
How Sure Can We Be about ML Methods-Based Evaluation of Compound Activity: Incorporation of Information about Prediction Uncertainty Using Deep Learning Techniques



Abstract
1. Introduction
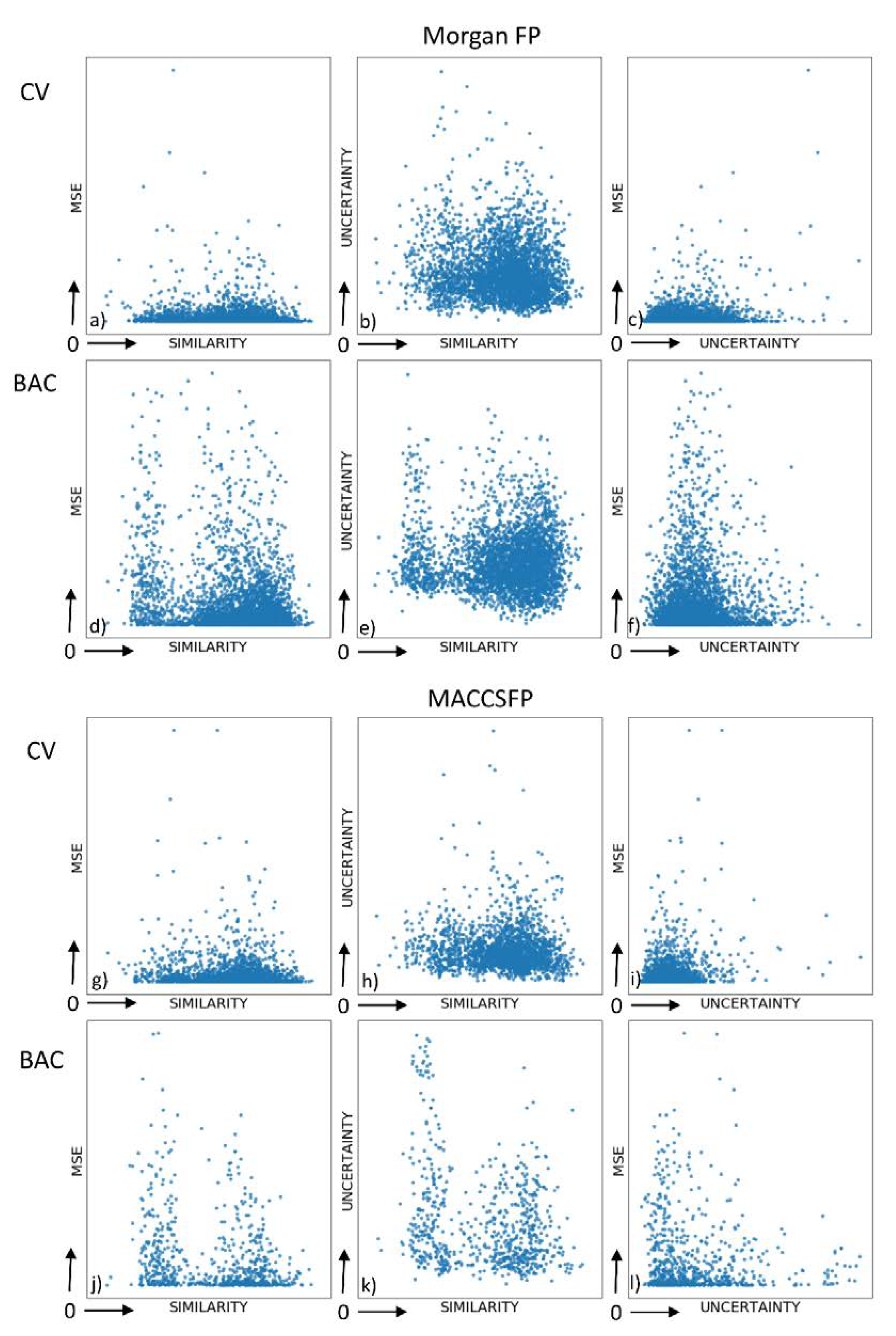
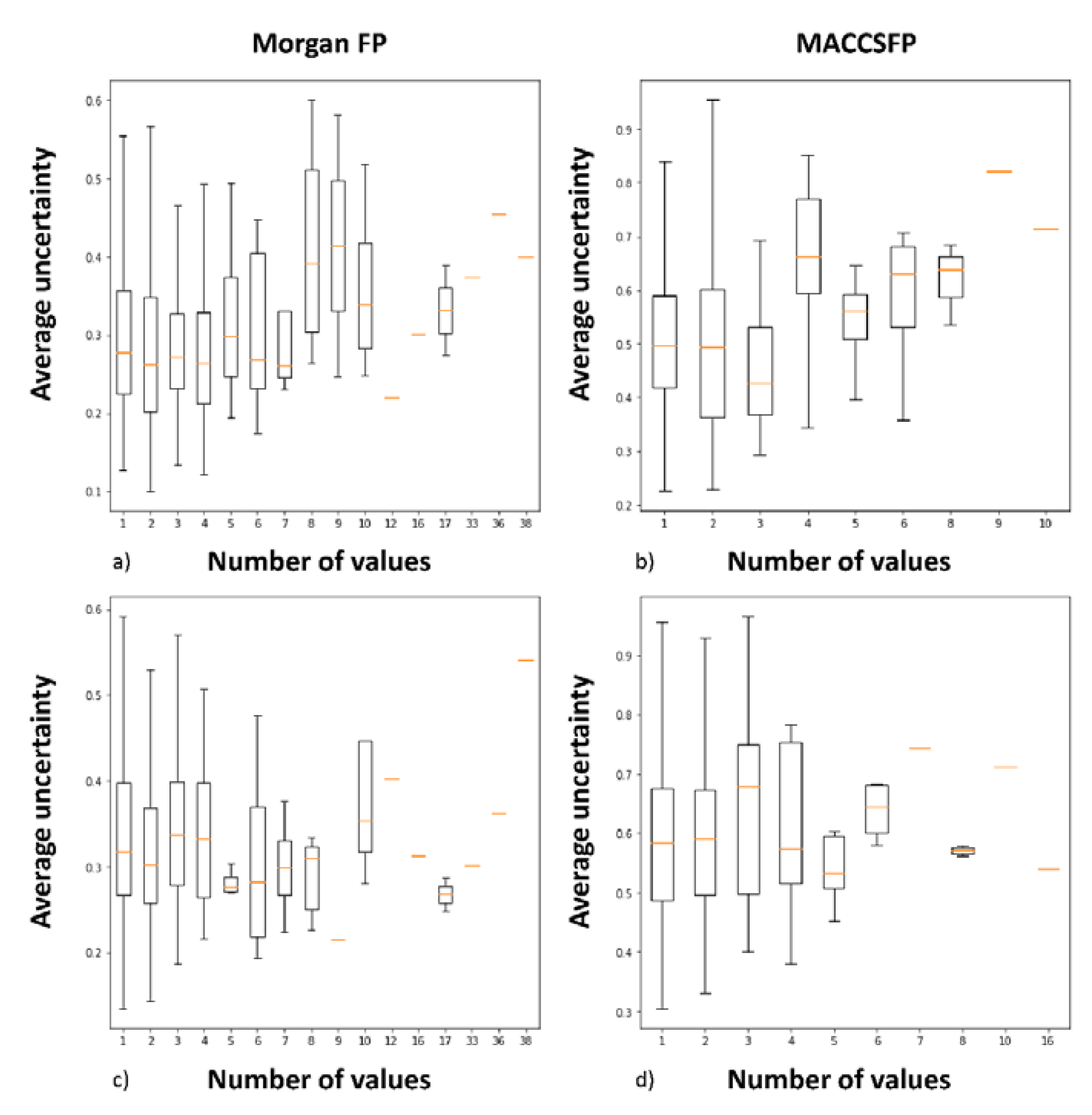
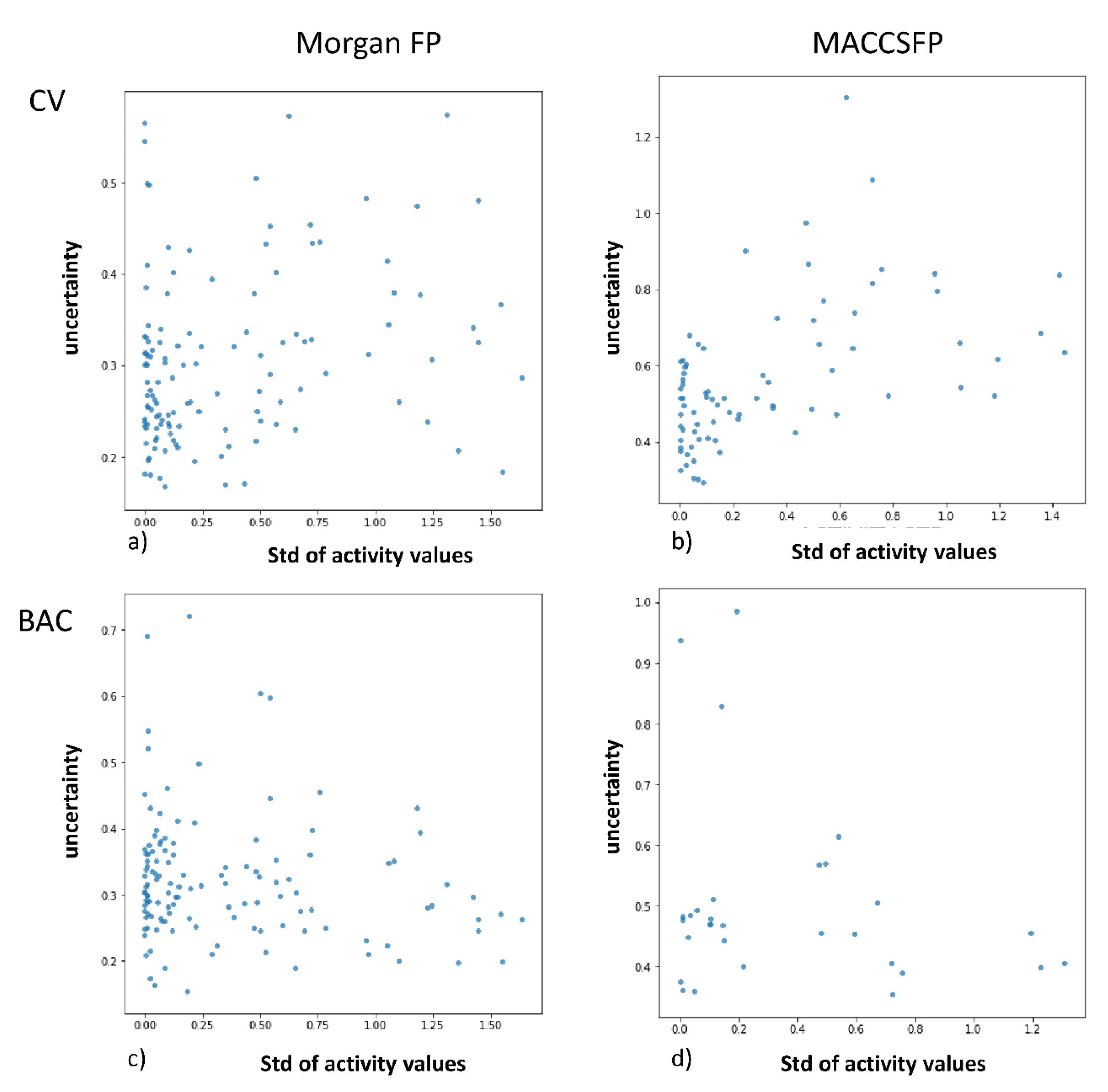
- the relationships between the prediction error, similarity to the training set and prediction uncertainty for the data from the test set were examined, together with analysis of correlation