3.2. Synthesis
3.2.1. Synthesis of 3-Methyl-4-(pent-4-yn-1-ylthio)benzonitrile 2
A mixture of 4-mercapto-3-methylbenzonitrile 1 (1 mol), finely divided K2CO3 (5 mol), KI (0.01 mol), 5-chloro-1-pentyne (1.5 mol), and N-methylpyrrolidone-2 was heated at 65 °C for 24 h. The cooled reaction mixture was treated by cold water and stirred for 3–4 h. The solid was collected and recrystallized from methanol. Light beige solid, yield 69%, m.p. 36–38 °C. MS (EI), m/z (Irelat.(%)): 215 [M]+ (67). Calc. 215.3140, C13H13NS. 1H-NMR (DMSO-d6): δ 1.94 (2H, quint, J = 7.3, CH2CH2CH2S), 2.25 (3H, s, CH3), 2.30 (2H, m, CH2CH2CH2S), 2.77 (1H, s, CHCCH2), 3.15 (2H, t, J = 7.3, CH2CH2CH2S), 7.46 (1H, d, J = 8.8, H6), 7.62 (1H, dd, J = 8.8, J = 0.5, H5), 7.63 (1H, s, H3) ppm.
3.2.2. General Procedure for the Synthesis of Compounds 3, 9, 16, 27a,b, 32
A mixture of benzonitriles 2, 8, 15, 25b, 26, and 31 (1 mmol), finely divided K2CO3 (5 mmol), and hydroxylamine hydrochloride (5 mmol) in absolute ethanol was refluxed for 24 h. The hot reaction mixture was filtered, and the remaining solids were washed with hot acetone. The combined filtrates were concentrated in vacuo. The residue was recrystallized from the corresponding solvent (in parentheses following mp data).
N′-Hydroxy-3-methyl-4-(pent-4-yn-1-ylthio)benzimidamide3, Light yellow solid, yield 92%, m.p. 64–66 °C (EtOH). MS (EI), m/z (Irelat.(%)): 248 [M]+ (54). Calc. 248.3439, C13H16N2OS. 1H-NMR (DMSO-d6): δ 1.86 (2H, quint, J = 7.3, CH2CH2CH2S), 2.37 (2H, t, J = 7.3, CH2CH2CH2S), 2.42 (3H, s, CH3Ph), 2.77 (1H, s, CHCCH2), 3.25 (2H, t, J = 7.3, CH2CH2CH2S), 4.96 (1H, s, NOH), 5.05 (2H, brs, NH2), 7.13 (1H, d, J = 8.8, H6), 7.29 (1H, dd, J = 8.8, J = 0.5, H5), 7.33 (1H, s, H3) ppm.
N′,4-dihydroxy-3-methylbenzimidamide9, Light beige solid, yield 35%, m.p. 70–71 °C (EtOH). MS (EI), m/z (Irelat.(%)): 166 [M]+ (100). Calc. 166.1772, C8H10N2O2. 1H-NMR (DMSO-d6): δ 2.30 (3H, s, CH3Ph), 4.99 (1H, s, NOH), 5.09 (2H, brs, NH2), 6.83 (1H, d, J = 7.5, H6), 7.34 (1H, d, J = 7.5, H5), 7.51 (1H, s, H3) ppm.
Tert-butyl (4-(N′-hydroxycarbamimidoyl)-2-methylphenyl)carbamate16, White solid, yield 58%, m.p. 74–76 °C (decomp.) (iPrOH). MS (EI), m/z (Irelat.(%)): 265 [M]+ (83). Calc. 265.3083, C13H19N3O3. 1H-NMR (DMSO-d6): δ 1.43 (9H, s, tBu), 2.22 (3H, s, CH3Ph), 4.94 (1H, s, NOH), 5.03 (2H, brs, NH2), 6.81 (1H, d, J = 7.5, H6), 7.24 (1H, d, J = 7.5, H5), 7.33 (1H, s, H3) ppm.
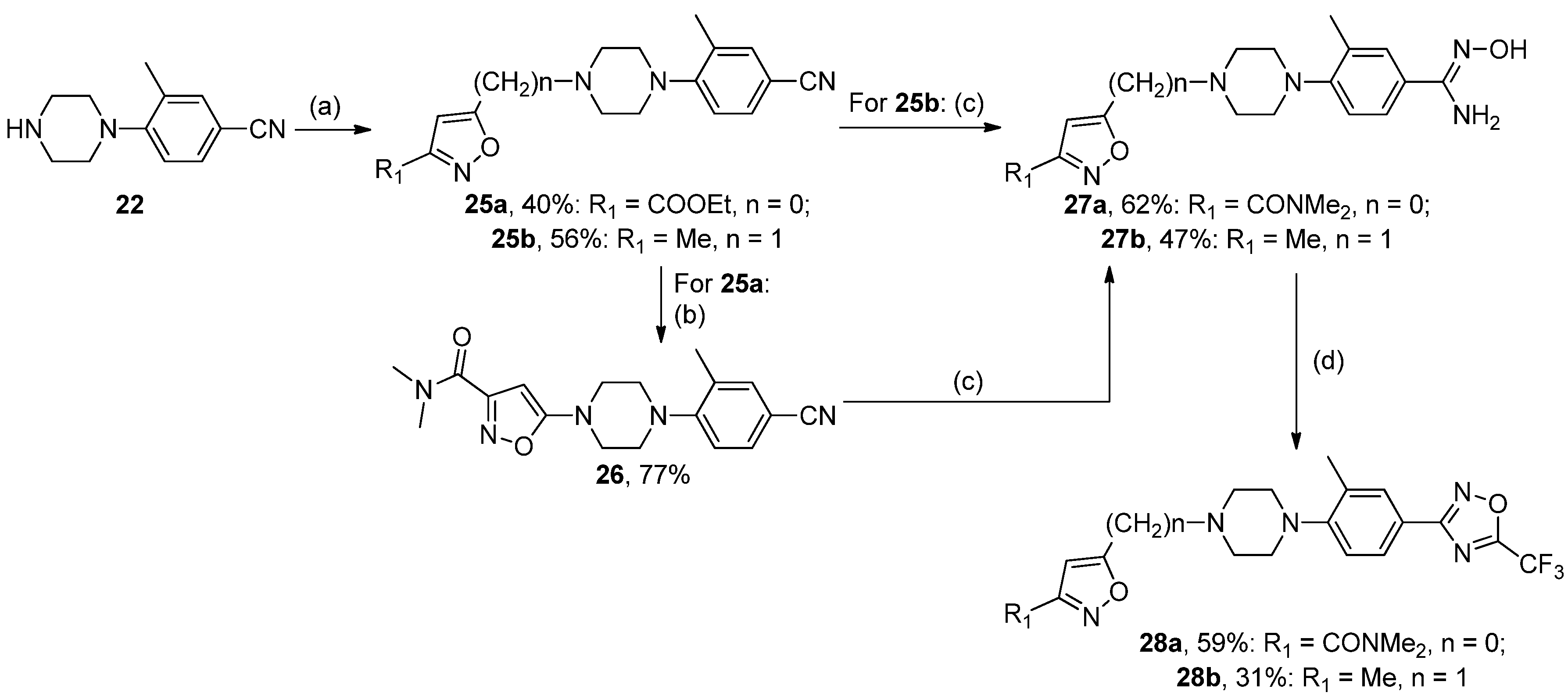
5-(4-(4-(N′-Hydroxycarbamimidoyl)-2-methylphenyl)piperazin-1-yl)-N,N-dimethylisoxazole-3-carboxamide27a, White solid, yield 62%, mp 203–205 °C (EtOH). MS (EI), m/z (Irelat.(%)): 372 [M]+ (67). Calc. 372.4216, C18H24N6O3. 1H-NMR (DMSO-d6): δ 2.22 (3H, s, CH3Ph), 2.73 (6H, s, N(CH3)2), 3.24 (4H, brt, N(CH2)2), 3.21 (4H, brt, N(CH2)2), 4.94 (1H, s, NOH), 5.01 (2H, brs, NH2), 5.80 (1H, s, isoxazole), 6.46 (1H, d, J = 8.0, H6), 7.26 (1H, d, J = 8.0, H5), 7.27 (1H, s, H3) ppm.
