The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties

 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
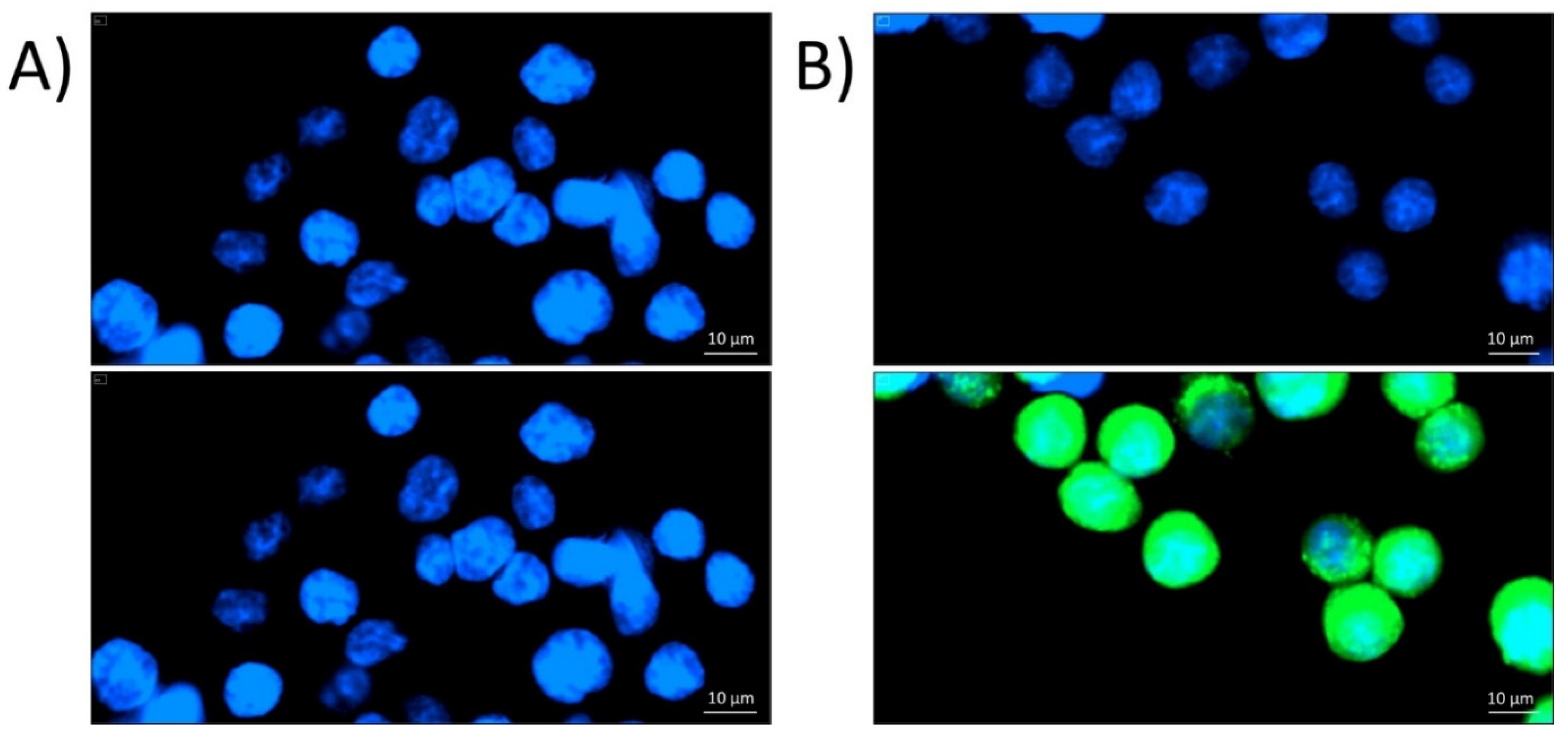
2.1. Localization of P8-D6 in Cellular Compartments
2.2. Stabilization of Topo–DNA Complexes
2.3. Genotoxicity
3. Discussion
4. Materials and Methods
4.1. Chemicals, Antibodies, and Enzymes
4.2. Cell Culture and Treatment
4.3. Fluorescence Imaging
4.4. Confocal Microscopy
4.5. In Vivo Complex of Enzyme (ICE) Assay
4.6. Single Cell Gel Electrophoreses (“Comet Assay”)
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dvorak, Z.; Kubáň, V.; Klejdus, B.; Hlaváč, J.; Vičar, J.; Ulrichová, J.; Simánek, V. Quaternary benzo[c]phenanthridines sanguinarine and chelerythrine: A review of investigations from chemical and biological studies. Heterocycles 2006, 68, 2403–2422. [Google Scholar] [CrossRef]
- El-Readi, M.Z.; Eid, S.; Ashour, M.L.; Tahrani, A.; Wink, M. Modulation of multidrug resistance in cancer cells by chelidonine and Chelidonium majus alkaloids. Phytomedicine 2013, 20, 282–294. [Google Scholar] [CrossRef]
- Larsen, A.K.; Grondard, L.; Couprie, J.; Desoize, B.; Comoe, L.; Jardillier, J.C.; Riou, J.F. The antileukemic alkaloid fagaronine is an inhibitor of DNA topoisomerases I and II. Biochem. Pharmacol. 1993, 46, 1403–1412. [Google Scholar] [CrossRef]
- Meier, C.; Kotthaus, J.; Stenzel, L.; Girreser, U.; Heber, D.; Clement, B. Synthesis and physicochemical characterization of novel 6-aminopyrido[3,4-c][1,9]phenanthrolines as aza-analogs of benzo[c]phenanthridines. Tetrahedron 2012, 68, 9105–9112. [Google Scholar] [CrossRef]
- Meier, C.; Steinhauer, T.N.; Koczian, F.; Plitzko, B.; Jarolim, K.; Girreser, U.; Braig, S.; Marko, D.; Vollmar, A.M.; Clement, B. A Dual Topoisomerase Inhibitor of Intense Pro-Apoptotic and Antileukemic Nature for Cancer Treatment. ChemMedChem 2017, 12, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Sun, Y.; Huang, S.N.; Nitiss, J.L. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat. Rev. Mol. Cell Biol. 2016, 17, 703–721. [Google Scholar] [CrossRef]
- Nitiss, J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer 2009, 9, 327–337. [Google Scholar] [CrossRef] [PubMed]