between uncertainty and the number of activity values provided—and also between uncertainty and standard deviation of activity values
- we tested whether incorporation of information about prediction uncertainty improves the compounds ranking on the basis of predicted activity
- uncertainty of predictions was used to search for the potential errors in the ChEMBL database.
2. Results and Discussion
2.1. General Observations
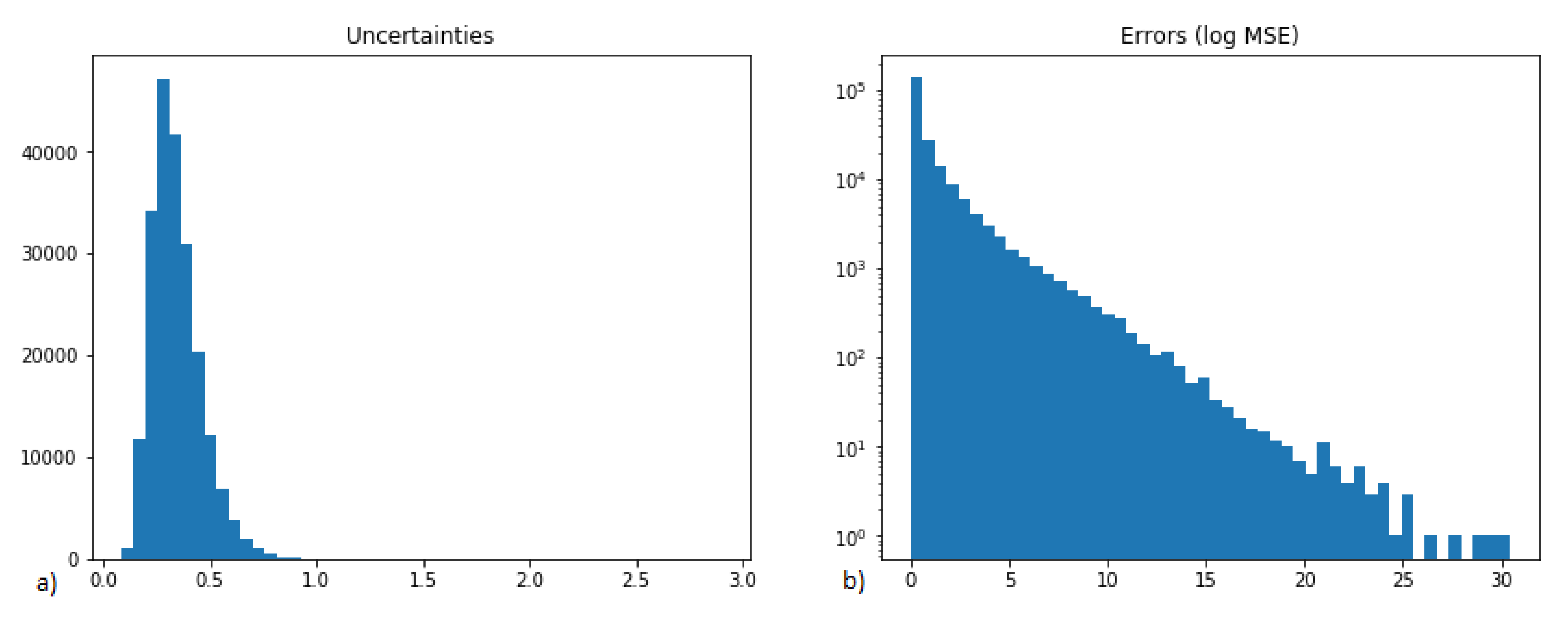
2.2. Analysis of Uncertainties, Errors and Compound Similarities