N′-hydroxy-3-methyl-4-(4-((3-methylisoxazol-5-yl)methyl)piperazin-1-yl)benzimidamide27b, White solid, yield 47%, m.p. 169–170 °C (EtOH). MS (EI), m/z (Irelat.(%)): 329 [M]+ (54). Calc. 329.3968, C17H23N5O2. 1H-NMR (DMSO-d6): δ 2.22 (3H, s, CH3), 2.30 (3H, s, CH3Ph), 2.73 (4H, m, N(CH2)2), 3.07 (4H, brt, N(CH2)2), 4.12 (2H, brs, NCH2), 4.96 (1H, s, NOH), 5.01 (2H, brs, NH2), 6.30 (1H, s, isoxazole), 6.46 (1H, d, J = 9.0, H6), 7.26 (1H, d, J = 9.0, H5), 7.27 (1H, s, H3) ppm.
4-(4-Acetylpiperazin-1-yl)-N′-hydroxy-3-methylbenzimidamide32, White solid, yield 50%, m.p. 230–232 °C (MeOH). MS (EI), m/z (Irelat.(%)): 276 [M]+ (61). Calc. 276.3342, C14H20N4O2. 1H-NMR (DMSO-d6): δ 1.93 (3H, s, CH3), 2.22 (3H, s, CH3Ph), 3.29 (4H, brs, N(CH2)2), 3.63 (4H, brs, N(CH2)2), 4.96 (1H, s, NOH), 5.03 (2H, brs, NH2), 6.46 (1H, d, J = 7.9, H6), 7.26 (1H, d, J = 7.9, H5), 7.27 (1H, s, H3) ppm.
3.2.3. General Procedure for the Synthesis of Compounds 4, 10, 17, 28a,b, 33
To a solution of 3, 9, 16, 27a,b, or 32 (1 mmol) in of pyridine heated to 80–90 °C carefully add dropwise trifluoroacetic anhydride (2 mmol) during 30 min. The reaction mixture was stored for 1 h at 85 °C. The cooled to rt mixture was diluted with water and extracted with ethyl acetate (3 times). The combined organic phases were washed with water (3 times), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was treated by water and stored in the refrigerator for 2–4 h. Crystals were collected and recrystallized from the corresponding solvent (in parentheses following mp data).
3-(3-Methyl-4-(pent-4-yn-1-ylthio)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole4, White solid, yield 54%, mp 49–50 °C (iPrOH). MS (EI), m/z (Irelat.(%)): 326 [M]+ (76). Calc. 326.3367, C15H13F3N2OS. 1H-NMR (DMSO-d6): δ 1.86 (2H, quint, J = 7.2, CH2CH2CH2S), 2.21 (3H, s, CH3Ph), 2.37 (2H, t, J = 7.2, CH2CH2CH2S), 2.77 (1H, s, CHCCH2), 3.25 (2H, t, J = 7.2, CH2CH2CH2S), 7.47 (1H, d, J = 7.5, H6), 7.59 (1H, d, J = 7.5, H5), 7.64 (1H, s, H3) ppm.
2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenol10, White solid, yield 43%, m.p. 64–65 °C (Hexane). MS (EI), m/z (Irelat.(%)): 244 [M]+ (64). Calc. 244.1700, C10H7F3N2O2. 1H-NMR (DMSO-d6): δ 2.19 (3H, s, CH3Ph), 7.05 (1H, d, J = 7.5, H6), 7.62 (1H, d, J = 7.5, H5), 7.99 (1H, s, H3) ppm.
Tert-butyl (2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)carbamate17, White solid, yield 48%, m.p. 104–106 °C (iPrOH). MS (EI), m/z (Irelat.(%)): 343 [M]+ (58). Calc. 343.3010, C15H16F3N3O3. 1H-NMR (DMSO-d6): δ 1.43 (9H, s, tBu), 2.16 (3H, s, CH3Ph), 7.53 (1H, d, J = 7.5, H6), 7.62 (1H, d, J = 7.5, H5), 7.65 (1H, s, H3) ppm.
