
Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects


 , ,
, ,  and
and 
Abstract
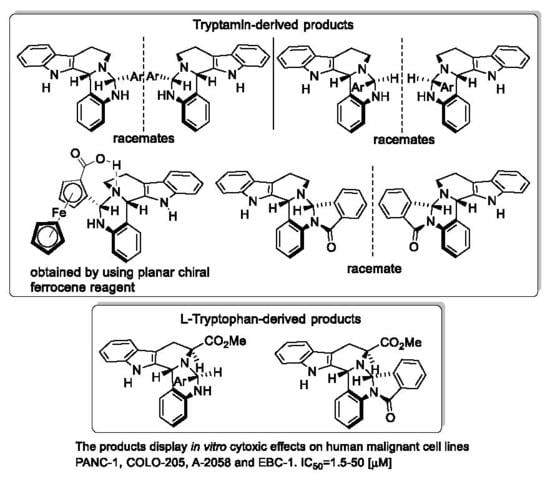
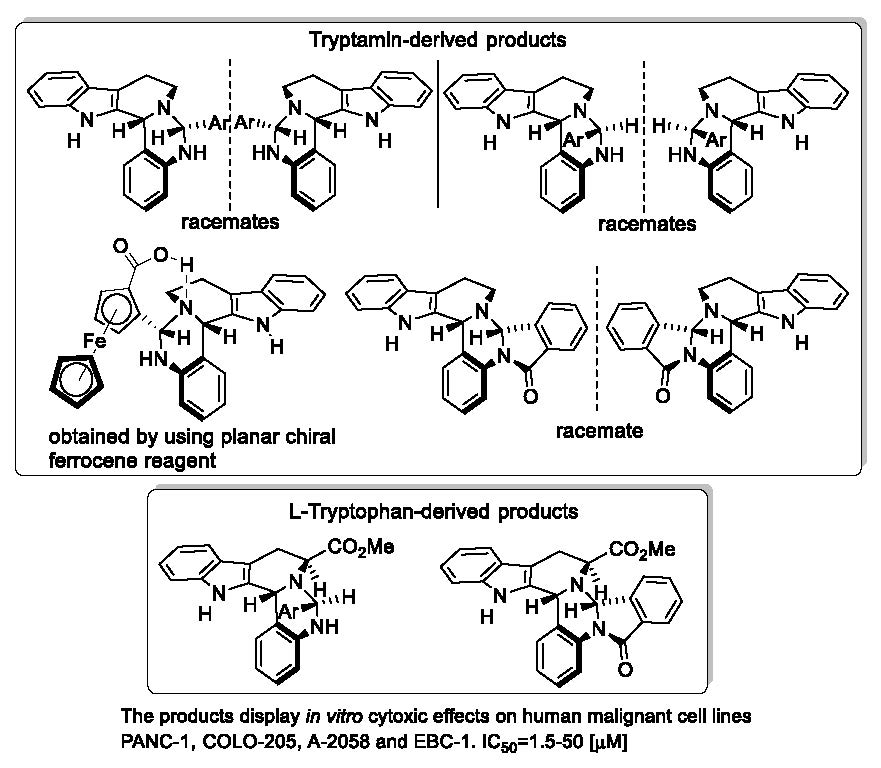
1. Introduction
2. Results and Discussion
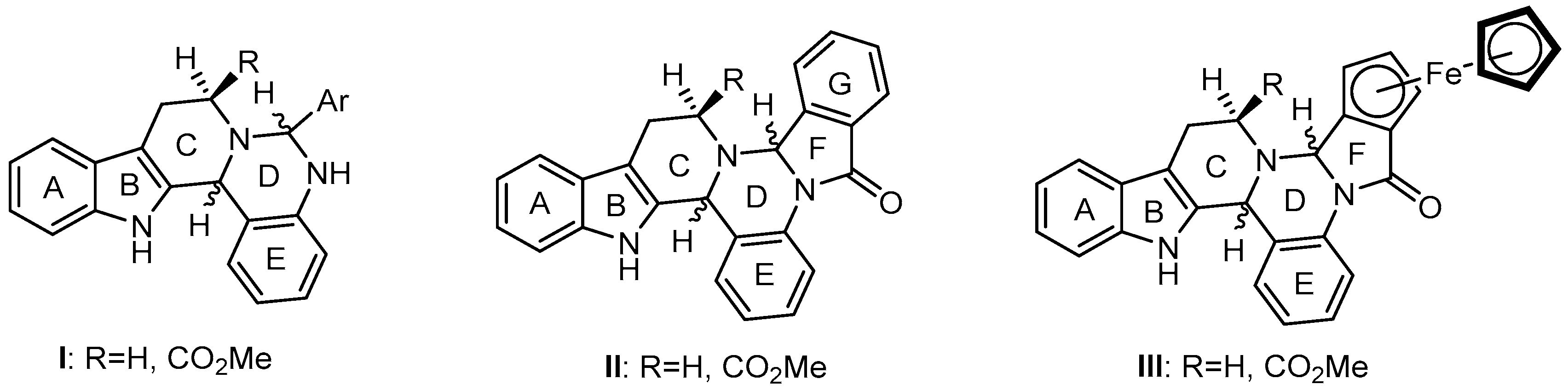
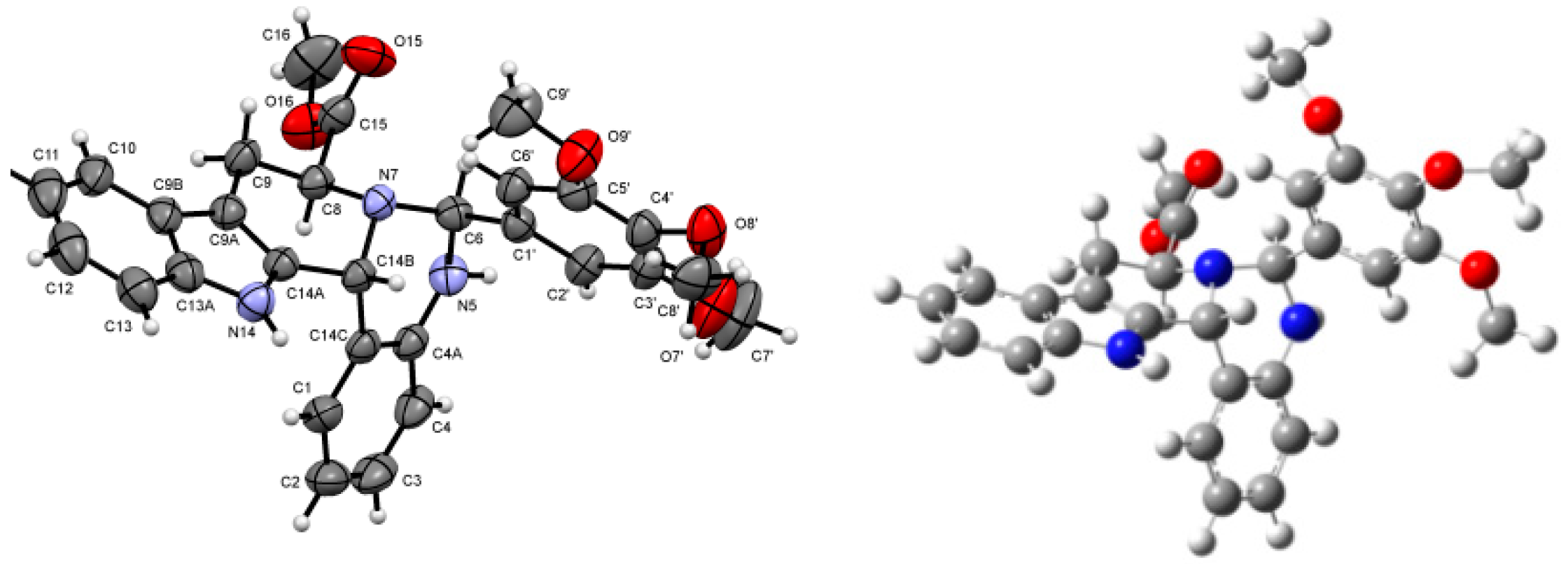
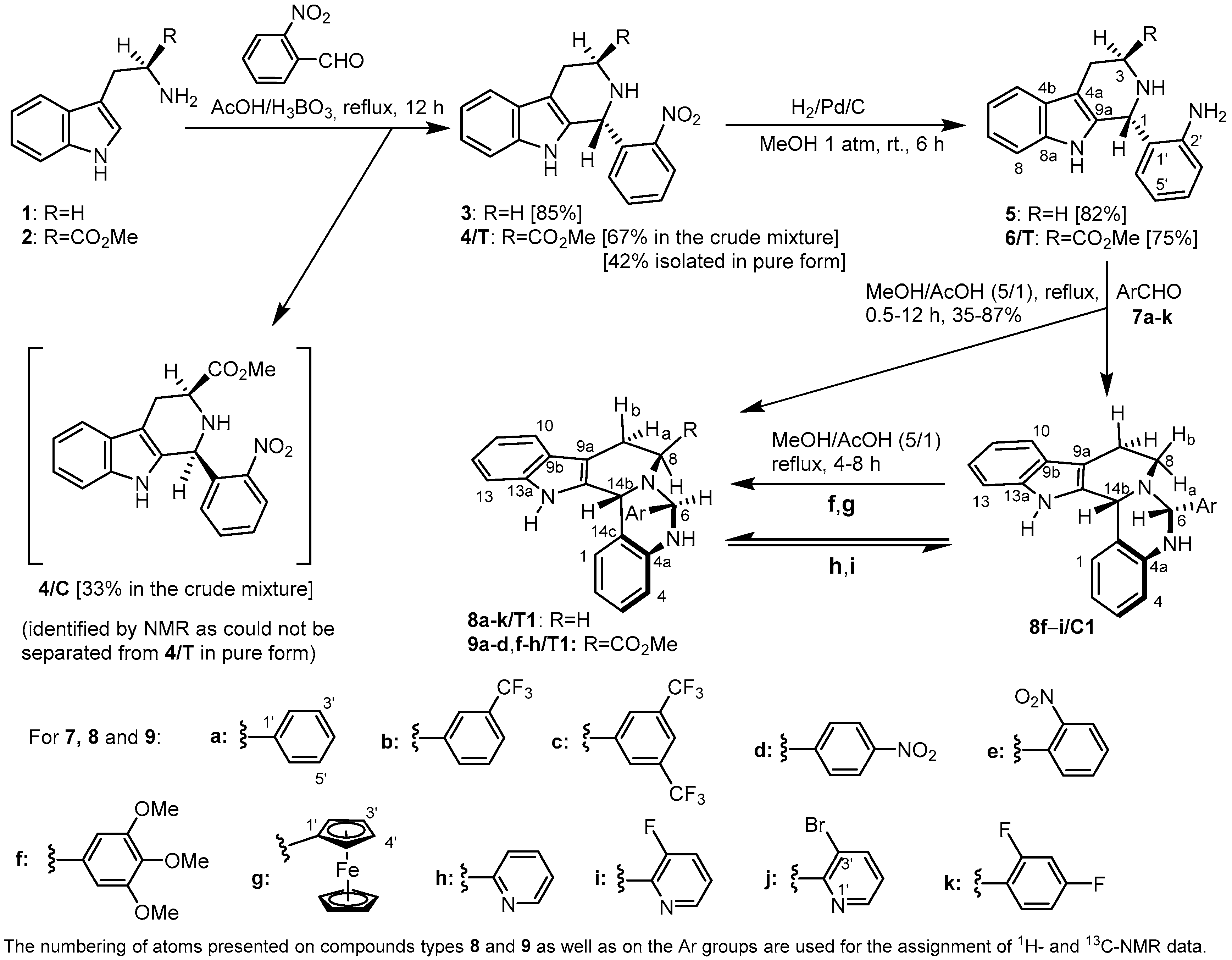
2.1. Synthesis and Structural Analysis of the 6-Aryl-Substituted 5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]-pyrido[1,2-c]quinazolines
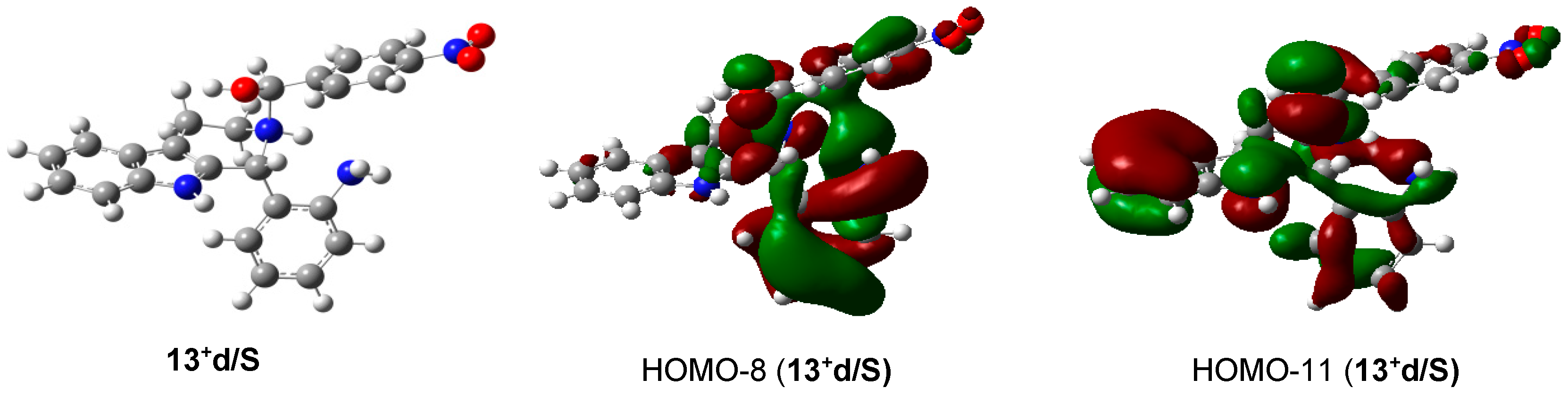
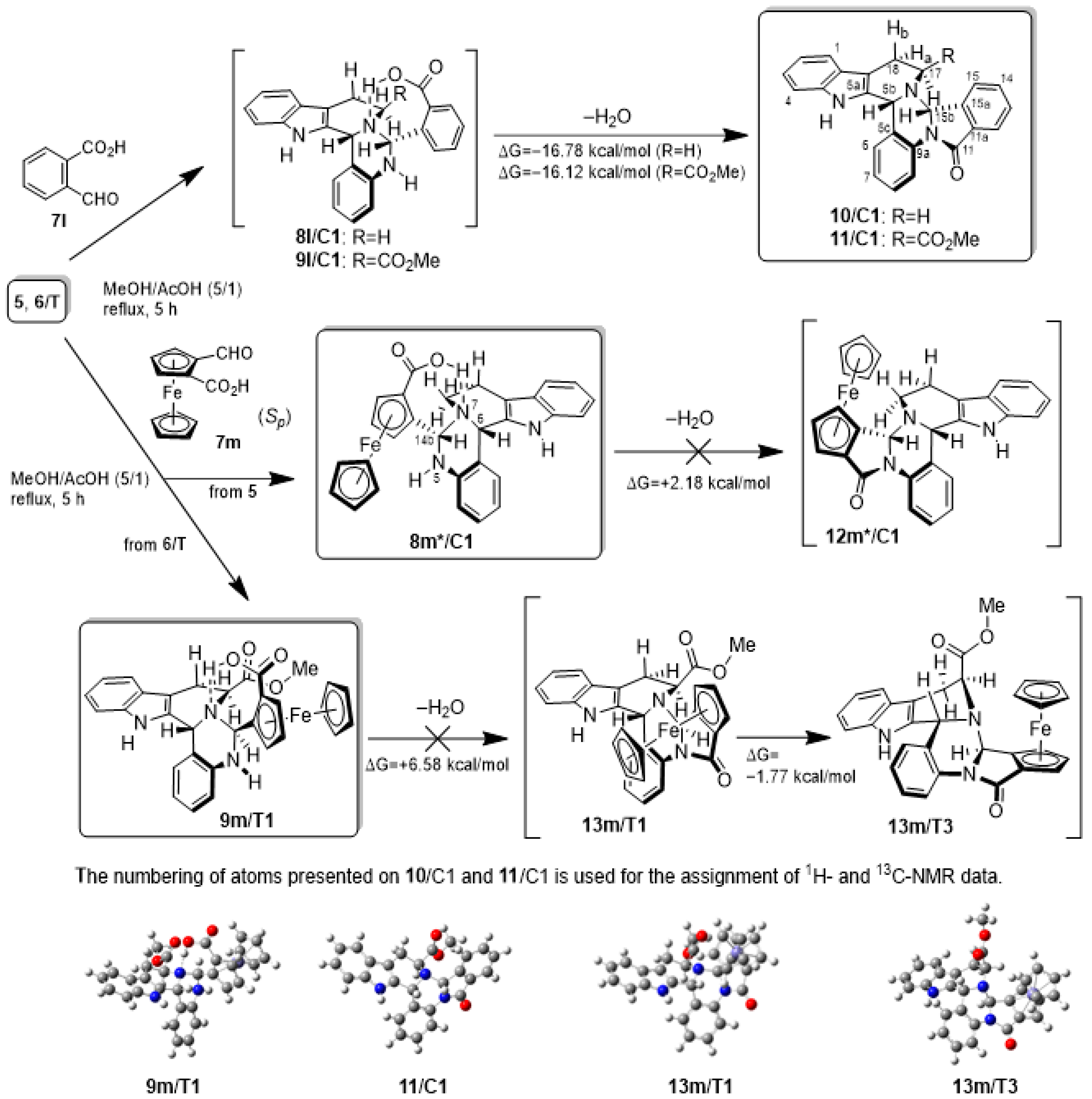
2.2. Preparative-, Spectral and Theoretical Studies on the Mechanism and Products of the Substituent-Controlled Annulations of 5 and 6/T Effected by Formyl-Substituted Carboxylic Acids
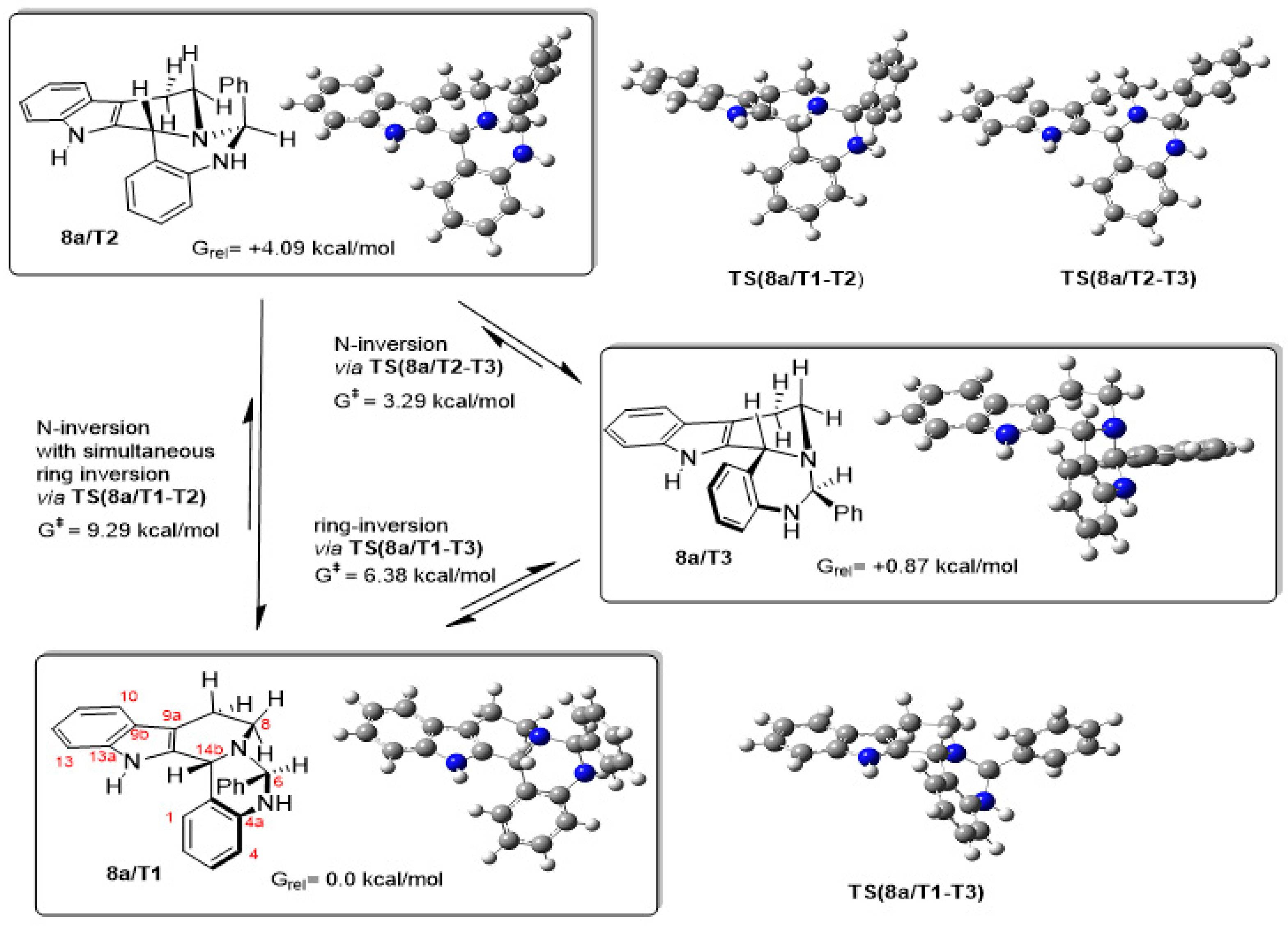
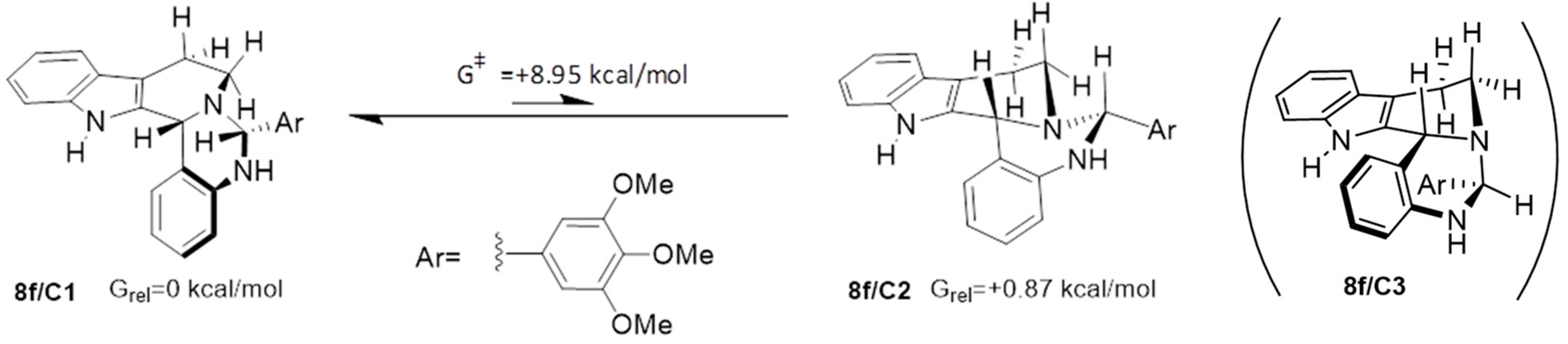
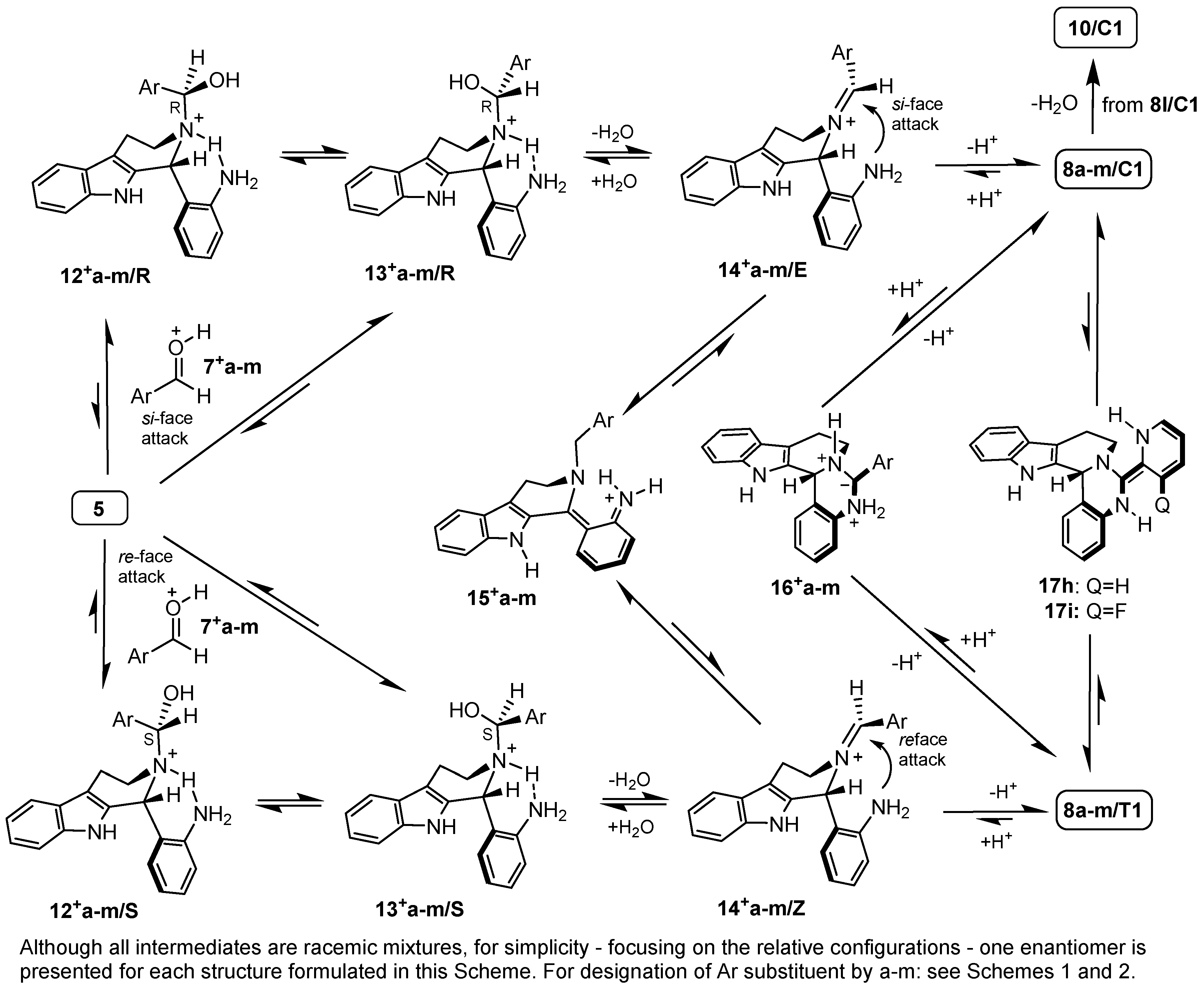