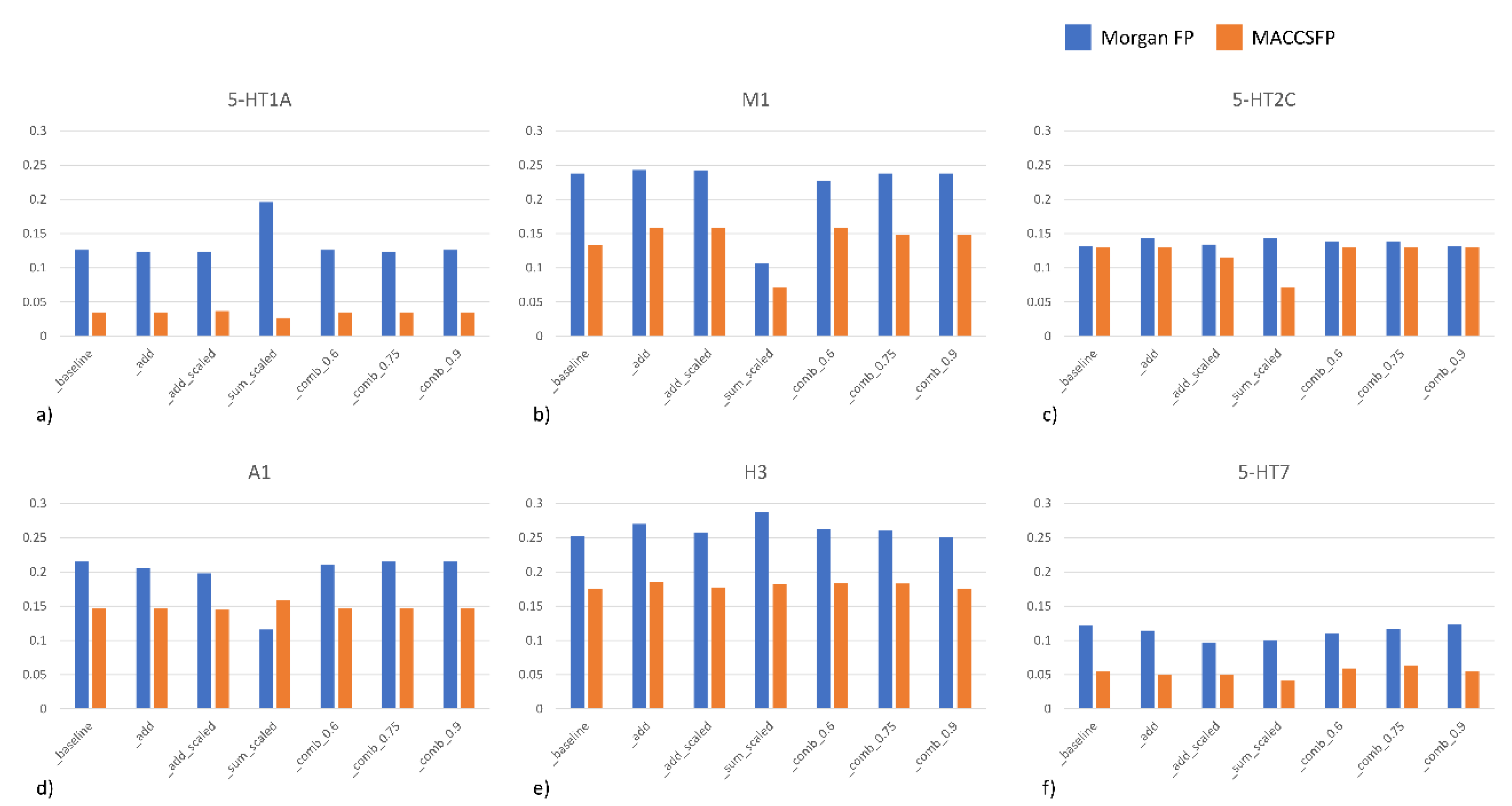
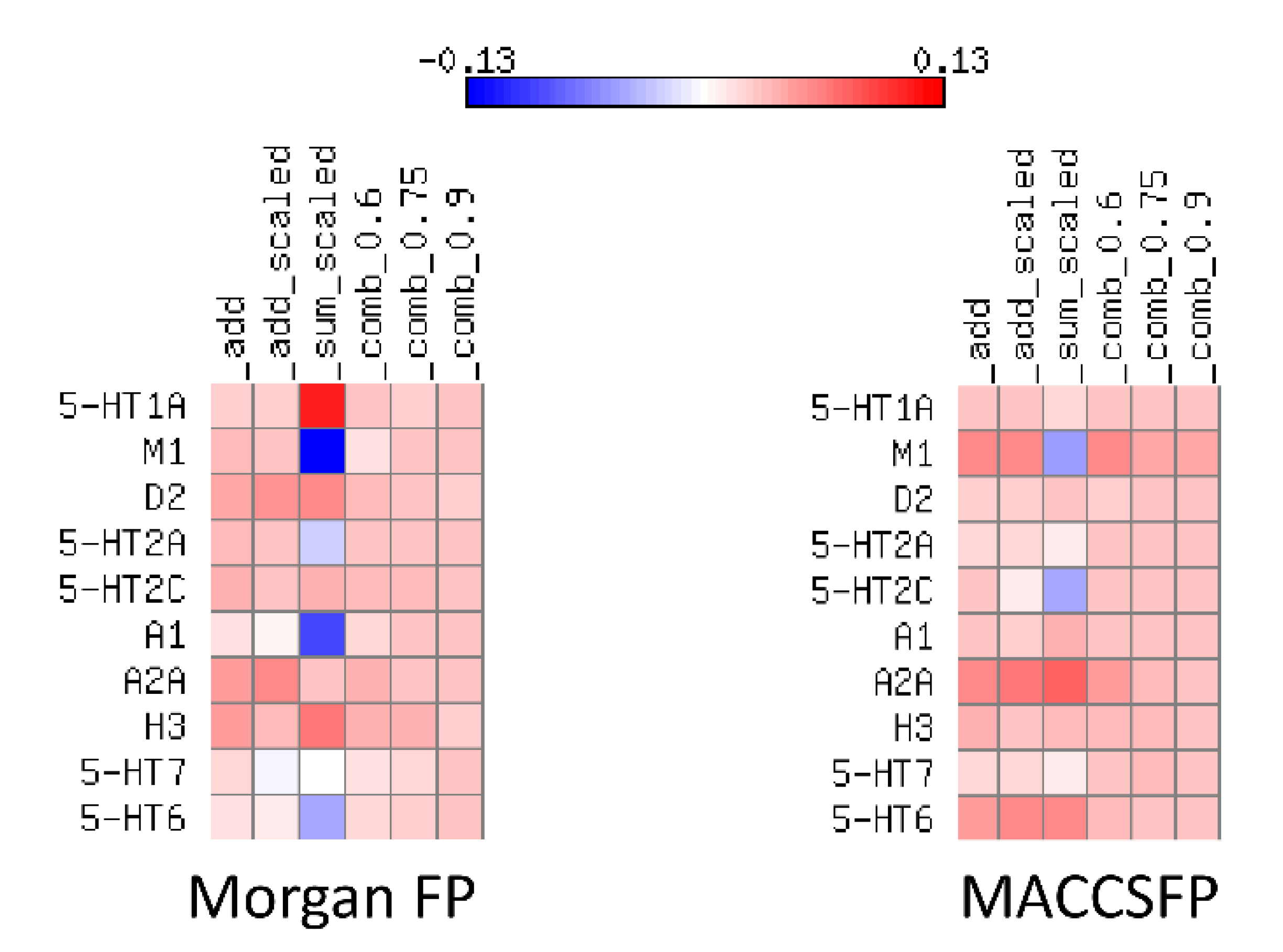
2.3. Compounds Ranking
- Baseline—only prediction of a model is taken into account
- Add—to model prediction, information about uncertainty is added directly; the less uncertain the model is about the sample, the better
- Scale—the uncertainty estimation is normalized to fit the range of [0,1]) and used to scale the predictionwhere is a normalized uncertainty based on the measures for the whole test set.