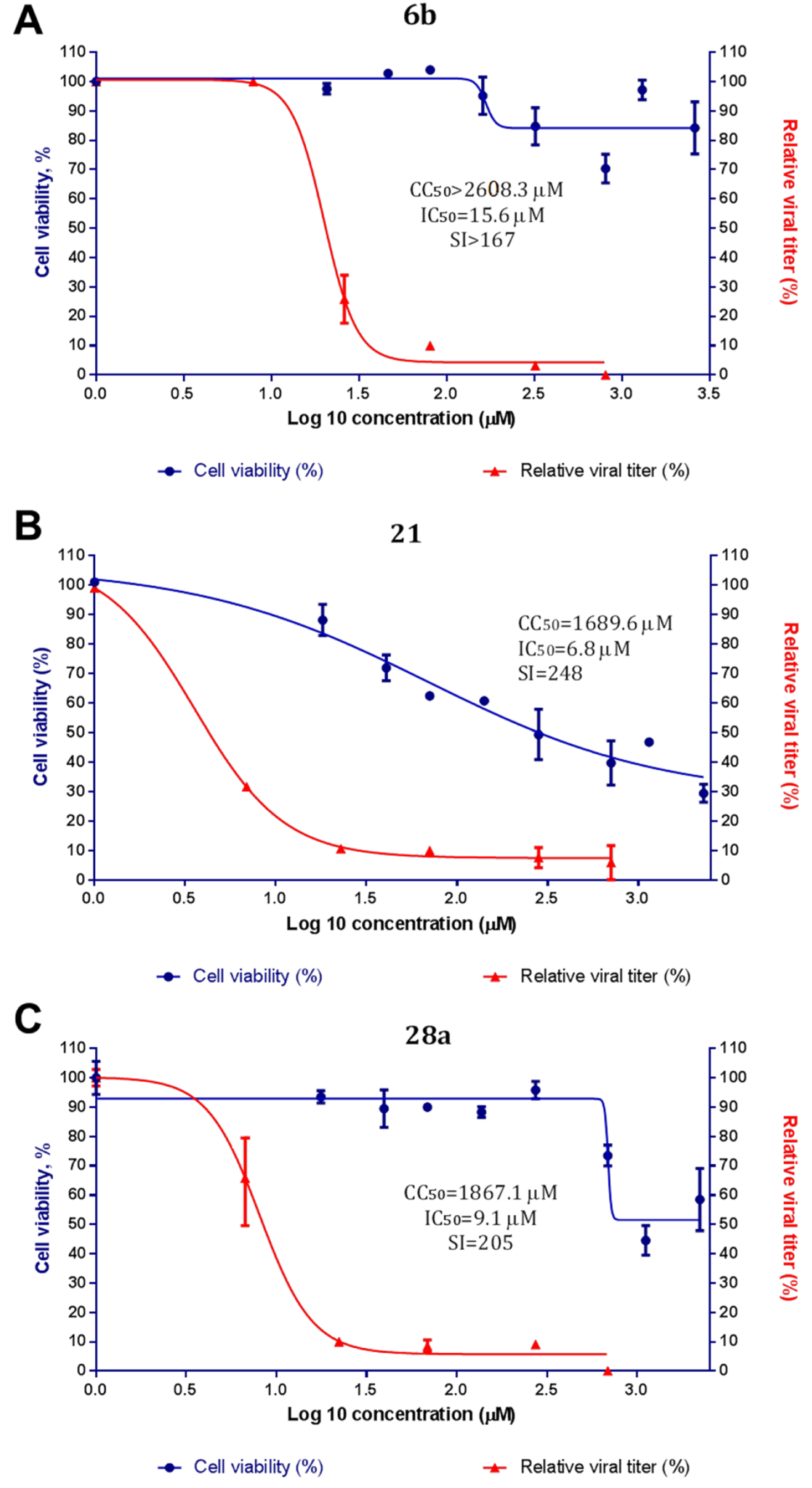
N,N-Dimethyl-5-(4-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)piperazin-1-yl)iso-xazole-3-carboxamide28a, White solid, yield 59%, m.p. 171–173 °C (EtOH). MS (EI), m/z (Irelat.(%)): 450 [M]+ (53). Calc. 450.4143, C20H21F3N6O3. 1H NMR (DMSO-d6): δ 2.39 (3H, s, CH3Ph), 2.98 (3H, s, NCH3), 3.04–3.11 (7H, m, N(CH2)2, NCH3), 3.52 (4H, brt, N(CH2)2), 5.54 (1H, s, isoxazole), 7.23 (1H, d, J = 8.0, H6), 7.86 (1H, d, J = 8.0, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 17.88, 35.15, 35.15, 46.54, 46.54, 49.96, 49.96, 86.16, 115.89 (q, J = 273.4), 118.10, 120.00, 126.97, 128.15, 129.03, 149.89, 156.44, 163.02, 166.00, 167.12 (q, J = 43.0), 171.16 ppm.
3-(3-Methyl-4-(4-((3-methylisoxazol-5-yl)methyl)piperazin-1-yl)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole28b, White solid, yield 31%, mp 94–96 °C (CCl4). MS (EI), m/z (Irelat.(%)): 407 [M]+ (48). Calc. 407.3896, C19H20F3N5O2. 1H-NMR (DMSO-d6): δ 2.28 (3H, s, CH3), 2.34 (3H, s, CH3Ph), 3.14 (4H, m, N(CH2)2), 3.52 (4H, brt, N(CH2)2), 4.37 (2H, brs, NCH2), 6.55 (1H, s, isoxazole), 7.21 (1H, d, J = 9.0, H6), 7.86 (1H, d, J = 9.0, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 11.26, 17.81, 50.00, 50.00, 52.11, 52.79, 52.79, 103.88, 115.23 (q, J = 273.1), 118.17, 120.00, 127.61, 128.68, 129.80, 149.91, 159.00, 165.99, 166.00, 167.15 (q, J = 43.5) ppm.
1-(4-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)piperazin-1-yl)ethanone33, White solid, yield 62%, m.p. 65–67 °C (Hexane). MS (EI), m/z (Irelat.(%)): 354 [M]+ (59). Calc. 354.3270, C16H17F3N4O2. 1H-NMR (DMSO-d6): δ 1.93 (3H, s, CH3), 2.21 (3H, s, CH3Ph), 3.30 (4H, brs, N(CH2)2), 3.63 (4H, brs, N(CH2)2), 7.00 (1H, d, J = 7.9, H6), 7.43 (1H, d, J = 7.9, H5), 7.56 (1H, s, H3) ppm.