2.3. Proposed Mechanism for the Rationalization of the Substituent- and Time-Dependent Interconversion of cis- and trans-Pentacycles 8/C1 and 8/T1 Based on the Results of DFT Modelling Studies
2.4. In vitro Cytotoxic Activity of the Novel Polycyclic β-Carbolines 8/C1, 8/T1, 9/T1, 10/C1 and 11/C1
3. Materials and Methods
3.1. (±)-1-(2-Nitrophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indole (3)
3.2. Methyl (1R,3S)-1-(2-Nitrophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indole-3-Carboxylate (4/T)
3.3. Hydrogenation of 1-(2-Nitrophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indoles 3 and 4/T
3.3.1. (±)-1-(2-Aminophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indole (5)
3.3.2. Methyl (1R,3S)-1-(2-Aminophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indole-3-Carboxylate (6/T)
3.4. Cyclization Reactions of 1-(2-Aminophenyl)-2,3,4,9-Tetrahydro-1H-Pyrido[3,4-b]indoles 5 and 6/T
3.4.1. (6R*,14bS*)-6-Phenyl-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8a/T1)
3.4.2. (6R*,14bS*)-6-(3-Trifluoromethylphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8b/T1)
3.4.3. (6R*,14bS*)-6-[3,5-Bis(trifluoromethyl)phenyl]-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido-[1,2-c]quinazoline (8c/T1)
3.4.4. (6R*,14bS*)-6-(4-Nitromethylphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-quinazoline (8d/T1)
3.4.5. (6R*,14bS*)-6-(2-Nitrophenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-quinazoline (8e/T1)
3.4.6. (6R*,14bR*)-6-(3,4,5-Trimethoxyphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8f/C1)
3.4.7. (6R*,14bS*)-6-(3,4,5-Trimethoxyphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8f/T1)
3.4.8. (6R*,14bR*)-6-Ferrocenyl-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8g/C1)
3.4.9. (6R*,14bS*)-6-Ferrocenyl-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8g/T1)
