
In Silico Screening for Novel Leucine Aminopeptidase Inhibitors with 3,4-Dihydroisoquinoline Scaffold



Abstract
1. Introduction
2. Results and Discussion
2.1. Database Search and Library Establishment
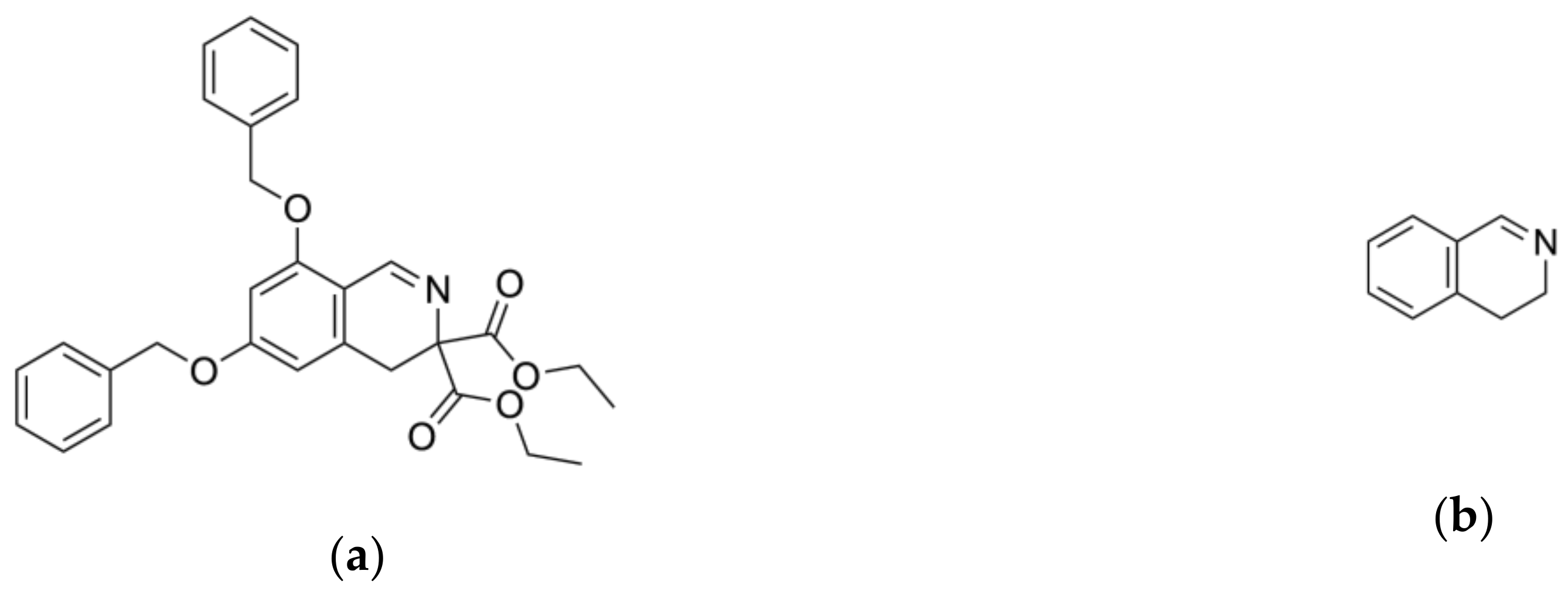
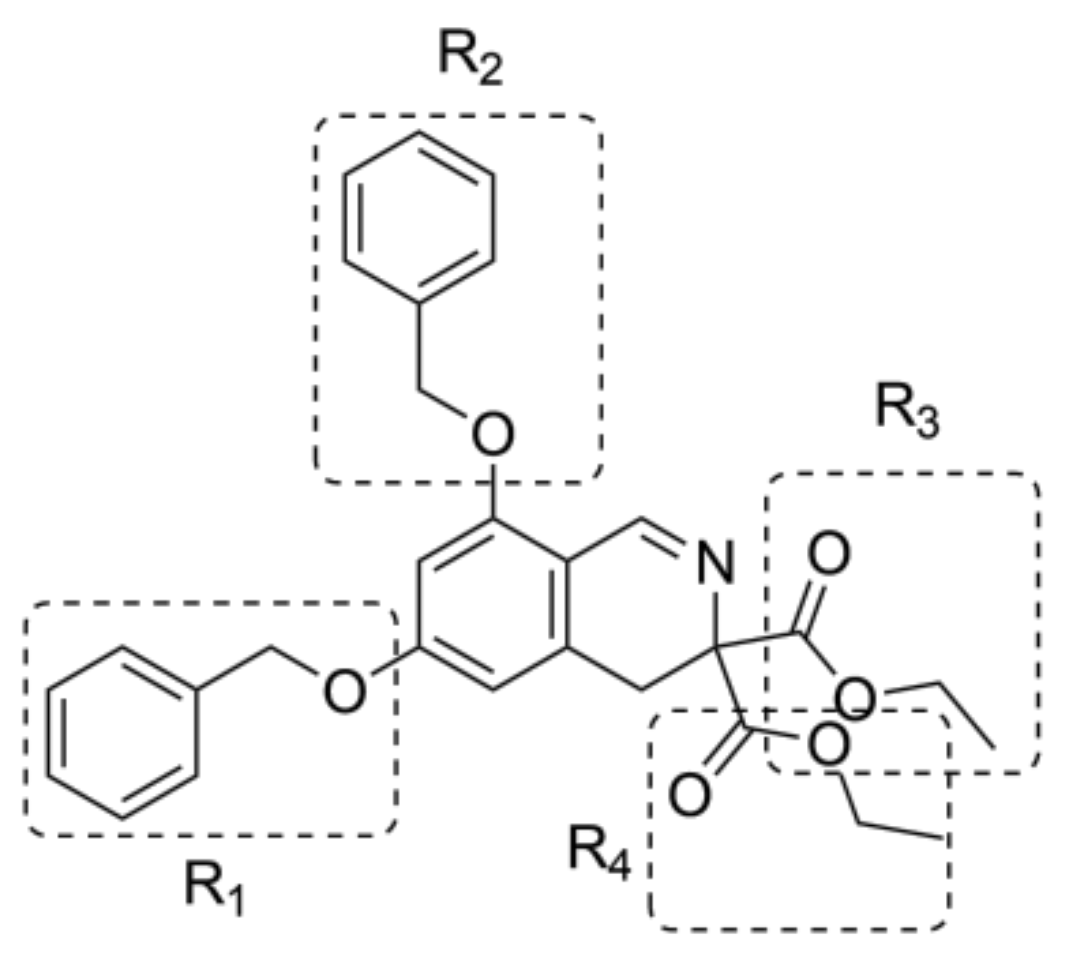
2.2. Ligand Growing Experiment
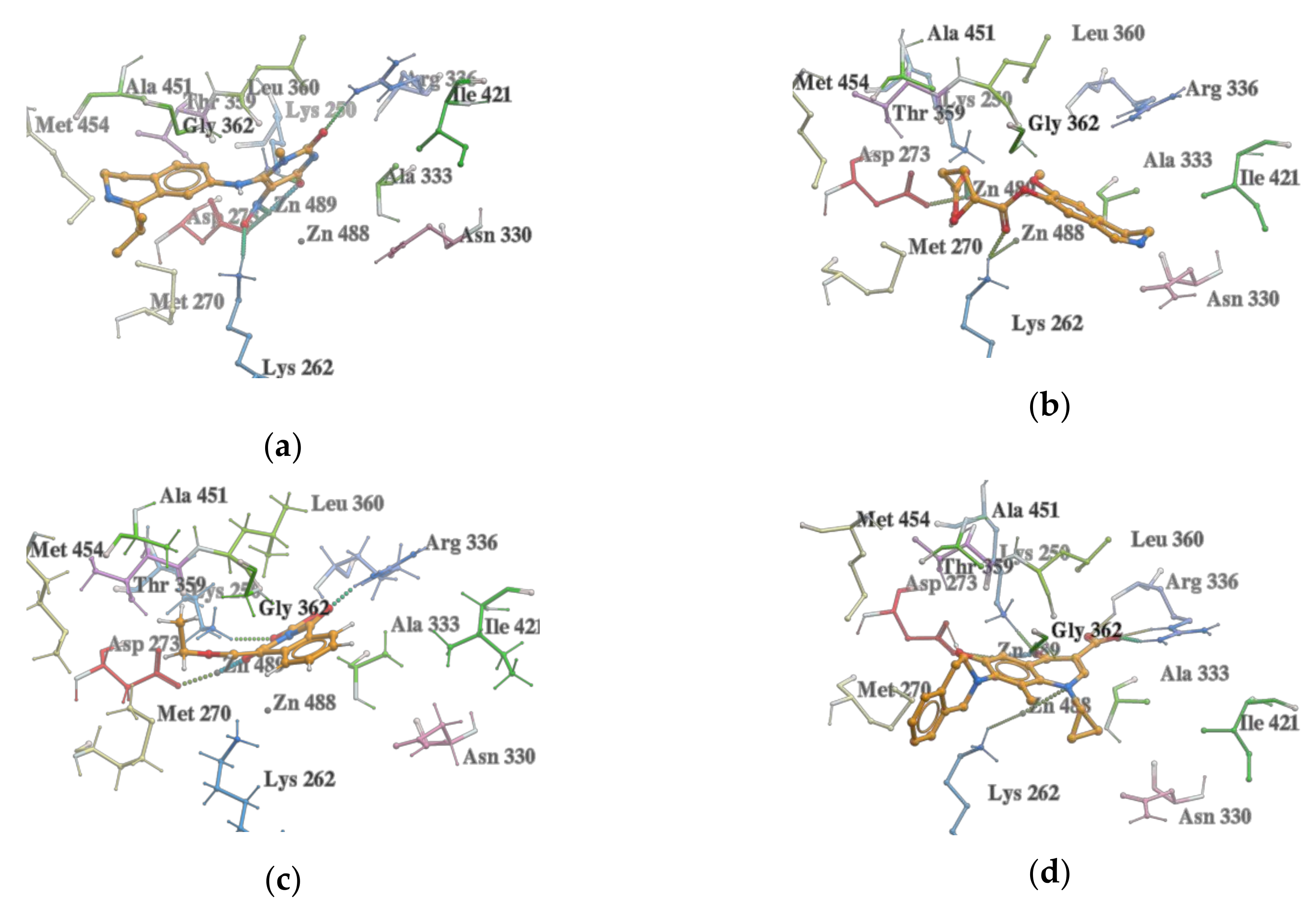
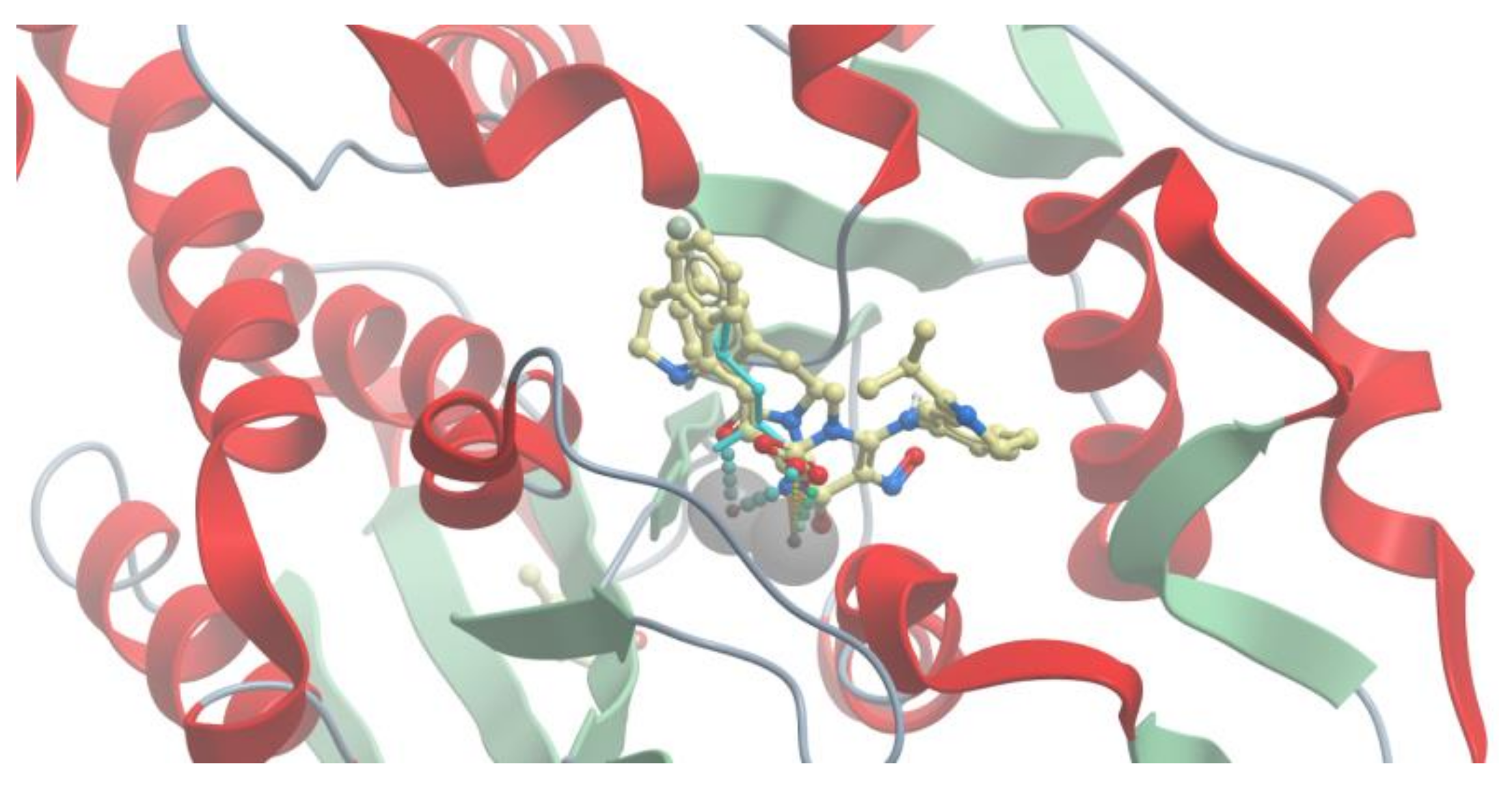
2.3. Molecular Docking
2.4. VS with Lead Finder
2.5. 3D-QSAR
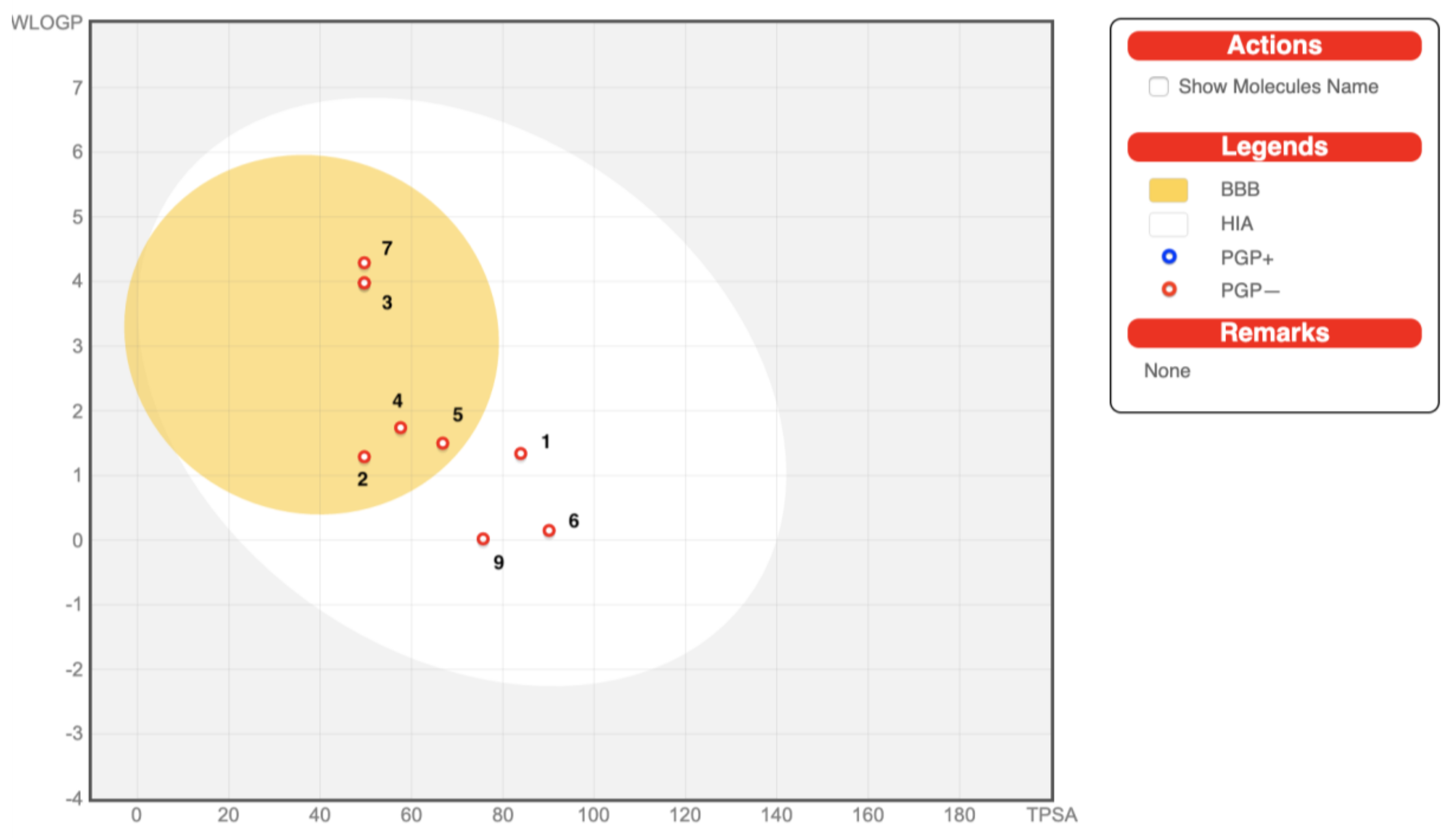
2.6. In Silico Drug-Likeness, Bioavailability, and Toxicology Prediction
3. Materials and Methods
3.1. Database Search and Library Establishment
3.2. Ligand Growing Experiment
3.3. Molecular Docking
3.4. VS with Lead Finder
3.5. 3D-QSAR
3.6. In Silico Drug-Likeness, Bioavailability, and Toxicology Prediction
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cao, B.; Soerjomataram, I.; Bray, F. The global cancer burden. In World Cancer Report: Cancer Research for Cancer Prevention; Wild, C.P., Weiderpass, E., Stewart, B.W., Eds.; International Agency for Research on Cancer: Lyon, France, 2020. [Google