3.2.4. General Procedure for the Synthesis of Compounds 6a, 12a, 20a, 28d
To a solution of 5 or 36 (3 mmol) in DMF, a solution of corresponding compounds 4, or 11, or 19, or 37 in DMF for 20–30 min was added and stirred at rt for 40–50 min. To the reaction solution, Et3N (3 mmol) in DMF was added at 80–90 °C for 2 h. The mixture was stirred at 80–90 °C for 1–2 h and at rt for 12 h. The reaction mixture was diluted with water and extracted with ethyl acetate (3-times). The combined organic phases were washed with water (3-times), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was treated by water and stored in the refrigerator for 2–4 h. Crystals were collected and recrystallized from the corresponding solvent (in parentheses following mp data).
3-(3-Methyl-4-((3-(3-carbethoxy-isoxazol-5-yl)propyl)thio)phenyl)-5-(trifluoromethyl)-1,2,4-oxadia-zole6a, White solid, yield 40%, m.p. 97–100 °C (MeOH). MS (EI), m/z (Irelat.(%)): 441 [M]+ (65). Calc. 441.4241, C19H18F3N3O4S. 1H-NMR (DMSO-d6): δ 1.27 (3H, t, J = 7.1, CH3CH2O), 2.05 (2H, quint, J = 7.5, J = 7.4, CH2CH2CH2S), 2.31 (3H, s, CH3Ph), 3.01 (2H, t, J = 7.5, CH2CH2CH2S), 3.11 (2H, t, J = 7.5, CH2CH2CH2S), 4.32 (2H, q, J = 7.1, CH3CH2O), 6.74 (1H, s, isoxazole), 7.47 (1H, d, J = 8.5, H6), 7.84 (2H, d, J = 8.5, H3, H5) ppm. 13C-NMR (DMSO-d6): δ 13.87, 19.49, 25.03, 25.97, 29.79, 61.67, 101.95, 115.76 (q, J = 273.7), 120.39, 125.38, 125.61, 128.15, 136.27, 141.90, 156.09, 159.50, 164.60 (q, J = 43.0), 168.24, 174.67 ppm.
Ethyl 5-(3-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenoxy)-3-oxopropyl)-isoxazole-3-carboxylate12a, White solid, yield 34%, m.p. 95–96 °C (Hexane). MS (EI), m/z (Irelat.(%)): 439 [M]+ (41). Calc. 439.3420, C19H16F3N3O6. 1H-NMR (DMSO-d6): δ 1.32 (3H, t, J = 7.1, CH3CH2O), 2.20 (3H, s, CH3Ph), 3.20–3.27 (4H, m, CH2CH2CO), 4.37 (2H, q, J = 7.1, CH3CH2O), 6.79 (1H, s, isoxazole), 7.33 (1H, d, J = 8.4, H6), 7.94 (1H, d, J = 8.4, H5), 8.01 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 13.64, 15.52, 21.43, 31.14, 60.95, 102.05, 115.84 (q, J = 273.6), 121.94, 122.20, 123.45, 126.23, 130.13, 131.78, 152.16, 159.88, 166.10 (q, J = 42.8), 167.84, 169.66, 171.15 ppm.
Ethyl 5-(3-((2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)amino)-3-oxopropyl)isoxa-zole-3-carboxylate20a, White solid, yield 78%, m.p. 166–168 °C (EtOH). MS (EI), m/z (Irelat.(%)): 438 [M]+ (56). Calc. 438.3573, C19H17F3N4O5. 1H-NMR (DMSO-d6):δ 1.31 (3H, t, J = 7.1, CH3CH2O), 2.31 (3H, s, CH3Ph), 2.89 (2H, t, J = 7.1, CH2CH2CO), 3.17 (2H, t, J = 7.1, CH2CH2CO), 4.36 (2H, q, J = 7.1, CH3CH2O), 6.70 (1H, s, isoxazole), 7.79 (1H, d, J = 8.8, H6), 7.87 (1H, dd, J = 1.8, 8.8, H5), 7.91 (1H, s, H3), 9.53 (1H, brs, NH) ppm. 13C-NMR (DMSO-d6): δ 13.88, 17.69, 21.97, 32.91, 61.70, 101.76, 115.73 (q, J = 273.4), 120.31, 124.54, 125.31, 129.29, 131.62, 140.21, 156.05, 159.50, 164.84 (q, J = 43.9), 168.14, 169.66, 174.86 ppm.
Methyl 5-(4-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)piperazin-1-yl)-isoxazole--3-carboxylate28d, White solid, yield 53%, m.p. 113–115 °C (EtOH). MS (EI), m/z (Irelat.(%)): 451 [M]+ (72). Calc. 451.3991, C20H20F3N5O4. 1H-NMR (DMSO-d6): δ 2.31 (3H, s, CH3Ph), 2.64 (4H, brs, N(CH2)2), 2.97 (4H, brs, N(CH2)2), 3.87 (2H, s, PhCH2N), 3.90 (3H, s, CH3O), 6.87 (1H, s, isoxazole), 7.17 (1H, d, J = 8.4, H6), 7.83 (1H, d, J = 8.4, H5), 7.84 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 17.89, 49.87, 49.87, 52.15, 52.81, 52.81, 53.11, 104.05, 115.34 (q, J = 273.2), 118.14, 120.03, 127.67, 128.57, 129.85, 150.23, 159.12, 160.31, 166.05, 166.14, 167.32 (q, J = 43.3) ppm.
3.2.5. General Procedure for the Synthesis of Compounds 6b, 12b, 20b
To a solution of NCS (2.5 mmol) and 1–2 drops of pyridine in DMF, a solution of acetaldoxime (2.5 mmol) in DMF was added for 30 min and stirred at rt for 1 h; then, to the solution, a solution of 4, or 11, or 19 (1 mmol) in DMF was added for 20 min. To the resulted mixture, Et3N (2.5 mmol) in DMF was added at 80–90 °C for 1 h and stirred at 80–90 °C for 3–4 h. The reaction mixture was diluted with water and extracted with ethyl acetate (3 times). The combined organic phases were washed with water (3 times), dried over anhydrous Na2SO4, and concentrated in vacuo. The residue was recrystallized from the corresponding solvent (in parentheses following mp data).