3.4.10. (6R*,14bR*)-6-(Pyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8h/C1)
3.4.11. (6R*,14bS*)-6-(Pyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-Quinazoline (8h/T1)
3.4.12. (6R*,14bR*)-6-(3-Fluoropyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8i/C1)
3.4.13. (6R*,14bS*)-6-(3-Fluoropyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-quinazoline (8i/T1)
3.4.14. (6R*,14bS*)-6-(3-Bromopyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8j/T1)
3.4.15. (6R*,14bS*)-6-[3,5-Difluorophenyl]-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazoline (8k/T1)
3.4.16. 2-(6S,14bS,Sp)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazolin-6-yl)-ferrocene-1-carboxylic acid (8m*/C1)
3.4.17. Methyl-(6S,8S,14bR)-6-Phenyl-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-Quinazoline-8-Carboxylate (9a/T1)
3.4.18. Methyl-(6S,8S,14bR)-6-(3-Trifluoromethylphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]-Pyrido[1,2-c]quinazoline-8-Carboxylate (9b/T1)
3.4.19. Methyl-(6S,8S,14bR)-6-[3,5-Bis(trifluoromethyl)phenyl]-5,6,8,9,14,14b-Hexahydroindolo-[2′,3′:3,4]pyrido[1,2-c] quinazoline-8-Carboxylate (9c/T1)
3.4.20. Methyl-(6S,8S,14bR)-6-(4-Nitrophenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-Quinazoline-8-Carboxylate (9d/T1)
3.4.21. Methyl-(6S,8S,14bR)-6-(3,4,5-Trimethoxyphenyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]-pyrido[1,2-c]quinazoline-8-Carboxylate (9f/T1)
3.4.22. Methyl-(6S,8S,14bR)-6-Ferrocenyl-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-Quinazoline-8-Carboxylate (9g/T1)
3.4.23. Methyl-(6S,8S,14bR)-6-(pyridine-2-yl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-Quinazoline-8-Carboxylate (9h/T1)
3.4.24. 2-((6S,8S,14bR,Sp)-8-(methoxycarbonyl)-5,6,8,9,14,14b-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]quinazolin-6-yl)-Ferrocene-1-Carboxylic acid (9m/T1).
