ionization tandem mass spectrometric method for the simultaneous measurement of trimethylamine and trimethylamine N-oxide in urine. J. Mass Spectrom. 2008, 43, 495–499. [Google Scholar] [CrossRef] [PubMed]
- Schlesinger, P. An improved gas--liquid chromatographic method for analysis of trimethylamine in urine. Anal. Biochem. 1979, 94, 358–359. [Google Scholar] [CrossRef]
- Matsushita, K.; Kato, K.; Ohsaka, A.; Kanazawa, M.; Aizawa, K. A simple and rapid method for detecting trimethylamine in human urine by proton NMR. Physiol. Chem. Phys. Med. NMR 1989, 21, 3–4. [Google Scholar]
- Zhang, A.Q.; Mitchell, S.C.; Ayesh, R.; Smith, R.L. Determination of trimethylamine and related aliphatic amines in human urine by head-space gas chromatography. J. Chromatogr. 1992, 584, 141–145. [Google Scholar] [CrossRef]
- Maschke, S.; Wahl, A.; Azaroual, N.; Boulet, O.; Crunelle, V.; Imbenotte, M.; Foulard, M.; Vermeersch, G.; Lhermitte, M. 1H-NMR analysis of trimethylamine in urine for the diagnosis of fish-odour syndrome. Clin. Chim. Acta 1997, 263, 139–146. [Google Scholar] [CrossRef]
- Mamer, O.A.; Choiniere, L.; Treacy, E.P. Measurement of trimethylamine and trimethylamine N-oxide independently in urine by fast atom bombardment mass spectrometry. Anal. Biochem. 1999, 276, 144–149. [Google Scholar] [CrossRef]
- Mills, G.A.; Walker, V.; Mughal, H. Quantitative determination of trimethylamine in urine by solid-phase microextraction and gas chromatography-mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1999, 723, 281–285. [Google Scholar] [CrossRef]
- Zhao, X.; Zeisel, S.H.; Zhang, S. Rapid LC-MRM-MS assay for simultaneous quantification of choline, betaine, trimethylamine, trimethylamine N-oxide, and creatinine in human plasma and urine. Electrophoresis 2015, 36, 2207–2214. [Google Scholar] [CrossRef] [PubMed]
- Awwad, H.M.; Geisel, J.; Obeid, R. Determination of trimethylamine, trimethylamine N-oxide, and taurine in human plasma and urine by UHPLC-MS/MS technique. J. Chromatogr. 2016, 1038, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Koeth, R.A.; Levison, B.S.; Culley, M.K.; Buffa, J.A.; Wang, Z.; Gregory, J.C.; Org, E.; Wu, Y.; Li, L.; Smith, J.D.; et al. Gamma-Butyrobetaine is a proatherogenic intermediate in gut microbial metabolism of L-carnitine to TMAO. Cell Metab. 2014, 20, 799–812. [Google Scholar] [CrossRef] [PubMed]
- Li, X.S.; Wang, Z.; Cajka, T.; Buffa, J.A.; Nemet, I.; Hurd, A.G.; Gu, X.; Skye, S.M.; Roberts, A.B.; Wu, Y.; et al. Untargeted metabolomics identifies trimethyllysine, a TMAO-producing nutrient precursor, as a predictor of incident cardiovascular disease risk. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Levison, B.S.; Hazen, J.E.; Donahue, L.; Li, X.M.; Hazen, S.L. Measurement of trimethylamine-N-oxide by stable isotope dilution liquid chromatography tandem mass spectrometry. Anal. Biochem. 2014, 455, 35–40. [Google Scholar] [CrossRef]
- Whiteside, S.A.; Razvi, H.; Dave, S.; Reid, G.; Burton, J.P. The microbiome of the urinary tract--a role beyond infection. Nat. Rev. Urol. 2015, 12, 81–90. [Google Scholar] [CrossRef]
- Tang, W.W.; Wang, Z.; Li, X.S.; Fan, Y.; Li, D.S.; Wu, Y.; Hazen, S.L. Increased Trimethylamine N-Oxide Portends High Mortality Risk Independent of Glycemic Control in Patients with Type 2 Diabetes Mellitus. Clin. Chem. 2017, 63, 297–306. [Google Scholar] [CrossRef]