3-(3-Methyl-4-((3-(3-methylisoxazol-5-yl)propyl)thio)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole6b, White solid, yield 32%, m.p. 60–63 °C (Hexane). MS (EI), m/z (Irelat.(%)): 383 [M]+ (41). Calc. 383.3880, C17H16F3N3O2S. 1H-NMR (DMSO-d6): δ 2.00 (2H, quint, J = 7.2, CH2CH2CH2S), 2.18 (3H, s, CH3), 2.35 (3H, s, CH3Ph), 2.89 (2H, t, J = 7.2, CH2CH2CH2S), 3.13 (2H, t, J = 7.2, CH2CH2CH2S), 6.14 (1H, s, isoxazole), 7.48 (1H, d, J = 7.5, H6), 7.85 (1H, d, J = 7.5, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 11.05, 19.53, 24.96, 26.04, 29.84, 101.76, 115.78 (q, J = 273.1), 120.38, 125.40, 125.62, 128.19, 136.25, 141.92, 159.95, 164.71 (q, J = 43.4), 168.25, 170.65 ppm.
2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl 3-(3-methylisoxazol-5-yl)propanoate12b, White solid, yield 29%, mp 67–69 °C (Hexane). MS (EI), m/z (Irelat.(%)): 381 [M]+ (53). Calc. 381.3060, C17H14F3N3O4. 1H-NMR (DMSO-d6):δ 2.18 (3H, s, CH3), 2.35 (3H, s, CH3Ph), 3.20–3.27 (4H, m, CH2CH2CO), 6.14 (1H, s, isoxazole), 7.48 (1H, d, J = 7.5, H6), 7.85 (1H, d, J = 7.5, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 10.89, 15.48, 21.45, 31.06, 102.16, 115.77 (q, J = 273.4), 122.15, 123.44, 126.34, 130.05, 131.67, 152.12, 159.46, 165.01 (q, J = 42.8), 167.92, 169.86, 170.97 ppm.
N-(2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)-3-(3-methylisoxazol-5-yl)propan-amide20b, White solid, yield 83%, m.p. 176–179 °C (EtOH). MS (EI), m/z (Irelat.(%)): 380 [M]+ (68). Calc. 380.3212, C17H15F3N4O3. 1H-NMR (DMSO-d6): δ 2.19 (3H, s, CH3), 2.31 (3H, s, CH3Ph), 2.82 (2H, t, J = 7.3, CH2CH2CO), 3.05 (2H, t, J = 7.3, CH2CH2CO), 6.13 (1H, s, isoxazole), 7.81 (1H, d, J = 8.5, H6), 7.87 (1H, dd, J = 1.8, 8.5, H5), 7.91 (1H, s, H3), 9.49 (1H, brs, NH) ppm. 13C-NMR (DMSO-d6): δ 11.15, 17.68, 21.87, 33.12, 100.97, 115.75 (q, J = 273.1), 120.33, 124.45, 125.30, 129.27, 121.67, 140.15, 159.86, 164.80 (q, J = 43.5), 168.10, 169.69, 173.97 ppm.
3.2.6. General Procedure for the Synthesis of Compounds 7, 21, 26
A mixture of 6a, or 20a, or 25a and dimethylamine solution 17 wt.% in dioxane was heated at 50–60 °C for 1–12 h. The cooled reaction mixture was concentrated in vacuo. The residue was treated by water and stored in the refrigerator for 12 h. Crystals were collected and recrystallized from the corresponding solvent (in parentheses following mp data).
N,N-dimethyl-5-(3-((2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)thio)propyl)isoxa-zole-3-carboxamide7, White solid, yield 57%, m.p. 95.5–97 °C (Hexane). MS (EI), m/z (Irelat.(%)): 440 [M]+ (84). Calc. 440.4394, C19H19F3N4O3S. 1H-NMR (DMSO-d6): δ2.05 (2H, quint, J = 7.2, CH2CH2CH2S), 2.34 (3H, s, CH3Ph), 2.99 (3H, s, NCH3), 3.05 (3H, s, NCH3), 3.02 (2H, t, J = 7.2, CH2CH2CH2S), 3.15 (2H, t, J = 7.2, CH2CH2CH2S), 6.52 (1H, s, isoxazole), 7.47 (1H, s, J = 7.5, H6), 7.85 (1H, d, J = 7.5, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 19.50, 24.94, 26.07, 29.86, 34.85, 37.95, 101.66, 115.77 (q, J = 273.3), 120.40, 125.40, 125.59, 128.17, 136.28, 141.94, 158.70, 160.58, 164.85 (q, J = 43.5), 168.24, 172.74 ppm.
N,N-dimethyl-5-(3-((2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)amino)-3-oxopro-pyl)isoxazole-3-carboxamide21, White solid, yield 63%, m.p. 169–171 °C (Hexane:EtOAc). MS (EI), m/z (Irelat.(%)): 437 [M]+ (63). Calc. 437.3725, C19H18F3N5O4. 1H-NMR (DMSO-d6): δ 2.31 (3H, s, CH3Ph), 2.88 (2H, t, J = 7.2, CH2CH2CO), 3.00 (3H, s, NCH3), 3.11 (3H, s, NCH3), 3.15 (2H, t, J = 7.2, CH2CH2CO), 6.49 (1H, s, isoxazole), 7.80 (1H, d, J = 8.3, H6), 7.87 (1H, dd, J = 2.0, 8.3, H5), 7.91 (1H, s, H3), 9.53 (1H, brs, NH) ppm. 13C-NMR (DMSO-d6): δ 17.66, 21.82, 32.99, 34.82, 37.86, 101.48, 115.72 (q, J = 273.2), 120.25, 124.49, 125.25, 129.22, 131.61, 140.17, 158.61, 160.48, 164.79 (q, J = 43.0), 168.09, 169.64, 172.79 ppm.