- Add scaled—the uncertainty estimation is normalized to fit into [0,1] and added directly to the prediction
- Sum scaled—both prediction of a model and its uncertainty are normalized and then summed upwhere is a normalized prediction based on the predictions over the whole dataset.
- Comb λ—linear combination of prediction and uncertainty with the λ coefficient (various λ values were tested).
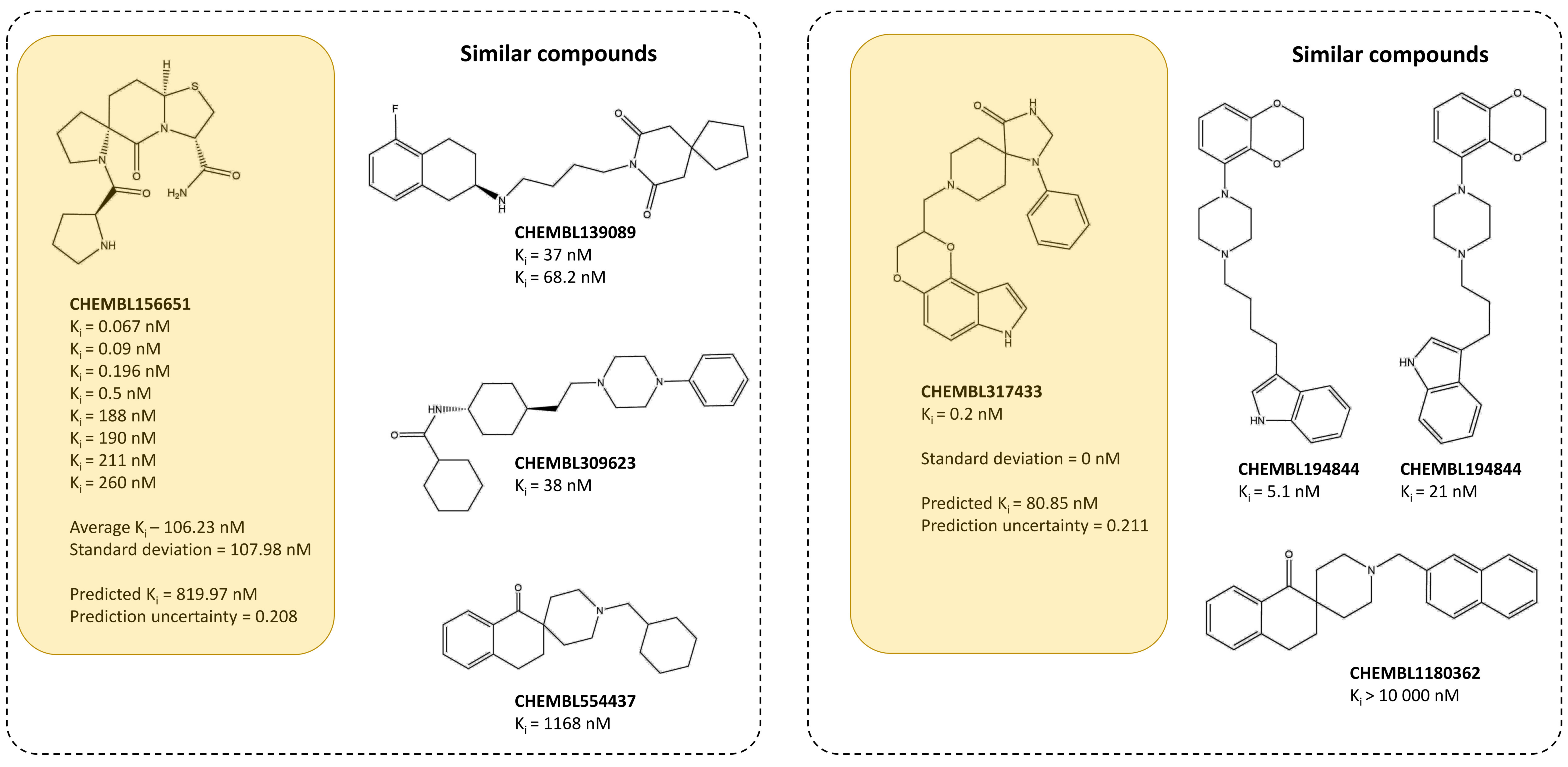
2.4. Detection of Potential Errors in Bioactivity Databases
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2013, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Reddy, A.S.; Pati, S.P.; Kumar, P.P.; Pradeep, H.; Sastry, G.N. Virtual screening in drug discovery—A computational perspective. Curr. Protein Pept. Sci. 2007, 8, 329–351. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, A. What do we know and when do we know it? J. Comput. Mol. Des. 2008, 22, 239–255. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.S.; Srinivas, K. Modern drug discovery process: An in silico approach. J. Bioinform. Seq. Anal. 2011, 2, 89–94. [Google Scholar]
- Egan, W.J.; Merz, K.M.; Baldwin, J.J. Prediction of drug absorption using multivariate statistics. J. Med. Chem. 2000, 43, 3867–3877. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv. Rev. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Ou-Yang, S.-S.; Lu, J.; Kong, X.-Q.; Liang, Z.-J.; Luo, C.; Jiang, H.-L. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131–1140. [Google Scholar] [CrossRef]
- Chiba, S.; Ikeda, K.; Ishida, T.; Gromiha, M.M.; Taguchi, Y.-H.; Iwadate, M.; Umeyama, H.; Hsin, K.-Y.; Kitano, H.; Yamamoto, K.; et al. Identification of potential inhibitors based on compound proposal contest: Tyrosine-protein kinase Yes as a target. Sci. Rep. 2015, 5, 17209. [Google Scholar] [CrossRef]
- Gaulton, A.; Bellis, L.; Bento, A.P.S.F.F.; Chambers, J.; Davies, M.; Hersey, A.; Light, Y.; McGlinchey, S.; Michalovich, D.; Al-Lazikani, B.; et al. ChEMBL: A large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2011, 40, D1100–1107. [Google Scholar] [CrossRef]
- Besnard, J.; Ruda, G.F.; Setola, V.; Abecassis, K.; Rodriguiz, R.M.; Huang, X.-P.; Norval, S.; Sassano, M.F.; Shin, A.I.; Webster, L.A.; et al. Automated design of ligands to polypharmacological profiles. Nature 2012, 492, 215–220. [Google Scholar] [CrossRef]
- Berman, H.M.; Bhat, T.N.; Bourne, P.; Feng, Z.; Gilliland, G.; Weissig, H.; Westbrook, J. The Protein Data Bank and the challenge of structural genomics. Nat. Genet. 2000, 7, 957–959. [Google Scholar]
- Wishart, D.S. DrugBank: A comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006, 34, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Wexler, P. TOXNET: An evolving web resource for toxicology and environmental health information. Toxicology 2001, 157, 3–10. [Google Scholar] [CrossRef]
- Melville, J.L.; Burke, E.; Hirst, J. Machine learning in virtual screening. Comb. Chem. High. Throughput Screen. 2009, 12, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Tao, L.; Zhang, P.; Qin, C.; Chen, S.; Zhang, C.; Chen, Z.; Zhu, F.; Yang, S.; Wei, Y.; Chen, Y.Z. Recent progresses in the exploration of machine learning methods as in-silico ADME prediction tools. Adv. Drug Deliv. Rev. 2015, 86, 83–100. [Google Scholar] [CrossRef] [PubMed]
- Fukunishi, Y. Structure-based drug screening and ligand-based drug screening with machine learning. Comb. Chem. High. Throughput Screen. 2009, 12, 397–408. [Google Scholar] [CrossRef]
- Agarwal, S.; Dugar, D.; Sengupta, S. Ranking Chemical Structures for Drug Discovery: A New Machine Learning Approach. J. Chem. Inf. Model. 2010, 50, 716–731. [Google Scholar] [CrossRef]
- Sakiyama, Y.; Yuki, H.; Moriya, T.; Hattori, K.; Suzuki, M.; Shimada, K.; Honma, T. Predicting human liver microsomal stability with machine learning techniques. J. Mol. Graph. Model. 2008, 26, 907–915. [Google Scholar] [CrossRef]
- Ma, X.H.; Jia, J.; Zhu, F.; Xue, Y.; Li, Z.R.; Chen, Y.Z. Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries. Comb. Chem. High. Throughput Screen. 2009, 12, 344–357. [Google Scholar] [CrossRef]
- Schwaighofer, A.; Schroeter, T.; Mika, S.; Blanchard, G. How wrong can we get? A review of machine learning approaches and error bars. Comb. Chem. High. Throughput Screen. 2009, 12, 453–468. [Google Scholar] [CrossRef]