3.4.25. (5bR*,15bS*)-5,5b,17,18-Tetrahydroindolo[2′,3′:3,4]pyrido[1,2-c]isoindolo[2,1-a]quinazolin-11(15bH)-One (10/C1)
3.4.26. Methyl (5bR,15bS,17S)-11-Oxo-5,5b,11,15b,17,18-Hexahydroindolo[2′,3′:3,4]pyrido[1,2-c]-isoindolo[2,1-a]quinazoline-17-Carboxylate (11/C1)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Gupta, L.; Srivastava, K.; Singh, S.; Puri, S.K.; Chauhan, P.M. Synthesis of 2-[3-(7-Chloro-quinolin-4-ylamino)-alkyl]-1-(substituted phenyl)-2,3,4,9-tetrahydro-1H-beta-carbolines as a new class of antimalarial agents. Bioorg. Med. Chem. Lett. 2008, 18, 3306–3309. [Google Scholar] [CrossRef]
- Tonin, L.T.; Barbosa, V.A.; Bocca, C.C.; Ramos, E.R.; Nakamura, C.V.; da Costa, W.F.; Basso, E.A.; Nakamura, T.U.; Sarragiotto, M.H. Comparative study of the trypanocidal activity of the methyl 1-nitrophenyl-1,2,3,4-9H-tetrahydro-beta-carboline-3-carboxylate derivatives and benznidazole using theoretical calculations and cyclic voltammetry. Eur. J. Med. Chem. 2009, 44, 1745–1750. [Google Scholar] [CrossRef]
- Riederer, P.; Foley, P.; Bringmann, G.; Feineis, D.; Bruckner, R.; Gerlach, M. Biochemical and pharmacological characterization of 1-trichloromethyl-1,2,3,4-tetrahydro-beta-carboline: A biologically relevant neurotoxin? Eur. J. Pharmacol. 2002, 442, 1–16. [Google Scholar] [CrossRef]
- Rabindran, S.K.; He, H.Y.; Singh, M.; Brown, E.; Collins, K.I.; Annable, T.; Greenberger, L.M. Reversal of a novel multidrug resistance mechanismin human colon carcinoma cells by fumitremorgin C. Cancer Res. 1998, 58, 5850–5858. [Google Scholar]
- He, H.Y.; Rabindran, S.G.; Greenberger, L.M.; Carter, G.T. Fumitremorgin C analogs that reverse mitoxantrone resistance in human colon carcinoma cells. Med. Chem. Res. 1999, 9, 424–437. [Google Scholar]
- Sunder-Plassmann, N.; Sarli, V.; Gartner, M.; Utz, M.; Seiler, J.; Huemmer, S.; Mayer, T.U.; Surrey, T.; Giannis, A. Synthesis and biological evaluation of new tetrahydro-beta-carbolines as inhibitors of the mitotic kinesin Eg5. Bioorg. Med. Chem. 2005, 13, 6094–6111. [Google Scholar] [CrossRef]
- Barsanti, P.A.; Wang, W.; Ni, Z.J.; Duhl, D.; Brammeier, N.; Martin, E.; Bussiere, D.; Walter, A.O. The discovery of tetrahydro-beta-carbolines as inhibitors of the kinesin Eg5. Bioorg. Med. Chem. Lett. 2010, 20, 157–160. [Google Scholar] [CrossRef]
- Song, Y.; Kesuma, D.; Wang, J.; Deng, Y.; Duan, J.; Wang, J.H.; Qi, R.Z. Specific inhibition of cyclin-dependent kinases and cell proliferation by harmine. Biochem. Biophys. Res. Commun. 2004, 317, 128–132. [Google Scholar] [CrossRef]
- Song, Y.; Wang, J.; Teng, S.F.; Kesuma, D.; Deng, Y.; Duan, J.; Wang, J.H.; Qi, R.Z.; Sim, M.M. β-Carbolines as specific inhibitors of cyclin-dependent kinases. Bioorg. Med. Chem. Lett. 2002, 12, 1129–1132. [Google Scholar] [CrossRef]
- Cao, R.; Peng, W.; Chen, H.; Ma, Y.; Liu, X.; Hou, X.; Guan, H.; Xu, A. DNA binding properties of 9-substituted harmine derivatives. Biochem. Biophys. Res. Commun. 2005, 338, 1557–1563. [Google Scholar] [CrossRef]
- Samundeeswari, S.; Chougala, B.; Holiyachi, M.; Shastri, L.; Kulkarni, M.; Dodamani, S.; Jalalpur, S.; Joshi, S.; Dixit, S.; Sunagar, V.; et al. Design and synthesis of novel phenyl -1, 4-beta-carboline-hybrid molecules as potential anticancer agents. Eur. J. Med. Chem. 2017, 128, 123–139. [Google Scholar] [CrossRef]
- Tamura, H.; Miwa, M. DNA Cleaving Activity and Cytotoxic Activity of Ferricenium Cations. Chem. Lett. 1997, 26, 1177–1178. [Google Scholar] [CrossRef]
- Houlton, A.; Roberts, R.M.G.; Silver, J. Studies on the anti-tumour activity of some iron sandwich compounds. J. Organomet. Chem. 1991, 418, 107–112. [Google Scholar] [CrossRef]
- Osella, D.; Ferrali, M.; Zanello, P.; Laschi, F.; Fontani, M.; Nervi, C.; Cavigiolio, G. On the mechanism of the antitumor activity of ferrocenium derivatives. Inorg. Chim. Acta 2000, 306, 42–48. [Google Scholar] [CrossRef]
- Simon, H.U.; Haj-Yehia, A.; Levi-Schaffer, F. Role of reactive oxygen species (ROS) in apoptosis induction. Apoptosis 2000, 5, 415–418. [Google Scholar] [CrossRef]
- Voltan, R.; Secchiero, P.; Casciano, F.; Milani, D.; Zauli, G.; Tisato, V. Redox signalling and oxidative stress: Cross talk with TNF-related apoptosis inducing ligand activity. Inter. J. Biochem. Cell Bio. 2016, 81, 364–374. [Google Scholar] [CrossRef]
- Ornelas, C. Application of ferrocene and its derivatives in cancer research. New J. Chem. 2011, 35, 1973–1985. [Google Scholar] [CrossRef]
- Braga, S.S.; Silva, A.M.S. A New Age for Iron: Antitumoral Ferrocenes. Organometallics 2013, 32, 5626–5639. [Google Scholar] [CrossRef]
- Jaouen, G.; Vessières, A.; Top, S. Ferrocifen type anti cancer drugs. Chem. Soc. Rev. 2015, 44, 8802–8817. [Google Scholar] [CrossRef]
- Kowalski, K. Recent developments in the chemistry of ferrocenyl secondary natural product conjugates. Coord. Chem. Rev. 2018, 366, 91–108. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 0066. [Google Scholar] [CrossRef]