Scholar]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Fang, C.; Zhang, J.; Yang, H.; Peng, L.; Wang, K.; Wang, Y.; Zhao, X.; Liu, H.; Dou, C.; Shi, L.; et al. Leucine aminopeptidase 3 promotes migration and invasion of breast cancer through upregulation of fascin and matrix metalloproteinases-2/9 expression. J. Cell Biochem. 2019, 120, 3611–3620. [Google Scholar] [CrossRef] [PubMed]
- Ziemska, J.; Solecka, J. Tyrosine kinase, aurora kinase and leucine aminopeptidase as attractive drug targets in anticancer therapy-characterisation of their inhibitors. Rocz. Panstw. Zakl. Hig. 2016, 67, 329–342. [Google Scholar]
- Tian, S.Y.; Chen, S.H.; Shao, B.F.; Cai, H.Y.; Zhou, Y.; Zhou, Y.L.; Xu, A.B. Expression of leucine aminopeptidase 3 (LAP3) correlates with prognosis and malignant development of human hepatocellular carcinoma (HCC). Int. J. Clin. Exp. Pathol. 2014, 7, 3752–3762. [Google Scholar]
- Wang, X.; Shi, L.; Deng, Y.; Qu, M.; Mao, S.; Xu, L.; Xu, W.; Fang, C. Inhibition of leucine aminopeptidase 3 supresses invasion of ovarian cancer cells through down-regulation of fascin and MMP-2/9. Eur. J. Pharmacol. 2015, 768, 116–122. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, X.; Shi, H.; Li, M.; Xue, Q.; Ren, H.; Yao, L.; Chen, X.; Zhang, J.; Wang, H. Overexpression of leucine aminopeptidase 3 contributes to malignant development of human esophageal squamous cell carcinoma. J. Mol. Histol. 2014, 45, 283–292. [Google Scholar] [CrossRef]
- Martínez, J.M.; Prieto, I.; Ramírez, M.J.; Cueva, C.; Alba, F.; Ramírez, M. Aminopeptidases activities in breast cancer tissue. Clin. Chem. 1999, 45, 1797–1802. [Google Scholar]
- Wang, X.; Song, K.; Li, L.; Chen, L. Structure-based drug design strategies and challenges. Curr. Top Med. Chem. 2018, 18, 998–1006. [Google Scholar] [CrossRef]
- Verma, J.; Khedkar, V.M.; Coutinho, E.C. 3D-QSAR in Drug Design—A review. Curr. Top Med. Chem. 2010, 10, 95–115. [Google Scholar] [CrossRef]
- Alam, S.; Khan, F. Virtual screening, docking, ADMET and system pharmacology studies on Garcinia caged xanthone derivatives for anticancer activity. Sci. Rep. 2018, 8, 5524. [Google Scholar]
- Zhang, Y.; Zhang, D.; Tian, H.; Jiao, Y.; Shi, Z.; Ran, T.; Liu, H.; Lu, S.; Xu, A.; Qiao, X.; et al. Identification of covalent binding sites targeting cysteines based on computational approaches. Mol. Pharm. 2016, 13, 3106–3118. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Malcolm, T.R.; McGowan, S. M17 aminopeptidases diversity function by moderating their macromolecular assemblies and active site environment. Biochimie 2019, 166, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Ziemska, J.; Guśpiel, A.; Jarosz, J.; Nasulewicz-Goldeman, A.; Wietrzyk, J.; Kawęcki, R.; Pypowski, K.; Jarończyk, M.; Solecka, J. Molecular docking studies, biological and toxicity evaluation of dihydroisoquinoline derivatives as potential anticancer agents. Bioorg. Med. Chem. 2016, 24, 5302–5314. [Google Scholar] [CrossRef] [PubMed]
- Bermejo, A.; Andreu, I.; Suvire, F.; Léonce, S.; Caignard, D.H.; Renard, P.; Pierré, A.; Enriz, R.D.; Cortes, D.; Cabedo, N. Syntheses and antitumor targeting G1 phase of the cell cycle of benzoyldihydroisoquinolines and related 1-substituted isoquinolines. J. Med. Chem. 2002, 45, 5058–5068. [Google Scholar] [CrossRef] [PubMed]
- Lejczak, B.; Kafarski, P.; Zygmunt, J. Inhibition of aminopeptidases by aminophosphonates. Biochemistry 1989, 28, 3549–3555. [Google Scholar] [CrossRef]