5-(4-(4-Cyano-2-methylphenyl)piperazin-1-yl)-N,N-dimethylisoxazole-3-carboxamide26, White solid, yield 77%, m.p. 145–147 °C (EtOH). MS (EI), m/z (Irelat.(%)): 339 [M]+ (72). Calc. 339.3916, C18H21N5O2. 1H-NMR (DMSO-d6): δ 2.29 (3H, s, CH3Ph), 2.73 (6H, s, N(CH3)2), 3.20 (4H, brt, N(CH2)2), 3.21 (4H, brt, N(CH2)2), 5.80 (1H, s, isoxazole), 6.60 (1H, d, J = 8.0, H6), 7.49 (1H, d, J = 8.0, H5), 7.64 (1H, s, H3) ppm.
3.2.7. Synthesis of 2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl pent-4-ynoate 11
A mixture of 10 (1 mmol), DCC (2 mmol), 4-pentynoic acid (2 mmol) in pyridine was stirred at rt for 12 h. The mixture was diluted with CHCl3, and precipitated urea was filtered off. The CHCl3 solution was washed with 3% aq. HCl and water (3 times) and then dried over Na2SO4. The solution was filtered off through a short silica gel column, and the solvent was concentrated in vacuo. White solid, yield 72%, m.p. 54–56 °C. MS (EI), m/z (Irelat.(%)): 324 [M]+ (78). Calc. 324.2546, C15H11F3N2O3. 1H-NMR (DMSO-d6): δ 2.22 (3H, s, CH3Ph), 2.51–2.56 (2H, quint, CH2CH2CO), 2.82 (2H, m, CH2CH2O), 2.86 (1H, s, CHCCH2), 7.32 (1H, d, J = 8.4, H6), 7.94 (1H, d, J = 8.4, H5), 8.01 (1H, s, H3) ppm.
3.2.8. Synthesis of tert-Butyl (4-cyano-2-methylphenyl)carbamate 15
A mixture of 14 (1 mol) and Boc2O (3 mol) was refluxed for 48 h. The reaction mixture was diluted with methanol, brought to the boil, and concentrated in vacuo. The procedure was repeated 3 times. The residue was treated by hexane and stored in the refrigerator for 4–6 h. Crystals were collected and recrystallized from ethanol. White solid, yield 93%, mp 89–91 °C. MS (EI), m/z (Irelat.(%)): 232 [M]+ (37). Calc. 232.2783, C13H16N2O2. 1H-NMR (DMSO-d6): δ 1.43 (9H, s, tBu), 2.28 (3H, s, CH3Ph), 6.88 (1H, d, J = 7.5, H6), 7.60 (1H, d, J = 7.5, H5), 7.74 (1H, s, H3) ppm.
3.2.9. Synthesis of 2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)aniline 18
To a solution of 17 (1 mmol) in DCM, TFA (10 mmol) was added at 0 °C. The reaction mixture was stirred at rt for 2–3 h, after which the solvents were removed in vacuo. The residue was triturated with water to provide the product as a solid. Crystals were collected and recrystallized from hexane:EtOAc. White solid, yield 99%, m.p. 118 °C (decomp.). MS (EI), m/z (Irelat.(%)): 243 [M]+ (81). Calc. 243.1852, C10H8F3N3O. 1H-NMR (DMSO-d6): δ 2.23 (3H, s, CH3Ph), 5.27 (2H, brs, NH2), 6.71 (1H, d, J = 7.5, H6), 7.58 (1H, d, J = 7.5, H5), 7.64 (1H, s, H3) ppm.
3.2.10. Synthesis of N-(2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)pent-4-ynamide 19
To a solution of 18 (1 mol) and DMAP (1.3 mol) in DCM, 4-pentynoic acid (1 mol) was added in one portion, followed by the addition of EDCI (1.3 mol) in one portion at rt. The reaction mixture was stirred at rt overnight, after which it was washed successively with 3% aq. HCl and water (3 times). The organic layer was dried over Na2SO4 and concentrated in vacuo. The residue was recrystallized from ethanol. White solid, yield 38%, m.p. 165–167 °C. MS (EI), m/z (Irelat.(%)): 323 [M]+ (65). Calc. 323.2699, C15H12F3N3O2. 1H-NMR (DMSO-d6): δ 2.16 (3H, s, CH3Ph), 2.50–2.57 (2H, quint, CH2CH2CO), 2.62 (2H, m, CH2CH2O), 2.78 (1H, s, CHCCH2), 7.32 (1H, d, J = 8.4, H6), 7.61 (1H, d, J = 8.4, H5), 7.70 (1H, s, H3), 9.12 (1H, brs, NH) ppm.
3.2.11. General Procedure for the Synthesis of Compounds 25a,b, 28c
A mixture of the corresponding phenyl-piperazine (1 mol), the corresponding isoxazole (1.4 mol), and finely divided K2CO3 (3 mol) in acetonitrile was refluxed for 24–48 h. The hot mixture was filtered, and the remaining solids were washed with acetonitrile. The combined filtrates were concentrated in vacuo. The residue was recrystallized from ethanol.
Ethyl 5-(4-(4-cyano-2-methylphenyl)piperazin-1-yl)isoxazole-3-carboxylate25a, Light beige solid, yield 40%, m.p. 124–125 °C. MS (EI), m/z (Irelat.(%)): 340 [M]+ (42). Calc. 340.3764, C18H20N4O3. 1H-NMR (DMSO-d6): δ 1.29 (3H, t, J = 7.1, CH3CH2O), 2.32 (3H, s, CH3Ph), 3.05 (4H, brs, N(CH2)2), 3.52 (4H, brs, N(CH2)2), 4.35 (2H, q, J = 7.1, CH3CH2O), 5.75 (1H, s, isoxazole), 7.19 (1H, d, J = 7.9, H6), 7.61 (1H, d, J = 7.9, H5), 7.63 (1H, s, H3) ppm.
3-Methyl-4-(4-((3-methylisoxazol-5-yl)methyl)piperazin-1-yl)benzonitrile25b, Yellow solid, yield 56%, m.p. 86–88 °C. MS (EI), m/z (Irelat.(%)): 296 [M]+ (57). Calc. 296.3669, C17H20N4O. 1H-NMR (DMSO-d6): δ 2.29 (3H, s, CH3), 2.30 (3H, s, CH3Ph), 2.72 (4H, m, N(CH2)2), 3.02 (4H, m, N(CH2)2), 4.12 (2H, brs, NCH2), 6.30 (1H, s, isoxazole), 6.60 (1H, d, J = 9.0, H6), 7.49 (1H, d, J = 9.0, H5), 7.64 (1H, s, H3) ppm.