- Douguet, M. Ligand-Based Approaches in Virtual Screening. Curr. Comput. Drug Des. 2008, 4, 180–190. [Google Scholar] [CrossRef]
- Chen, B.; Harrison, R.; Papadatos, G.; Willett, P.; Wood, D.J.; Lewell, X.Q.; Greenidge, P.; Stiefl, N. Evaluation of machine-learning methods for ligand-based virtual screening. J. Comput. Mol. Des. 2007, 21, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J. Machine learning methods in chemoinformatics. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 468–481. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Ji, L.; Chen, Y.; Tang, K.; Wang, H.; Zhu, R.; Wei, J.; Cao, Z.; Liu, Q. When drug discovery meets web search: Learning to Rank for ligand-based virtual screening. J. Cheminformatics 2015, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Alberga, D.; Trisciuzzi, D.; Montaruli, M.; Leonetti, F.; Mangiatordi, G.F.; Nicolotti, O. A New Approach for Drug Target and Bioactivity Prediction: The Multifingerprint Similarity Search Algorithm (MuSSeL). J. Chem. Inf. Model. 2018, 59, 586–596. [Google Scholar] [CrossRef]
- Acharya, C.; Coop, A.; Polli, J.E.; MacKerell, A.D. Recent advances in ligand-based drug design: Relevance and utility of the conformationally sampled pharmacophore approach. Curr. Comput. Drug Des. 2011, 7, 10–22. [Google Scholar] [CrossRef]
- Yasuo, N.; Sekijima, M. Improved Method of Structure-Based Virtual Screening via Interaction-Energy-Based Learning. J. Chem. Inf. Model. 2019, 59, 1050–1061. [Google Scholar] [CrossRef]
- Duan, J.; Sastry, M.; Dixon, S.; Lowrie, J.; Sherman, W. Analysis and comparison of 2D fingerprints: Insights into database screening performance using eight fingerprint methods. J. Chemin- 2011, 3, P1. [Google Scholar] [CrossRef][Green Version]
- Nisius, B.; Bajorath, J. Molecular Fingerprint Recombination: Generating Hybrid Fingerprints for Similarity Searching from Different Fingerprint Types. ChemMedChem 2009, 4, 1859–1863. [Google Scholar] [CrossRef]
- Gardiner, E.J.; Gillet, V.J.; Haranczyk, M.; Hert, J.; Holliday, J.D.; Malim, N.H.A.H.; Patel, Y.; Willett, P. Turbo similarity searching: Effect of fingerprint and dataset on virtual-screening performance. Stat. Anal. Data Mining: ASA Data Sci. J. 2009, 2, 103–114. [Google Scholar] [CrossRef]
- Heikamp, K.; Bajorath, J. How Do 2D Fingerprints Detect Structurally Diverse Active Compounds? Revealing Compound Subset-Specific Fingerprint Features through Systematic Selection. J. Chem. Inf. Model. 2011, 51, 2254–2265. [Google Scholar] [CrossRef] [PubMed]
- Sastry, M.; Lowrie, J.F.; Dixon, S.L.; Sherman, W. Large-Scale Systematic Analysis of 2D Fingerprint Methods and Parameters to Improve Virtual Screening Enrichments. J. Chem. Inf. Model. 2010, 50, 771–784. [Google Scholar] [CrossRef] [PubMed]
- Leśniak, D.; Jastrzębski, S.; Podlewska, S.; Czarnecki, W.M.; Bojarski, A. Quo vadis G protein-coupled receptor ligands? A tool for analysis of the emergence of new groups of compounds over time. Bioorganic Med. Chem. Lett. 2017, 27, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Wallach, I.; Heifets, A. Most Ligand-Based Classification Benchmarks Reward Memorization Rather than Generalization. J. Chem. Inf. Model. 2018, 58, 916–932. [Google Scholar] [CrossRef]
- Leśniak, D.; Podlewska, S.; Jastrzębski, S.; Sieradzki, I.; Bojarski, A.; Tabor, J. Development of New Methods Needs Proper Evaluation—Benchmarking Sets for Machine Learning Experiments for Class A GPCRs. J. Chem. Inf. Model. 2019, 59, 4974–4992. [Google Scholar] [CrossRef]
- Smusz, S.; Czarnecki, W.M.; Warszycki, D.; Bojarski, A. Exploiting uncertainty measures in compounds activity prediction using support vector machines. Bioorganic Med. Chem. Lett. 2015, 25, 100–105. [Google Scholar] [CrossRef]
- Der Kiureghian, A.; Ditlevsen, O. Aleatory or epistemic? Does it matter? Struct. Saf. 2009, 31, 105–112. [Google Scholar] [CrossRef]
- Unterthiner, T.; Mayr, A.; Klambauer, G.; Steijaert, M.; Ceulemans, H.; Wegner, J.; Hochreiter, S. Deep Learning as an Opportunity in Virtual Screening. In Proceedings of the NIPS Workshop on Deep Learning and Representation Learning, Montreal, QC, Canada, 8–13 December 2014; pp. 1058–1066. Available online: http://www.bioinf.at/publications/2014/NIPS2014a.pdf (accessed on 5 December 2019).