- McClendon, A.K.; Osheroff, N. DNA topoisomerase II, genotoxicity, and cancer. Mutat. Res. 2007, 623, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Larsen, A.K.; Escargueil, A.E.; Skladanowski, A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol. Ther. 2003, 99, 167–181. [Google Scholar] [CrossRef]
- Nitiss, J.L. Targeting DNA topoisomerase II in cancer chemotherapy. Nat. Rev. Cancer 2009, 9, 338–350. [Google Scholar] [CrossRef]
- Pommier, Y. Topoisomerase I inhibitors: Camptothecins and beyond. Nat. Rev. Cancer 2006, 6, 789–802. [Google Scholar] [CrossRef] [PubMed]
- Salerno, S.; Da Settimo, F.; Taliani, S.; Simorini, F.; La Motta, C.; Fornaciari, G.; Marini, A.M. Recent advances in the development of dual topoisomerase I and II inhibitors as anticancer drugs. Curr. Med. Chem. 2010, 17, 4270–4290. [Google Scholar] [CrossRef] [PubMed]
- Esselen, M.; Boettler, U.; Teller, N.; Bachler, S.; Hutter, M.; Rufer, C.E.; Skrbek, S.; Marko, D. Anthocyanin-rich blackberry extract suppresses the DNA-damaging properties of topoisomerase I and II poisons in colon carcinoma cells. J. Agric. Food Chem. 2011, 59, 6966–6973. [Google Scholar] [CrossRef] [PubMed]
- Esselen, M.; Barth, S.W.; Winkler, S.; Baechler, S.; Briviba, K.; Watzl, B.; Skrbek, S.; Marko, D. Anthocyanins suppress the cleavable complex formation by irinotecan and diminish its DNA-strand-breaking activity in the colon of Wistar rats. Carcinogenesis 2013, 34, 835–840. [Google Scholar] [CrossRef]
- Aichinger, G.; Puntscher, H.; Beisl, J.; Kutt, M.L.; Warth, B.; Marko, D. Delphinidin protects colon carcinoma cells against the genotoxic effects of the mycotoxin altertoxin II. Toxicol. Lett. 2018, 284, 136–142. [Google Scholar] [CrossRef]
- Subramanian, D.; Furbee, C.S.; Muller, M.T. ICE bioassay. Isolating in vivo complexes of enzyme to DNA. Methods Mol. Biol. 2001, 95, 137–147. [Google Scholar]
- Nitiss, J.L.; Soans, E.; Rogojina, A.; Seth, A.; Mishina, M. Topoisomerase Assays. Curr. Protoc. Pharmacol. 2012, 57, 3.3.1–3.3.27. [Google Scholar] [CrossRef]
- Patel, A.G.; Flatten, K.S.; Peterson, K.L.; Beito, T.G.; Schneider, P.A.; Perkins, A.L.; Harki, D.A.; Kaufmann, S.H. Immunodetection of human topoisomerase I-DNA covalent complexes. Nucleic Acids Res. 2016, 44, 2816–2826. [Google Scholar] [CrossRef]
- Gromova, I.I.; Kjeldsen, E.; Svejstrup, J.Q.; Alsner, J.; Christiansen, K.; Westergaard, O. Characterization of an altered DNA catalysis of a camptothecin-resistant eukaryotic topoisomerase I. Nucleic Acids Res. 1993, 21, 593–600. [Google Scholar] [CrossRef]