Ethyl 5-(4-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)piperazin-1-yl)isoxazole-3-carboxylate28c, White solid, yield 80%, m.p. 121–123 °C. MS (EI), m/z (Irelat.(%)): 451 [M]+ (46). Calc. 451.3991, C20H20F3N5O4. 1H-NMR (DMSO-d6):δ 1.30 (3H, t, J = 7.1, CH3CH2O), 2.38 (3H, s, CH3Ph), 3.07 (4H, brs, N(CH2)2), 3.54 (4H, brs, N(CH2)2), 4.32 (2H, q, J = 7.1, CH3CH2O), 5.80 (1H, s, isoxazole), 7.23 (1H, d, J = 7.9, H6), 7.86 (1H, d, J = 7.9, H5), 7.87 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 13.89, 17.82, 46.58, 46.58, 49.94, 49.94, 61.75, 87.21, 115.73 (q, J = 273.2), 118.15, 120.05, 126.94, 128.19, 128.95, 150.07, 156.42, 160.34, 162.95, 167.10 (q, J = 43.1), 171.05 ppm.
3.2.12. Synthesis of 1-(4-(4-Bromo-2-methylphenyl)piperazin-1-yl)ethanone 30
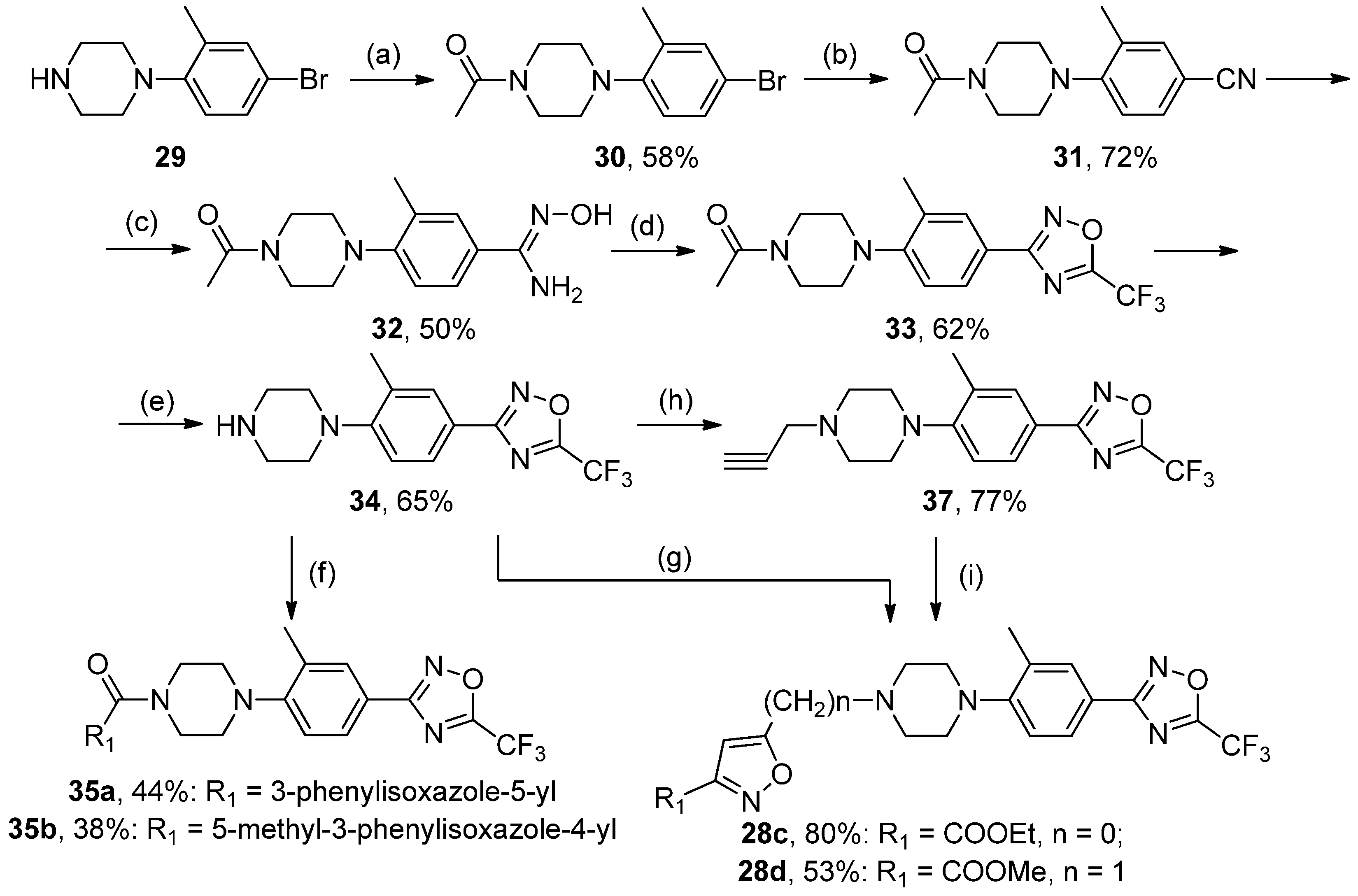
A mixture of 19 (1 mol) and Ac2O (5 mol) was heated at 60–65 °C for 4–5 h. The reaction mixture was poured into cold water and stirred for 2 h. Precipitate was collected and recrystallized from methanol. White solid, yield 58%, m.p. 120–121 °C. MS (EI), m/z (Irelat.(%)): 297 [M]+ (73). Calc. 297.1909, C13H17BrN2O. 1H-NMR (DMSO-d6): δ 1.93 (3H, s, CH3), 2.14 (3H, s, CH3Ph), 3.12 (4H, brs, N(CH2)2), 3.63 (4H, brs, N(CH2)2), 6.47 (1H, d, J = 7.9, H6), 6.83 (1H, d, J = 7.9, H5), 6.85 (1H, s, H3) ppm.