- Lusci, A.; Pollastri, G.; Baldi, P. Deep Architectures and Deep Learning in Chemoinformatics: The Prediction of Aqueous Solubility for Drug-Like Molecules. J. Chem. Inf. Model. 2013, 53, 1563–1575. [Google Scholar] [CrossRef]
- Ekins, S. The Next Era: Deep Learning in Pharmaceutical Research. Pharm. Res. 2016, 33, 2594–2603. [Google Scholar] [CrossRef]
- Kim, I.-W.; Oh, J.M. Deep learning: From chemoinformatics to precision medicine. J. Pharm. Investig. 2017, 13, 317–323. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. J. Chemin- 2017, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Dai, Z.; Chen, F.; Gao, S.; Pei, J.; Lai, L. Deep Learning for Drug-Induced Liver Injury. J. Chem. Inf. Model. 2015, 55, 2085–2093. [Google Scholar] [CrossRef]
- Ma, J.; Sheridan, R.P.; Liaw, A.; Dahl, G.E.; Svetnik, V. Deep Neural Nets as a Method for Quantitative Structure–Activity Relationships. J. Chem. Inf. Model. 2015, 55, 263–274. [Google Scholar] [CrossRef] [PubMed]
- Ragoza, M.; Hochuli, J.; Idrobo, E.; Sunseri, J.; Koes, D.R. Protein–Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model. 2017, 57, 942–957. [Google Scholar] [CrossRef] [PubMed]
- Gal, Y.; Ghahramani, Z. Dropout dropout as a Bayesian approximation: Representing model uncertainty in deep learning. In Proceedings of the International Conference on Machine Learning, New York, NY, USA, 19–24 June 2016; pp. 1050–1059. [Google Scholar]
- Srivastava, N.; Hinton, G.; Krizhevsky, A.; Sutskever, I.; Salakhutdinov, R. Dropout dropout: A Simple Way to Prevent Neural Networks from Overfitting. J. Mach. Learn. Res. 2014, 15, 1929–1958. [Google Scholar]
- Hernández-Lobato, J.M.; Adams, R. Probabilistic backpropagation for scalable learning of bayesian neural networks. In Proceedings of the International Conference on Machine Learning (ICML 2015), Lille, France, 6–11 July 2015. [Google Scholar]
- Graves, A. Practical variational inference for neural networks. Adv. Neural Inf. Process. Syst. 2011, 24, 2348–2356. [Google Scholar]
- Oh, S.; Ha, H.-J.; Chi, D.; Lee, H. Serotonin Receptor and Transporter Ligands - Current Status. Curr. Med. Chem. 2001, 8, 999–1034. [Google Scholar] [CrossRef]
- Westkaemper, R.B.; Roth, B.L. Structure and Function Reveal Insights in the Pharmacology of 5-HT Receptor Subtypes. In The Serotonin Receptors; Humana Press: Totowa, NJ, USA, 2008; pp. 39–58. [Google Scholar]
- Glennon, R.A. Higher-End serotonin receptors: 5-HT5, 5-HT6, and 5-HT7. J. Med. Chem. 2003, 46, 2795–2812. [Google Scholar] [CrossRef]
- Wang, C.; Jiang, Y.; Ma, J.; Wu, H.; Wacker, D.; Katritch, V.; Han, G.W.; Liu, W.; Huang, X.-P.; Vardy, E.; et al. Structural Basis for Molecular Recognition at Serotonin Receptors. Science 2013, 340, 610–614. [Google Scholar] [CrossRef]
- Eglen, R.M.; Choppin, A.; Watson, N. Therapeutic opportunities from muscarinic receptor research. Trends Pharmacol. Sci. 2001, 22, 409–414. [Google Scholar] [CrossRef]
- Hocher, B. Adenosine A1 receptor antagonists in clinical research and development. Kidney Int. 2010, 78, 438–445. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moreau, J.-L.; Huber, G. Central adenosine A2A receptors: An overview. Brain Res. Rev. 1999, 31, 65–82. [Google Scholar] [CrossRef]
- Xu, F.; Wu, H.; Katritch, V.; Han, G.W.; Jacobson, K.A.; Gao, Z.-G.; Cherezov, V.; Stevens, R.C. Structure of an Agonist-Bound Human A2A Adenosine Receptor. Science 2011, 332, 322–327. [Google Scholar] [CrossRef] [PubMed]
- Passani, M.B.; Lin, J.-S.; Hancock, A.; Crochet, S.; Blandina, P. The histamine H3 receptor as a novel therapeutic target for cognitive and sleep disorders. Trends Pharmacol. Sci. 2004, 25, 618–625. [Google Scholar] [CrossRef]
- Missale, C.; Nash, S.R.; Robinson, S.W.; Jaber, M.; Caron, M.G. Dopamine receptors: From structure to function. Physiol. Rev. 1998, 78, 189–225. [Google Scholar] [CrossRef]
- Wang, S.; Che, T.; Levit, A.; Shoichet, B.K.; Wacker, D.; Roth, B.L. Structure of the D2 dopamine receptor bound to the atypical antipsychotic drug risperidone. Nature 2018, 555, 269–273. [Google Scholar] [CrossRef]
- Qadri, F.; Bader, M. Kinin B1 receptors as a therapeutic target for inflammation. Expert Opin. Ther. Targets 2017, 22, 31–44. [Google Scholar] [CrossRef]
- Cai, M.; Hruby, V.J. The Melanocortin Receptor System: A Target for Multiple Degenerative Diseases. Curr. Protein Pept. Sci. 2016, 17, 488–496. [Google Scholar] [CrossRef]
- Lalanne, L.; Ayranci, G.; Kieffer, B.L.; Lutz, P.-E. The Kappa Opioid Receptor: From Addiction to Depression, and Back. Front. Psychol. 2014, 5, 170. [Google Scholar] [CrossRef]
- Valentino, R.J.; Volkow, N.D. Untangling the complexity of opioid receptor function. Neuropsychopharmacol. 2018, 43, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Scammell, T.E.; Winrow, C.J. Orexin receptors: Pharmacology and therapeutic opportunities. Annu. Rev. Pharmacol. Toxicol. 2011, 51, 243–266. [Google Scholar] [CrossRef]
- Zou, S.; Kumar, U. Cannabinoid Receptors and the Endocannabinoid System: Signaling and Function in the Central Nervous System. Int. J. Mol. Sci. 2018, 19, 833. [Google Scholar]
- Liu, J.; Clough, S.J.; Hutchinson, A.J.; Adamah-Biassi, E.B.; Popovska-Gorevski, M.; Dubocovich, M.L. MT1 and MT2 Melatonin Receptors: A Therapeutic Perspective. Annu. Rev. Pharmacol. Toxicol. 2015, 56, 361–383. [Google Scholar] [CrossRef] [PubMed]
- Crupi, R.; Impellizzeri, D.; Cuzzocrea, S. Role of Metabotropic Glutamate Receptors in Neurological Disorders. Front. Mol. Neurosci. 2019, 12, 20. [Google Scholar] [CrossRef] [PubMed]
- Hughes, C.E.; Nibbs, R.J.B. A guide to chemokines and their receptors. FEBS J. 2018, 285, 2944–2971. [Google Scholar] [CrossRef]
- Griffith, J.W.; Sokol, C.L.; Luster, A.D. Chemokines and Chemokine Receptors: Positioning Cells for Host Defense and Immunity. Annu. Rev. Immunol. 2014, 32, 659–702. [Google Scholar] [CrossRef]
- Morgan, H.L. The Generation of a Unique Machine Description for Chemical Structures-A Technique Developed at Chemical Abstracts Service. J. Chem. Doc. 1965, 5, 107–113. [Google Scholar] [CrossRef]
- Accelrys, MACCS Structural Keys. Available online: http://www.3dsbiovia.com (accessed on 5 December 2019).