- Subramanian, D.; Kraut, E.; Staubus, A.; Young, D.C.; Muller, M.T. Analysis of topoisomerase I/DNA complexes in patients administered topotecan. Cancer Res. 1995, 55, 2097–2103. [Google Scholar]
- Baldwin, E.L.; Osheroff, N. Etoposide, topoisomerase II and cancer. Curr. Med. Chem. Anticancer Agents 2005, 5, 363–372. [Google Scholar] [CrossRef] [PubMed]
- Pommier, Y.; Leo, E.; Zhang, H.; Marchand, C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010, 17, 421–433. [Google Scholar] [CrossRef] [PubMed]
- Swift, L.H.; Golsteyn, R.M. Genotoxic anti-cancer agents and their relationship to DNA damage, mitosis, and checkpoint adaptation in proliferating cancer cells. Int. J. Mol. 2014, 15, 3403–3431. [Google Scholar] [CrossRef] [PubMed]
- van Gijn, R.; Lendfers, R.; Schellens, J.; Bult, A.; Beijnen, J. Dual topoisomerase I/II inhibitors. J. Oncol. Pharm. Pract 2000, 6, 92–108. [Google Scholar] [CrossRef]
- Poddevin, B.; Riou, J.F.; Lavelle, F.; Pommier, Y. Dual topoisomerase I and II inhibition by intoplicine (RP-60475), a new antitumor agent in early clinical trials. Mol. Pharmacol. 1993, 44, 767–774. [Google Scholar] [PubMed]
- Kluza, J.; Lansiaux, A.; Wattez, N.; Mahieu, C.; Osheroff, N.; Bailly, C. Apoptotic response of HL-60 human leukemia cells to the antitumor drug TAS-103. Cancer Res. 2000, 60, 4077–4084. [Google Scholar]
- Adjei, A.A.; Charron, M.; Rowinsky, E.K.; Svingen, P.A.; Miller, J.; Reid, J.M.; Sebolt-Leopold, J.; Ames, M.M.; Kaufmann, S.H. Effect of pyrazoloacridine (NSC 366140) on DNA topoisomerases I and II. Clin. Cancer Res. 1998, 4, 683–691. [Google Scholar]
- Padget, K.; Stewart, A.; Charlton, P.; Tilby, M.J.; Austin, C.A. An investigation into the formation of N- [2-(dimethylamino)ethyl]acridine-4-carboxamide (DACA) and 6-[2-(dimethylamino)ethylamino]- 3-hydroxy-7H-indeno[2,1-C]quinolin-7-one dihydrochloride (TAS-103) stabilised DNA topoisomerase I and II cleavable complexes in human leukaemia cells. Biochem. Pharmacol. 2000, 60, 817–821. [Google Scholar]
- Newman, R.A.; Kim, J.; Newman, B.M.; Bruno, R.; Bayssas, M.; Klink-Alaki, M.; Pazdur, R. Phase I trial of intoplicine (RP 60475) administered as a 72 h infusion every 3 weeks in patients with solid tumors. Anti-Cancer Drugs 1999, 10, 889–894. [Google Scholar] [CrossRef]