3.2.13. Synthesis of 4-(4-Acetylpiperazin-1-yl)-3-methylbenzonitrile 31
A mixture of 30 (1 mol) and CuCN (1.4. mol) in NMP was heated at 150 °C for 4 h. The cool mixture was poured into 3% aq. HCl and diluted with ethyl acetate (3 times). The combined organic layers were washed with water (3 times), dried over Na2SO4, and concentrated in vacuo. The residue was triturated with water and collected. White solid, yield 72%, m.p. 230–232 °C. MS (EI), m/z (Irelat.(%)): 243 [M]+ (69). Calc. 243.3043, C14H17N3O. 1H NMR (DMSO-d6): δ 1.93 (3H, s, CH3), 2.29 (3H, s, CH3Ph), 3.32 (4H, brs, N(CH2)2), 3.63 (4H, brs, N(CH2)2), 6.60 (1H, d, J = 7.9, H6), 7.49 (1H, d, J = 7.9, H5), 7.64 (1H, s, H3) ppm.
3.2.14. Synthesis of 3-(3-Methyl-4-(piperazin-1-yl)phenyl)-5-(trifluoromethyl)-1,2,4-oxadiazole 34
To a solution of 33 (1 mmol) in EtOH, concentrated HCl (3 mL) was added. The reaction mixture was refluxed for 4 h. The mixture was dissolved in water, neutralized with saturated aq. NaHCO3, and extracted with EtOAc (3 times). The combined organics were dried (Na2SO4) and concentrated in vacuo. The residue was purified by column chromatography (eluent CHCl3:MeOH = 10:1), Rf = 0.55. White solid, yield 65%, m.p. 78–80 °C. MS (EI), m/z (Irelat.(%)): 312 [M]+ (52). Calc. 312.2903, C14H15F3N4O. 1H NMR (DMSO-d6): δ 1.87 (1H, s, NH), 2.23 (3H, s, CH3Ph), 2.82 (4H, brs, N(CH2)2), 3.08 (4H, brs, N(CH2)2), 7.00 (1H, d, J = 7.9, H6), 7.43 (1H, d, J = 7.9, H5), 7.56 (1H, s, H3) ppm.
3.2.15. General Procedure for the Synthesis of Compounds 35a,b
To a solution of 34 (1 mol) and DMAP (1.3 mol) in DCM, the corresponding acid (1 mol) was added in one portion, followed by the addition of EDCI (1.3 mol) in one portion at rt. The reaction mixture was stirred at rt overnight, after which it was washed successively with 3% aq. HCl and water (3 times). The organic layer was dried and concentrated in vacuo. The residue was triturated with water to provide the product as solid and recrystallized from ethanol.
(4-(2-Methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)piperazin-1-yl)(3-phenylisoxazol--5-yl)methanone35a, White solid, yield 44%, m.p. 134–136 °C. MS (EI), m/z (Irelat.(%)): 483 [M]+ (52). Calc. 483.4425, C24H20F3N5O3. 1H-NMR (DMSO-d6): δ 2.39 (3H, s, CH3Ph), 3.05 (4H, m, N(CH2)2), 3.84 (4H, brt, CON(CH2)2), 4.37 (2H, brs, NCH2), 7.24 (1H, д, J = 8.5, H6), 7.55 (3H, m, m, p-Ph), 7.59 (1H, s, isoxazole), 7.86 (1H, d, J = 8.5, H5), 7.87 (1H, s, H3), 7.96 (2H, m, o-Ph) ppm. 13C-NMR (DMSO-d6): δ 17.68, 46.67, 46.67, 50.24, 50.24, 100.78, 115.90 (q, J = 273.2), 118.51, 120.05, 127.10, 127.10, 127.32, 128.17, 128.70, 128.75, 129.71, 129.71, 130.45, 150.10, 155.66, 159.67, 162.39, 166.10 (q, J = 43.3), 167.93 ppm.
(5-Methyl-3-phenylisoxazol-4-yl)(4-(2-methyl-4-(5-(trifluoromethyl)-1,2,4-oxadiazol-3-yl)phenyl)pi-perazin-1-yl)methanone35b, White solid, yield 38%, m.p. 119–121 °C. MS (EI), m/z (Irelat.(%)): 497 [M]+ (64). Calc. 497.4691, C25H22F3N5O3. 1H-NMR (CDCl3): δ 2.39 (3H, s, CH3Ph), 2.57 (3H, s, CH3), 2.87–4.07 (8H, m, N(CH2)2), 6.92 (1H, d, J = 7.86, H6), 7.51 (3H, m, m, p-Ph), 7.52 (1H, s, isoxazole), 7.70 (2H, m, o-Ph), 7.92 (1H, d, J = 8.5, H5), 7.94 (1H, s, H3) ppm. 13C-NMR (DMSO-d6): δ 12.40, 17.94, 46.75, 46.75, 50.32, 50.32, 112.0, 115.70 (q, J = 273.4), 118.43, 120.00, 127.12, 128.42, 128.65, 128.89, 129.72, 129.72, 130.00, 130.00, 130.67, 149.74, 161.87, 164.00, 166.02 (q, J = 43.4), 167.31, 169.98 ppm.
3.2.16. Synthesis of 3-(3-Methyl-4-(4-(prop-2-yn-1-yl)piperazin-1-yl)phenyl)-5-(trifluoromethyl)-1,2,4-oxa-diazole 37
A mixture of 34 (1 mmol), propargyl bromide (1.3 mmol), finely divided K2CO3 (3 mmol), and KI (0.1 mmol) in acetonitrile was heated at 50 °C for 2 h. The hot mixture was filtered, and the remaining solids were washed with acetonitrile. The combined organic filtrates were concentrated in vacuo. The residue was triturated with water, and precipitate was collected. Yellow solid, yield 77%, m.p. 75–78 °C. MS (EI), m/z (Irelat.(%)): 350 [M]+ (71). Calc. 350.3383, C17H17F3N4O. 1H-NMR (DMSO-d6): δ 2.23 (3H, s, CH3Ph), 2.54 (4H, brs, N(CH2)2), 2.79 (1H, s, CHCCH2), 3.12 (4H, brs, N(CH2)2), 3.33 (2H, s, NCH2),7.00 (1H, d, J = 7.9, H6), 7.43 (1H, d, J = 7.9, H5), 7.56 (1H, s, H3) ppm.

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