- Grembecka, J.; Sokalski, W.A.; Kafarski, P. Computer-aided design and activity prediction of leucine aminopeptidase inhibitors. J. Comput. Aided Mol. Des. 2000, 14, 531–544. [Google Scholar] [CrossRef]
- Ziemska, J.; Solecka, J.; Jaronczyk, M. QSAR, docking studies and toxicology prediction of isoquinoline derivatives as leucine aminopeptidase inhibitors. Chem. Pap. 2017, 71, 2557–2568. [Google Scholar] [CrossRef]
- Sterling, T.; Irwin, J.J. ZINC 15-Ligand discovery for everyone. J. Chem. Inf. Model. 2015, 55, 2324–2337. [Google Scholar] [CrossRef]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.A.; Thiessen, P.A.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]
- Probst, D.; Reymond, J.L. Exploring DrugBank in virtual reality chemical space. J. Chem. Inf. Model. 2018, 58, 1731–1735. [Google Scholar] [CrossRef]
- Lipinski, C.A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharm. Toxicol Methods. 2000, 44, 235–249. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Spark, Version 10.5.6; Cresset: Litlington, Cambridgeshire, UK, 2020; Available online: http://www.cresset-group.com/spark/ (accessed on 18 March 2019).
- Cheeseright, T.; Mackey, M.; Rose, S.; Vinter, A. Molecular field extrema as descriptors of biological activity: Definition and validation. J. Chem. Inf. Model. 2006, 46, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Shi, C.Y.; Xie, J.; Dai, J.H.; Tian, Y. Identification of potential dipeptidyl peptidase (DPP)-IV inhibitors among Moringa oleifera, phytochemicals by virtual screening, molecular docking analysis, ADME/T-based prediction, and in vitro analyses. Molecules 2020, 25, 189. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M. Biased probability Monte Carlo conformational searches and electrostatic calculations for peptides and proteins. J. Mol. Biol. 1994, 235, 983–1002. [Google Scholar] [CrossRef]
- Abagyan, R.; Totrov, M.; Kuznetsov, D. ICM—A new method for protein modelling and design. Applications to docking and structure prediction from the distorted native conformation. J. Comp. Chem. 1994, 15, 488–506. [Google Scholar] [CrossRef]
- RSCB Protein Data Bank Website. Available online: http://www.rcsb.org/pdb/ (accessed on 5 February 2020).