- Ramaswamy, B.; Mrozek, E.; Kuebler, J.P.; Bekaii-Saab, T.; Kraut, E.H. Phase II trial of pyrazoloacridine (NSC#366140) in patients with metastatic breast cancer. Investig. New Drugs 2011, 29, 347–351. [Google Scholar]
- Clement, B.; Girreser, U.; Steinhauer, T.N.; Meier, C.; Marko, D.; Aichinger, G.; Kaltefleiter, I.; Stenzel, L.; Heber, D.; Weide, M.; et al. 11-Substituted Benzo c phenanthridines: New Structures and Insight into Their Mode of Antiproliferative Action. ChemMedChem 2016, 11, 2155–2170. [Google Scholar] [CrossRef] [PubMed]
- Tice, R.R.; Agurell, E.; Anderson, D.; Burlinson, B.; Hartmann, A.; Kobayashi, H.; Miyamae, Y.; Rojas, E.; Ryu, J.C.; Sasaki, Y.F. Single cell gel/comet assay: Guidelines for in vitro and in vivo genetic toxicology testing. Environ. Mol. Mutagen. 2000, 35, 206–221. [Google Scholar] [CrossRef]
- Aichinger, G.; Beisl, J.; Marko, D. Genistein and delphinidin antagonize the genotoxic effects of the mycotoxin alternariol in human colon carcinoma cells. Mol. Nutr. Food Res. 2017, 61. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compound P8-D6 are available from the authors. |






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aichinger, G.; Lichtenberger, F.-B.; Steinhauer, T.N.; Flörkemeier, I.; Del Favero, G.; Clement, B.; Marko, D. The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties. Molecules 2020, 25, 1524. https://doi.org/10.3390/molecules25071524
Aichinger G, Lichtenberger F-B, Steinhauer TN, Flörkemeier I, Del Favero G, Clement B, Marko D. The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties. Molecules. 2020; 25(7):1524. https://doi.org/10.3390/molecules25071524
Chicago/Turabian StyleAichinger, Georg, Falk-Bach Lichtenberger, Tamara N. Steinhauer, Inken Flörkemeier, Giorgia Del Favero, Bernd Clement, and Doris Marko. 2020. "The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties" Molecules 25, no. 7: 1524. https://doi.org/10.3390/molecules25071524
APA StyleAichinger, G., Lichtenberger, F.-B., Steinhauer, T. N., Flörkemeier, I., Del Favero, G., Clement, B., & Marko, D. (2020). The Aza-Analogous Benzo[c]phenanthridine P8-D6 Acts as a Dual Topoisomerase I and II Poison, thus Exhibiting Potent Genotoxic Properties. Molecules, 25(7), 1524. https://doi.org/10.3390/molecules25071524