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org. (accessed on 5 December 2019).
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. Available online: https://arxiv.org/abs/1412.6980v9 (accessed on 5 December 2019).
- Ramsundar, B.; Eastman, P.; Walters, P.; Pande, V. Deep Learning for the Life Sciences: Applying Deep Learning to Genomics, Microscopy, Drug Discovery, and More; O’Reilly Media, Inc.: Sebastopol, CA, USA, 2019. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Morgan | MACCSFP | ||||
|---|---|---|---|---|---|---|
| MSE | Dropout MSE | Uncertainty | MSE | Dropout MSE | Uncertainty | |
| 5-HT1A | 0.416 ± 0.02 | 0.406 ± 0.02 | 0.318 ± 0.01 | 0.568 ± 0.04 | 0.552 ± 0.04 | 0.580 ± 0.00 |
| ACM1 | 0.665 ± 0.18 | 0.660 ± 0.17 | 0.352 ± 0.01 | 0.781 ± 0.16 | 0.763 ± 0.15 | 0.567 ± 0.01 |
| D2 | 0.352 ± 0.03 | 0.344 ± 0.03 | 0.298 ± 0.01 | 0.436 ± 0.00 | 0.420 ± 0.00 | 0.510 ± 0.00 |
| 5-HT2A | 0.467 ± 0.03 | 0.459 ± 0.03 | 0.319 ± 0.01 | 0.559 ± 0.06 | 0.551 ± 0.05 | 0.523 ± 0.01 |
| 5-HT2C | 0.494 ± 0.02 | 0.488 ± 0.02 | 0.304 ± 0.01 | 0.532 ± 0.03 | 0.518 ± 0.02 | 0.535 ± 0.04 |
| A1 | 0.429 ± 0.03 | 0.421 ± 0.03 | 0.308 ± 0.00 | 0.479 ± 0.02 | 0.474 ± 0.02 | 0.547 ± 0.03 |
| A2A | 0.412 ± 0.03 | 0.406 ± 0.03 | 0.326 ± 0.01 | 0.528 ± 0.03 | 0.515 ± 0.02 | 0.561 ± 0.02 |
| H3 | 0.407 ± 0.04 | 0.399 ± 0.04 | 0.300 ± 0.01 | 0.504 ± 0.02 | 0.488 ± 0.002 | 0.518 ± 0.02 |
| 5-HT7 | 0.455 ± 0.04 | 0.450 ± 0.03 | 0.302 ± 0.02 | 0.899 ± 0.04 | 0.902 ± 0.03 | 0.523 ± 0.02 |
| 5-HT6 | 0.461 ± 0.03 | 0.455 ± 0.02 | 0.309 ± 0.00 | 0.520 ± 0.00 | 0.507 ± 0.00 | 0.544 ± 0.01 |
| MT1A | 0.630 ± 0.12 | 0.622 ± 0.13 | 0.365 ± 0.02 | 0.769 ± 0.10 | 0.761 ± 0.10 | 0.596 ± 0.03 |
| MT1B | 0.630 ± 0.06 | 0.623 ± 0.05 | 0.351 ± 0.02 | 0.856 ± 0.13 | 0.839 ± 0.12 | 0.586 ± 0.02 |
| CB1 | 0.494 ± 0.05 | 0.486 ± 0.05 | 0.343 ± 0.01 | 0.584 ± 0.03 | 0.578 ± 0.03 | 0.565 ± 0.03 |
| MOR | 0.538 ± 0.06 | 0.528 ± 0.06 | 0.380 ± 0.01 | 0.663 ± 0.06 | 0.643 ± 0.06 | 0.645 ± 0.02 |
| DOR | 0.473 ± 0.01 | 0.466 ± 0.01 | 0.380 ± 0.00 | 0.614 ± 0.03 | 0.605 ± 0.03 | 0.636 ± 0.01 |
| KOR | 0.513 ± 0.04 | 0.500 ± 0.04 | 0.375 ± 0.01 | 0.651 ± 0.04 | 0.636 ± 0.04 | 0.629 ± 0.03 |
| CB2 | 0.542 ± 0.03 | 0.525 ± 0.03 | 0.343 ± 0.01 | 0.641 ± 0.03 | 0.627 ± 0.03 | 0.601 ± 0.02 |
| MC4 | 0.407 ± 0.04 | 0.396 ± 0.04 | 0.361 ± 0.00 | 0.540 ± 0.03 | 0.526 ± 0.04 | 0.589 ± 0.01 |
| mGluR5 | 0.628 ± 0.11 | 0.627 ± 0.11 | 0.332 ± 0.02 | 0.720 ± 0.09 | 0.713 ± 0.09 | 0.524 ± 0.03 |
| CCR2 | 0.417 ± 0.08 | 0.409 ± 0.08 | 0.285 ± 0.02 | 0.546 ± 0.20 | 0.539 ± 0.20 | 0.534 ± 0.03 |
| B1 | 0.578 ± 0.15 | 0.576 ± 0.16 | 0.320 ± 0.03 | 0.722 ± 0.18 | 0.722 ± 0.17 | 0.618 ± 0.04 |
| MC5 | 0.484 ± 0.11 | 0.471 ± 0.10 | 0.433 ± 0.03 | 0.428 ± 0.11 | 0.423 ± 0.10 | 0.578 ± 0.04 |
| MC3 | 0.339 ± 0.06 | 0.343 ± 0.06 | 0.413 ± 0.02 | 0.422 ± 0.09 | 0.418 ± 0.08 | 0.542 ± 0.02 |
| OX2R | 0.445 ± 0.04 | 0.439 ± 0.04 | 0.307 ± 0.01 | 0.562 ± 0.06 | 0.550 ± 0.06 | 0.599 ± 0.01 |
| OX1R | 0.340 ± 0.04 | 0.336 ± 0.03 | 0.325 ± 0.01 | 0.510 ± 0.05 | 0.500 ± 0.05 | 0.570 ± 0.01 |
| Target | Morgan FP | MACCSFP | ||||
|---|---|---|---|---|---|---|
| MSE | Dropout MSE | Uncertainty | MSE | Dropout MSE | Uncertainty | |
| 5-HT1A | 1.323 ± 0.21 | 1.284 ± 0.18 | 0.347 ± 0.04 | 1.879 ± 0.10 | 1.746 ± 0.03 | 0.563 ± 0.07 |
| ACM1 | 1.788 ± 0.43 | 1.766 ± 0.41 | 0.389 ± 0.07 | 2.792 ± 0.80 | 2.706 ± 0.73 | 0.594 ± 0.09 |
| D2 | 0.967 ± 0.16 | 0.954 ± 0.16 | 0.342 ± 0.02 | 1.424 ± 0.26 | 1.304 ± 0.15 | 0.588 ± 0.04 |
| 5-HT2A | 1.667 ± 0.56 | 1.609 ± 0.57 | 0.395 ± 0.04 | 1.496 ± 0.00 | 1.449 ± 0.00 | 0.568 ± 0.00 |
| 5-HT2C | 1.534 ± 0.75 | 1.502 ± 0.72 | 0.338 ± 0.04 | 1.714 ± 0.44 | 1.671 ± 0.44 | 0.517 ± 0.01 |
| A1 | 1.378 ± 0.56 | 1.359 ± 0.56 | 0.337 ± 0.03 | 1.602 ± 0.00 | 1.537 ± 0.00 | 0.476 ± 0.00 |
| A2A | 1.445 ± 0.37 | 1.430 ± 0.36 | 0.367 ± 0.03 | 1.264 ± 0.19 | 1.207 ± 0.19 | 0.567 ± 0.02 |
| H3 | 1.001 ± 0.13 | 0.961 ± 0.13 | 0.354 ± 0.02 | 1.284 ± 0.46 | 1.205 ± 0.43 | 0.547 ± 0.05 |
| 5-HT7 | 1.267 ± 0.53 | 1.252 ± 0.54 | 0.316 ± 0.03 | 2.027 ± 0.65 | 1.956 ± 0.63 | 0.534 ± 0.04 |