- Sträter, N.; Lipscomb, W.N. Two-metal ion mechanism of bovine lens leucine aminopeptidase: Active site solvent structure and binding mode of L-leucinal, a gem-diolate transition state analogue, by X-ray crystallography. Biochemistry. 1995, 34, 14792–14800. [Google Scholar] [CrossRef]
- Floresta, G.; Gentile, D.; Perrini, G.; Patamia, V.; Rescifina, A. Computational tools in the discovery of FABP4 ligands: A statistical and molecular modelling approach. Mar. Drugs. 2019, 17, 624. [Google Scholar] [CrossRef]
- Grembecka, J.; Kafarski, P. Leucine aminopeptidase as a target for inhibitor design. Mini. Rev. Med. Chem. 2001, 1, 133–144. [Google Scholar] [CrossRef]
- Stroganov, O.V.; Novikov, F.N.; Stroylov, V.S.; Kulkov, V.; Chilov, G.G. Lead Finder: An approach to improve accuracy of protein−ligand docking, binding energy estimation, and virtual screening. J. Chem. Inf. Model. 2008, 48, 2371–2385. [Google Scholar] [CrossRef]
- Lead Finder, Software Package v 1.1.13; BioMolTech: Toronto, ON, Canada, 2020; Available online: http://www.cresset-group.com/lead-finder/ (accessed on 5 February 2020).
- Forge Software (v10.6.0); Cresset: Litlington, Cambridgeshire, UK, 2020.
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Potts, R.O. and Guy, R.H. Predicting skin permeability. Pharm. Res. 1992, 09, 663–669. [Google Scholar] [CrossRef] [PubMed]
- Meyer, U.A. Cytochrome P450 enzymes. Drug Metab Interact. 2012, 27, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Teague, S.J.; Davis, A.M.; Leeson, P.D.; Oprea, T. The design of leadlike combinatorial libraries. Angew. Chem. Int. Ed. Engl. 1999, 38, 3743–3748. [Google Scholar] [CrossRef]
- Ghose, A.K.; Viswanadhan, V.N.; Wendoloski, J.J. A knowledge-based approach in designing combinatorial or medicinal chemistry libraries for drug discovery. 1. A qualitative and quantitative characterization of known drug databases. J. Comb. Chem. 1999, 1, 55–68. [Google Scholar] [CrossRef]
- Veber, D.F.; Johnson, S.R.; Cheng, H.Y.; Smith, B.R.; Ward, K.W.; Kopple, K.D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. [Google Scholar] [CrossRef]
- Egan, W.J.; Merz Jr, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Muegge, I.; Heald, S.L.; Brittelli, D. Simple selection criteria for drug-like chemical matter. J. Med. Chem. 2001, 44, 1841–1846. [Google Scholar] [CrossRef]
- Yang, H.; Lou, C.; Sun, L.; Li, J.; Cai, Y.; Wang, Z.; Li, W.; Liu, G.; Tang, Y. admetSAR 2.0: Web-service for prediction and optimization of chemical ADMET properties. Bioinformatics 2019, 35, 1067–1069. [Google Scholar] [CrossRef]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Warren, G.L.; Do, T.D.; Kelley, B.P.; Nicholls, A.; Warren, S.D. Essential considerations for using protein–ligand structures in drug discovery. Drug Discov. Tod. 2012, 17, 1270–1281. [Google Scholar] [CrossRef] [PubMed]
- Dervisis, N.; Klahn, S. Therapeutic innovations: Tyrosine kinase inhibitors in cancer. Vet. Sci. 2016, 3, 4. [Google Scholar] [CrossRef] [PubMed]
- Longo, D.L. Imatinib changed everything. N. Engl. J. Med. 2017, 376, 982–983. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound’s number/ID | Chemical Name | Chemical Structure | pIC50 | ICM Score | LF Score |
|---|---|---|---|---|---|
| 1/ PUBCHEM 101710591 | 1,3-dioxo-4-(ethoxymethylene)-3,4-dihydroisoquinoline-2(1H)-carboxylic acid |  | 4.8 | −51.73 | −12.63 |
| 2/ ZINC 238690488 | (E)-3-(3,4-dihydroisoquinolin-1-yl)acrylic acid |  | 4.6 | −40.09 | −10.63 |
| 3/ ZINC 1243196903 | 5-(1-isopropyl-3,4-dihydroisoquinolin-7-yl)-2-methylbenzoic acid |  | 4.3 | −35.99 | −10.23 |