- Károlyi, B.I.; Bősze, S.; Orbán, E.; Sohár, P.; Drahos, L.; Gál, E.; Csámpai, A. Acylated mono-, bis- and tris- Cinchona-Based Amines Containing Ferrocene or Organic Residues: Synthesis, Structure and in vitro Antitumor Activity on Selected Human Cancer Cell Lines. Molecules 2012, 17, 2316–2329. [Google Scholar] [CrossRef] [PubMed]
- Csókás, D.; Zupkó, I.; Károlyi, B.I.; Drahos, L.; Holczbauer, T.; Palló, A.; Czugler, M.; Csámpai, A. Synthesis, spectroscopy, X-ray analysis and in vitro antiproliferative effect of ferrocenylmethylene-hydrazinylpyridazin-3(2H)-ones and related ferroceno[d]pyridazin-1(2H)-ones. J. Organomet. Chem. 2014, 743, 130–138. [Google Scholar] [CrossRef]
- Csókás, D.; Károlyi, B.I.; Bősze, S.; Szabó, I.; Báti, G.; Drahos, L.; Csámpai, A. 2,3-Dihydroimidazo[1,2-b]ferroceno[d]pyridazines and a 3,4-dihydro-2H-pyrimido[1,2-b]ferro- ceno[d]pyridazine: Synthesis, structure and in vitro antiproliferation activity on selected human cancer cell lines. J. Organomet. Chem. 2014, 750, 41–48. [Google Scholar] [CrossRef]
- Kocsis, L.; Szabó, I.; Bősze, S.; Jernei, T.; Hudecz, F.; Csámpai, A. Synthesis, structure and in vitro cytostatic activity of ferrocene—Cinchona hybrids. Bioorg. Med. Chem. Lett. 2016, 26, 946–949. [Google Scholar] [CrossRef]
- Podolski-Renić, A.; Bősze, S.; Dinić, J.; Kocsis, L.; Hudecz, F.; Csámpai, A.; Pešić, M. Ferrocene–cinchona hybrids with triazolyl-chalcone linkers act as pro-oxidants and sensitize human cancer cell lines to paclitaxel. Metallomics 2017, 9, 1132–1141. [Google Scholar] [CrossRef]
- Jernei, T.; Bősze, S.; Szabó, R.; Hudecz, F.; Majrik, K.; Csámpai, A. N-ferrocenylpyridazinones and new organic analogues: Synthesis, cyclic voltammetry, DFT analysis and in vitro antiproliferative activity associated with ROS-generation. Tetrahedron 2017, 73, 6181–6192. [Google Scholar] [CrossRef]
- Bárány, P.; Szabó-Oláh, R.; Kovács, I.; Czuczi, T.; Szabó, C.L.; Takács, A.; Lajkó, E.; Láng, O.; Kőhidai, L.; Schlosser, G.; et al. Ferrocene-Containing Impiridone (ONC201) Hybrids: Synthesis, DFT Modelling, In vitro Evaluation, and Structure–Activity Relationships. Molecules 2018, 23, 2248. [Google Scholar] [CrossRef]
- Jernei, T.; Duró, C.; Dembo, A.; Lajkó, E.; Takács, A.; Kőhidai, L.; Schlosser, G.; Csámpai, A. Synthesis, Structure and In vitro Cytotoxic Activity of Novel Cinchona—Chalcone Hybrids with 1,4-Disubstituted- and 1,5-Disubstituted 1,2,3-Triazole Linkers. Molecules 2019, 24, 4077. [Google Scholar] [CrossRef]
- Fodor, K.J.; Kocsis, V.L.; Kiss, K.; Károlyi, B.I.; Szabolcs, Á.; Silaghi-Dumitrescu, L.; Csámpai, A. Comparative evaluation of a Pictet–Spengler protocol in microwave-assisted conversions of tryptamine with aryl- and carboxyaryl aldehydes: Role of ring strain in cyclocondensation of the primarily formed carboxyaryl-substituted β-carbolines. Monat. Chem. 2013, 144, 1381–1387. [Google Scholar] [CrossRef]
- Perdew, J.P.; Wang, Y. Accurate and simple analytic representation of the electron-gas correlation energy. Phys. Rev. B. 1992, 45, 13244–13249. [Google Scholar] [CrossRef] [PubMed]
- Godbout, N.; Salahub, D.R.; Andzelm, J.; Wimmer, E. Optimization of Gaussian-type basis sets for local spin density functional calculations. Part I. Boron through neon, optimization technique and validation. Can. J. Chem. 1992, 70, 560–571. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. (Theochem) 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Paier, J.; Marsman, M.; Kresse, G. Why does the B3LYP hybrid functional fail for metals? J. Chem. Phys. 2007, 127, 024103. [Google Scholar] [CrossRef]
- Wolinski, K.; Hilton, J.F.; Pulay, P. Efficient implementation of the gauge-independent atomic orbital method for NMR chemical shift calculations. J. Am. Chem. Soc. 1990, 112, 8251–8260. [Google Scholar] [CrossRef]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys. 1993, 98, 1372–1377. [Google Scholar] [CrossRef]
- Krishan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. (Eds.) Gaussian 09, Revision A.02; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
Sample Availability: Samples of the compounds reported in this contribution are available from the authors. |









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | Yield (Reaction Time) | Product | Yield (Reaction Time) |
|---|---|---|---|
| 8a/T1 | 37% (0.5 h)/77% (12 h) | 8i/C1/T1 | 80% (0.5 h) e/35% (12 h) e |
| 8b/T1 | 75% (1 h)/72% (12 h) | 8j/T1 | 80% (1 h)/87% (12 h) |
| 8c/T1 | 79% (1 h)/76% (12 h) | 8k/T1 | 69% (1 h)/71% (12 h) |
| 8d/T1 | 74% (1 h)/78% (12 h) | 9a/T1 | 64% (2 h) |
| 8e/T1 | 80% (1 h)/75% (12 h) | 9b/T1 | 70% (2 h) |
| 8f/C1 | 63% (0.5 h) | 9c/T1 | 62% (2 h) |
| 8f/C1/T1 | 80% (4 h) a/75% (8 h) b | 9d/T1 | 73% (2 h) |