- Wang, Z.; Roberts, A.B.; Buffa, J.A.; Levison, B.S.; Zhu, W.; Org, E.; Gu, X.; Huang, Y.; Zamanian-Daryoush, M.; Culley, M.K.; et al. Non-lethal Inhibition of Gut Microbial Trimethylamine Production for the Treatment of Atherosclerosis. Cell 2015, 163, 1585–1595. [Google Scholar] [CrossRef]
- Roberts, A.B.; Gu, X.; Buffa, J.A.; Hurd, A.G.; Wang, Z.; Zhu, W.; Gupta, N.; Skye, S.M.; Cody, D.B.; Levison, B.S.; et al. Development of a gut microbe-targeted nonlethal therapeutic to inhibit thrombosis potential. Nat. Med. 2018, 24, 1407–1417. [Google Scholar] [CrossRef]
- Wenzl, T.; Haedrich, J.; Schaechtele, A.; Robouch, P.; Stroka, J. Guidance Document on the Estimation of LOD and LOQ for Measurements in the Field of Contaminants in Feed and Food; Publications Office of the European Union: Luxemburg, 2016. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | TMA | TMAO | ||
|---|---|---|---|---|
| LLOD | 0.26 µM | 0.57 µM | ||
| LLOQ | 0.40 µM | 5.0 µM | ||
| ULOQ | >50 µM | >10 mM | ||
| Accuracy (%) | 0.5 µM | 96.2 | 10 µM | 98.4 |
| 2 µM | 97.0 | 50 µM | 102.8 | |
| 5 µM | 101.1 | 100 µM | 102.1 | |
| 10 µM | 102.0 | 250 µM | 102.1 | |
| 20 µM | 102.3 | 500 µM | 100.6 | |
| 40 µM | 101.3 | 2000 µM | 101.3 | |
| Urine | TMA | TMAO | ||||
|---|---|---|---|---|---|---|
| Mean ± SD | Intraday CV% | Interday CV% | Mean ± SD | Intraday CV% | Interday CV% | |
| 1 | 0.99 ± 0.09 | 3.4 | 17.6 | 305.6 ± 22.7 | 5.2 | 12.4 |
| 2 | 2.41 ± 0.10 | 2.2 | 8.6 | 236.8 ± 15.9 | 5.1 | 10.5 |
| 3 | 2.28 ± 0.14 | 2.1 | 12.3 | 303.3 ± 11.8 | 2.9 | 6.1 |
| 4 | 4.60 ± 0.18 | 2.5 | 7.0 | 393.3 ± 30.3 | 8.5 | 2.3 |
| 5 | 19.54 ± 0.65 | 2.1 | 6.2 | 825.2 ± 68.1 | 7.8 | 10.1 |
| 6 | 9.96 ± 0.34 | 2.5 | 5.9 | 1215.2 ± 47.3 | 2.6 | 7.3 |
| 0% | 25% | 50% | 75% | 100% | |
|---|---|---|---|---|---|
| TMA (μM) | 0.70 | 1.37 | 2.07 | 2.62 | 4.39 |
| TMAO (μM) | 52.0 | 243.7 | 379.0 | 648.1 | 1141.0 |
| TMAO/TMA (mol/mol) | 66 | 132 | 208 | 315 | 506 |
| TMA/creatinine (mmol/mol) | 0.05 | 0.09 | 0.13 | 0.23 | 1.43 |
| TMAO/creatinine (mmol/mol) | 11.3 | 22.5 | 33.8 | 45.6 | 106.4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jia, X.; Osborn, L.J.; Wang, Z. Simultaneous Measurement of Urinary Trimethylamine (TMA) and Trimethylamine N-Oxide (TMAO) by Liquid Chromatography–Mass Spectrometry. Molecules 2020, 25, 1862. https://doi.org/10.3390/molecules25081862
Jia X, Osborn LJ, Wang Z. Simultaneous Measurement of Urinary Trimethylamine (TMA) and Trimethylamine N-Oxide (TMAO) by Liquid Chromatography–Mass Spectrometry. Molecules. 2020; 25(8):1862. https://doi.org/10.3390/molecules25081862
Chicago/Turabian StyleJia, Xun, Lucas J. Osborn, and Zeneng Wang. 2020. "Simultaneous Measurement of Urinary Trimethylamine (TMA) and Trimethylamine N-Oxide (TMAO) by Liquid Chromatography–Mass Spectrometry" Molecules 25, no. 8: 1862. https://doi.org/10.3390/molecules25081862
APA StyleJia, X., Osborn, L. J., & Wang, Z. (2020). Simultaneous Measurement of Urinary Trimethylamine (TMA) and Trimethylamine N-Oxide (TMAO) by Liquid Chromatography–Mass Spectrometry. Molecules, 25(8), 1862. https://doi.org/10.3390/molecules25081862