| 4/ PUBCHEM 67293279 | 6-bromo-1-oxo-3,4-dihydroisoquinoline-2-carboxylic acid |  | 4.9 | −35.37 | −12.97 |
| 5/ PUBCHEM 82579683 | 3-methyl-1-oxo-2-(oxolan-2-ylmethyl)-3,4-dihydroisoquinoline-4-carboxylic acid |  | 4.5 | −34.91 | −10.80 |
| 6/ PUBCHEM 135927986 | (3S)-6,7-dihydroxy-3,4-dihydroisoquinoline-3-carboxylic acid |  | 4.8 | −33.99 | −11.39 |
| 7/ ZINC 1206051829 | 2,3-difluoro-5-(1-isopropyl-3,4-dihydroisoquinolin-7-yl)benzoic acid |  | 4.6 | −33.92 | −11.06 |
| 8/ ZINC 1243180093 | 3-(1-isopropyl-3,4-dihydroisoquinolin-7-yl)-5-methylbenzoic acid |  | 4.5 | −32.84 | −12.23 |
| 9/ ZINC 34115917 | (3S)-1-amino-3,4-dihydroisoquinoline-3-carboxylic acid |  | 4.4 | −32.12 | −10.73 |
| Pharmacokinetic Parameters | Compounds | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |
| GI Absorption a | high | high | high | high | high | high | high | high | high |
| BBB b Barrier Permeation | no | yes | yes | yes | yes | no | yes | yes | no |
| PGP c Substrate | no | no | no | no | no | no | no | no | no |
| CYP d 1A2 Inhibitor | no | yes | yes | yes | no | no | yes | yes | no |
| CYP d 2C19 Inhibitor | no | no | no | no | no | no | yes | yes | no |
| CYP d 2C9 Inhibitor | no | no | yes | no | no | no | no | yes | no |
| CYP d 2D6 Inhibitor | no | no | yes | no | no | no | no | yes | no |
| CYP d 3A4 Inhibitor | no | no | no | no | no | no | no | no | no |
| Log Kp (Skin Permeation e) | −6.57 | −6.53 | −5.19 | −6.16 | −7.01 | −7.27 | −5.44 | −5.19 | −7.08 |
| Leadlikeness | yes | no | no | yes | yes | no | no | no | no |
| Bioavailability Score | 0.56 | 0.56 | 0.56 | 0.56 | 0.56 | 0.56 | 0.56 | 0.56 | 0.55 |
| PAINSBrenk | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| 3 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | |
| Compounds | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 |
|---|---|---|---|---|---|---|---|---|---|
| Carcinogenicity | −(0.73) | −(0.83) | −(0.89) | −(0.84) | −(0.81) | −(0.76) | −(0.86) | −(0.89) | −(0.87) |
| Eye corrosion | −(0.98) | −(0.95) | −(0.98) | −(0.98) | −(0.99) | −(0.99) | −(0.98) | −(0.98) | −(0.99) |
| Eye irritation | +(0.85) | +(0.94) | −(0.96) | +(0.85) | −(0.96) | +(0.86) | −(0.98) | −(0.92) | −(0.79) |
| Ames mutagenesis | −(0.61) | −(0.82) | −(0.65) | −(0.74) | +(0.51) | −(0.75) | −(0.63) | −(0.74) | −(0.66) |
| Hepatotoxicity | +(0.63) | +(0.73) | +(0.83) | +(0.55) | −(0.55) | −(0.53) | +(0.83) | +(0.80) | +(0.53) |
| Acute oral toxicity | III (0.63) | II (0.42) | III (0.51) | III (0.60) | III (0.73) | III (0.41) | III (0.53) | III (0.53) | II (0.37) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ziemska, J.; Solecka, J.; Jarończyk, M. In Silico Screening for Novel Leucine Aminopeptidase Inhibitors with 3,4-Dihydroisoquinoline Scaffold. Molecules 2020, 25, 1753. https://doi.org/10.3390/molecules25071753
Ziemska J, Solecka J, Jarończyk M. In Silico Screening for Novel Leucine Aminopeptidase Inhibitors with 3,4-Dihydroisoquinoline Scaffold. Molecules. 2020; 25(7):1753. https://doi.org/10.3390/molecules25071753
Chicago/Turabian StyleZiemska, Joanna, Jolanta Solecka, and Małgorzata Jarończyk. 2020. "In Silico Screening for Novel Leucine Aminopeptidase Inhibitors with 3,4-Dihydroisoquinoline Scaffold" Molecules 25, no. 7: 1753. https://doi.org/10.3390/molecules25071753
APA StyleZiemska, J., Solecka, J., & Jarończyk, M. (2020). In Silico Screening for Novel Leucine Aminopeptidase Inhibitors with 3,4-Dihydroisoquinoline Scaffold. Molecules, 25(7), 1753. https://doi.org/10.3390/molecules25071753