| 8g/C1 | 39% (0.5 h) | 9f/T1 | 71% (2 h) |
| 8g/C1/T1 | 52% (4 h) c | 9g/T1 | 52% (2 h) |
| 8h/C1/T1 | 72% (0.5 h) d/25% (12 h) d | 9h/T1 | 56% (2 h) |
| ΔG(T1-C1) [kcal/mol] | ΔG(T1-C1) [kcal/mol] | ΔG(T1-C1) [kcal/mol] | |||
|---|---|---|---|---|---|
| 8a | −1.13 | 8e | −3.06 | 8i | +0.19 |
| 8b | −1.65 | 8f | −1.52 | 8j | −0.75 |
| 8c | −1.58 | 8g | −0.47 | 8k | −1.73 |
| 8d | −1.48 | 8h | +0.34 |
| Relative Stability of Intermediate Pairs Expressed in ΔG [kcal/mol] | ||||
|---|---|---|---|---|
| Diastereoselectivity of primary coupling to 12+ | 12+a/R−12+a/S −0.68 | 12+d/R−12+d/S +0.17 | 12+f/R−12+f/S −0.70 | 12+g/R−12+g/S −0.97 |
| Diastereoselectivity of primary coupling to 13+ | 13+a/R−13+a/S −0.21 | 13+d/R−13+d/S +2.83 | 13+f/R−13+f/S −3.41 | 13+g/R−13+g/S −0.92 |
| Readiness of rotation 12+/R→13+/R a | 13+a/R−12+a/R −1.48 | 13+d/R−12+d/R +0.79 | 13+f/R−12+f/R −2.21 | 13+g/R−12+g/R −1.77 |
| Readiness of rotation 12+/S→13+/S a | 13+a/S−12+a/S −1.95 | 13+d/S−12+d/S −1.87 | 13+f/S−12+f/S +0.50 | 13+g/S−12+g/S −1.82 |
| Readiness of isomerisation 14+/E→15+ | 15+a−14+a/E +2.42 | 15+d−14+d/E +1.04 | 15+f−14+f/E +10.24 | 15+g−14+g/E +13.19 |
| Readiness of isomerisation 15+→14+/Z | 14+a/Z−15+a −1.61 | 14+d/Z−15+d +0.86 | 14+f/Z−15+f −8.06 | 14+g/Z−15+g −12.04 |
| Preference of isomerization pathway via 15+ over that via 16+ | 16+a−14+a +46.10 b/+48.02 c | 16+d−14+d +35.54 b/+36.76 c | 16+f−14+f +50.63 b/+52.24 c | 16+g−14+g +51.93 b/+53.35 c |
| IC50 [M] | ||||
|---|---|---|---|---|
| PANC-1 | COLO-205 | A2058 | EBC-1 | |
| 8a/T1 | 7.0 × 10−6 ± 1.9 × 10−6 | 5.6 × 10−6 ± 5.6 × 10−7 | 1.4 × 10−5 ± 3.2 × 10−6 | 6.4 × 10−6 ± 1.1 × 10−6 |
| 9a/T1 | > 10−4 | > 10−4 | > 10−4 | > 10−4 |
| 8b/T1 | 2.8 × 10−6 ± 1.0 × 10−6 | 1.5 × 10−6 ± 5.9 × 10−7 | 2.5 × 10−6 ± 4.0 × 10−7 | 4.7 × 10−6 ± 4.1 × 10−7 |
| 9b/T1 | > 10−4 | > 10−4 | > 10−4 | > 10−4 |
| 8c/T1 | 2.3 × 10−5 ± 1.1 × 10−6 | 5.0 × 10−5 ± 1.9 × 10−6 | 2.5 × 10−5 ± 9.5 × 10−7 | 2.5 × 10−5 ± 5.8 × 10−7 |
| 9c/T1 | 2.5 × 10−5 ± 1.2 × 10−6 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8d/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 9d/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8e/T1 | 2.6 × 10−5 ± 1.2 × 10−6 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8f/C1 | 5.2 × 10−6 ± 2.3 × 10−6 | 2.5 × 10−6 ± 9.4 × 10−8 | 2.1 × 10−6 ± 1.9 × 10−7 | 4.1 × 10−6 ± 7.8 × 10−7 |
| 8f/C1/T1 b | > 5.0 × 10−5 | > 5.0 × 10−5 | 3.6 × 10−5 ± 1.2510−6 | 1.6 × 10−5 ± 3.0 × 10−6 |
| 9f/T1 | 1.3 × 10−5 ± 1.2 × 10−5 | 2.0 × 10−5 ± 7.6 × 10−7 | 2.6 × 10−5 ± 1.6 × 10−6 | 1.1 × 10−5 ± 5.2 × 10−6 |
| 8g/C1 | > 10−4 | > 10−4 | > 10−4 | > 10−4 |
| 8g/C1/T1 c | > 5.0 × 10−5 | 3.9 × 10−5 ± 1.1 × 10−6 | 5.0 × 10−5 ± 5.4 × 10−6 | 2.3 × 10−5 ± 1.1 × 10−6 |
| 9g/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8h/C1/T1 d | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | 4.7 × 10−5 ± 2.1 × 10−6 |
| 9h/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8i/C1/T1 e | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8j/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | 1.4 × 10−5 ± 3.2 × 10−6 | 5.5 × 10−6 ± 1.9 × 10−7 |
| 8k/T1 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 | > 5.0 × 10−5 |
| 8m*/C1 | > 5.0 × 10−5 | > 5.0 × 10−5 | 3.8 × 10−5 ± 8.8 × 10−7 | 3.2 × 10−5 ± 2.5 × 10−5 |
| 10/C1 | > 10−4 | > 10−4 | > 10−4 | > 10−5 |
| 11/C1 | 2.9 × 10−5 ± 2.4 × 10−6 | > 5.0 × 10−5 | 5.6 × 10−6 ± 1.3 × 10−7 | 2.1 × 10−5 ± 1.5 × 10−6 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fodor, K.J.; Hutai, D.; Jernei, T.; Takács, A.; Szász, Z.; Sulyok-Eiler, M.; Harmat, V.; Oláh Szabó, R.; Schlosser, G.; Hudecz, F.; et al. Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects. Molecules 2020, 25, 1599. https://doi.org/10.3390/molecules25071599
Fodor KJ, Hutai D, Jernei T, Takács A, Szász Z, Sulyok-Eiler M, Harmat V, Oláh Szabó R, Schlosser G, Hudecz F, et al. Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects. Molecules. 2020; 25(7):1599. https://doi.org/10.3390/molecules25071599
Chicago/Turabian StyleFodor, Kinga Judit, Dániel Hutai, Tamás Jernei, Angéla Takács, Zsófia Szász, Máté Sulyok-Eiler, Veronika Harmat, Rita Oláh Szabó, Gitta Schlosser, Ferenc Hudecz, and et al. 2020. "Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects" Molecules 25, no. 7: 1599. https://doi.org/10.3390/molecules25071599
APA StyleFodor, K. J., Hutai, D., Jernei, T., Takács, A., Szász, Z., Sulyok-Eiler, M., Harmat, V., Oláh Szabó, R., Schlosser, G., Hudecz, F., Kőhidai, L., & Csámpai, A. (2020). Novel Polycondensed Partly Saturated β-Carbolines Including Ferrocene Derivatives: Synthesis, DFT-Supported Structural Analysis, Mechanism of Some Diastereoselective Transformations and a Preliminary Study of their In Vitro Antiproliferative Effects. Molecules, 25(7), 1599. https://doi.org/10.3390/molecules25071599