| 5-HT6 | 1.337 ± 0.39 | 1.305 ± 0.38 | 0.323 ± 0.03 | 1.597 ± 0.41 | 1.453 ± 0.44 | 0.564 ± 0.03 |
| MT1A | 1.989 ± 0.54 | 1.989 ± 0.55 | 0.367 ± 0.06 | 2.101 ± 0.60 | 2.071 ± 0.59 | 0.532 ± 0.04 |
| MT1B | 1.921 ± 0.27 | 1.914 ± 0.28 | 0.340 ± 0.06 | 1.925 ± 0.42 | 1.899 ± 0.43 | 0.565 ± 0.06 |
| CB1 | 1.413 ± 0.44 | 1.399 ± 0.41 | 0.351 ± 0.03 | 1.595 ± 0.49 | 1.511 ± 0.43 | 0.563 ± 0.02 |
| MOR | 1.602 ± 0.30 | 1.546 ± 0.30 | 0.405 ± 0.02 | 1.994 ± 0.38 | 1.878 ± 0.38 | 0.653 ± 0.05 |
| DOR | 1.921 ± 1.01 | 1.895 ± 0.99 | 0.360 ± 0.02 | 2.393 ± 1.07 | 2.251 ± 0.94 | 0.607 ± 0.05 |
| KOR | 1.653 ± 0.43 | 1.613 ± 0.42 | 0.401 ± 0.03 | 2.159 ± 0.86 | 2.052 ± 0.79 | 0.651 ± 0.05 |
| CB2 | 1.601 ± 0.41 | 1.577 ± 0.43 | 0.350 ± 0.02 | 1.552 ± 0.41 | 1.508 ± 0.38 | 0.619 ± 0.08 |
| MC4 | 1.652 ± 0.87 | 1.561 ± 0.78 | 0.392 ± 0.07 | 1.406 ± 0.49 | 1.371 ± 0.48 | 0.577 ± 0.07 |
| mGluR5 | 2.289 ± 1.08 | 2.279 ± 1.08 | 0.336 ± 0.05 | 1.844 ± 0.71 | 1.839 ± 0.73 | 0.488 ± 0.07 |
| CCR2 | 0.792 ± 0.43 | 0.789 ± 0.43 | 0.288 ± 0.05 | 1.480 ± 0.60 | 1.428 ± 0.57 | 0.411 ± 0.04 |
| B1 | 1.903 ± 0.65 | 1.894 ± 0.66 | 0.353 ± 0.05 | 1.503 ± 0.42 | 1.478 ± 0.42 | 0.597 ± 0.16 |
| MC5 | 1.221 ± 1.12 | 1.237 ± 1.13 | 0.441 ± 0.04 | 1.230 ± 1.35 | 1.207 ± 1.31 | 0.504 ± 0.02 |
| MC3 | 0.947 ± 0.41 | 0.913 ± 0.39 | 0.482 ± 0.05 | 0.917 ± 0.43 | 0.895 ± 0.42 | 0.533 ± 0.05 |
| OX2R | 1.192 ± 0.64 | 1.183 ± 0.63 | 0.291 ± 0.02 | 1.573 ± 0.84 | 1.582 ± 0.87 | 0.547 ± 0.05 |
| OX1R | 1.178 ± 0.57 | 1.164 ± 0.56 | 0.345 ± 0.05 | 1.833 ± 0.14 | 1.778 ± 0.13 | 0.569 ± 0.08 |
| Target CHEMBLID | Target Name | Training Set Size CV | Test Set Size CV | Training Set Size BAC | Test Set Size BAC |
|---|---|---|---|---|---|
| CHEMBL214 | 5-HT1A | 2599 | 649 | 2495 | 753 |
| CHEMBL216 | ACM1 | 676 | 168 | 652 | 192 |
| CHEMBL217 | D2 | 4496 | 1124 | 4243 | 1379 |
| CHEMBL224 | 5-HT2A | 2385 | 596 | 2025 | 956 |
| CHEMBL225 | 5-HT2C | 1559 | 389 | 1590 | 358 |
| CHEMBL226 | Adenosine A1 | 2782 | 695 | 2758 | 719 |
| CHEMBL251 | Adenosine A2A | 3165 | 791 | 2915 | 1041 |
| CHEMBL264 | Histamine H3 | 2546 | 636 | 2689 | 493 |
| CHEMBL3155 | 5-HT7 | 1209 | 302 | 1070 | 441 |
| CHEMBL3371 | 5-HT6 | 2074 | 518 | 1979 | 613 |
| CHEMBL1945 | MT 1A | 573 | 143 | 622 | 94 |
| CHEMBL1946 | MT 1B | 572 | 142 | 524 | 190 |
| CHEMBL218 | CB1 | 1945 | 486 | 1950 | 481 |
| CHEMBL233 | MOR | 2815 | 703 | 2997 | 521 |
| CHEMBL236 | DOR | 2457 | 614 | 2070 | 1001 |
| CHEMBL237 | KOR | 2381 | 595 | 2147 | 829 |
| CHEMBL253 | CB2 | 2611 | 652 | 2687 | 576 |
| CHEMBL259 | MC4 | 1450 | 362 | 1364 | 448 |
| CHEMBL3227 | mGluR5 | 272 | 67 | 283 | 56 |
| CHEMBL4015 | CCR2 | 168 | 42 | 180 | 30 |
| CHEMBL4308 | B1 | 444 | 110 | 431 | 123 |
| CHEMBL4608 | MC5 | 314 | 78 | 334 | 58 |
| CHEMBL4644 | MC3 | 400 | 99 | 410 | 89 |
| CHEMBL4792 | OX2R | 1269 | 317 | 1175 | 411 |
| CHEMBL5113 | OX1R | 1087 | 271 | 1182 | 176 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sieradzki, I.; Leśniak, D.; Podlewska, S. How Sure Can We Be about ML Methods-Based Evaluation of Compound Activity: Incorporation of Information about Prediction Uncertainty Using Deep Learning Techniques. Molecules 2020, 25, 1452. https://doi.org/10.3390/molecules25061452
Sieradzki I, Leśniak D, Podlewska S. How Sure Can We Be about ML Methods-Based Evaluation of Compound Activity: Incorporation of Information about Prediction Uncertainty Using Deep Learning Techniques. Molecules. 2020; 25(6):1452. https://doi.org/10.3390/molecules25061452
Chicago/Turabian StyleSieradzki, Igor, Damian Leśniak, and Sabina Podlewska. 2020. "How Sure Can We Be about ML Methods-Based Evaluation of Compound Activity: Incorporation of Information about Prediction Uncertainty Using Deep Learning Techniques" Molecules 25, no. 6: 1452. https://doi.org/10.3390/molecules25061452
APA StyleSieradzki, I., Leśniak, D., & Podlewska, S. (2020). How Sure Can We Be about ML Methods-Based Evaluation of Compound Activity: Incorporation of Information about Prediction Uncertainty Using Deep Learning Techniques. Molecules, 25(6), 1452. https://doi.org/10.3390/molecules25061452