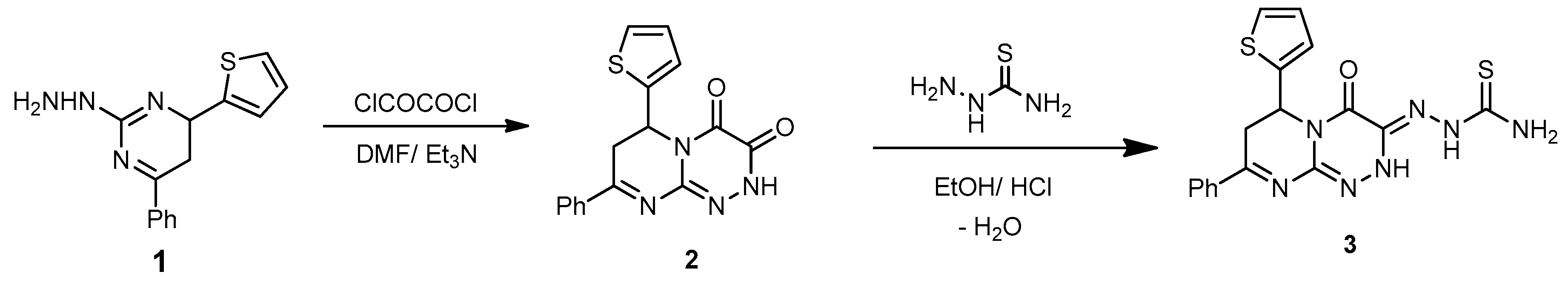
3.2. Synthesis
8-Phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazine-3,4-dione (2). A mixture of compound 1 (0.27 g, 1 mmol) and oxalyl chloride (0.12 g, 1 mmol) in DMF (15 mL) containing Et3N (0.5 mL) was refluxed for 4 h. After cooling, the reaction mixture was poured onto ice/H2O. The formed solid was collected by filtration and recrystallized from EtOH to afford compound 2 as a light yellow powder with a 75% yield; mp 252–254 °C; IR (KBr): (cm−1) 689 (C–S–C), 1610–1620 (2 C=N), 1645–1685 (2 amidic C=O), 3235 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.82 (dd, J = 13.7 and 4.7 Hz, 1H), 2.91 (dd, J = 9.5 and 13.7 Hz, 1H), 5.09 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.05 (d, J = 4.5, 1H, Thioph.(C5)-H), 10.56 (s, 1H, NH Deutr. Exch); MS (m/z, %): 324.07 (M+, 10). Anal. Calcd for C16H12N4O2S (324.36): C, 59.25; H, 3.73; N, 17.27%. Found: C, 59.02; H, 3.12; N, 17.11%.
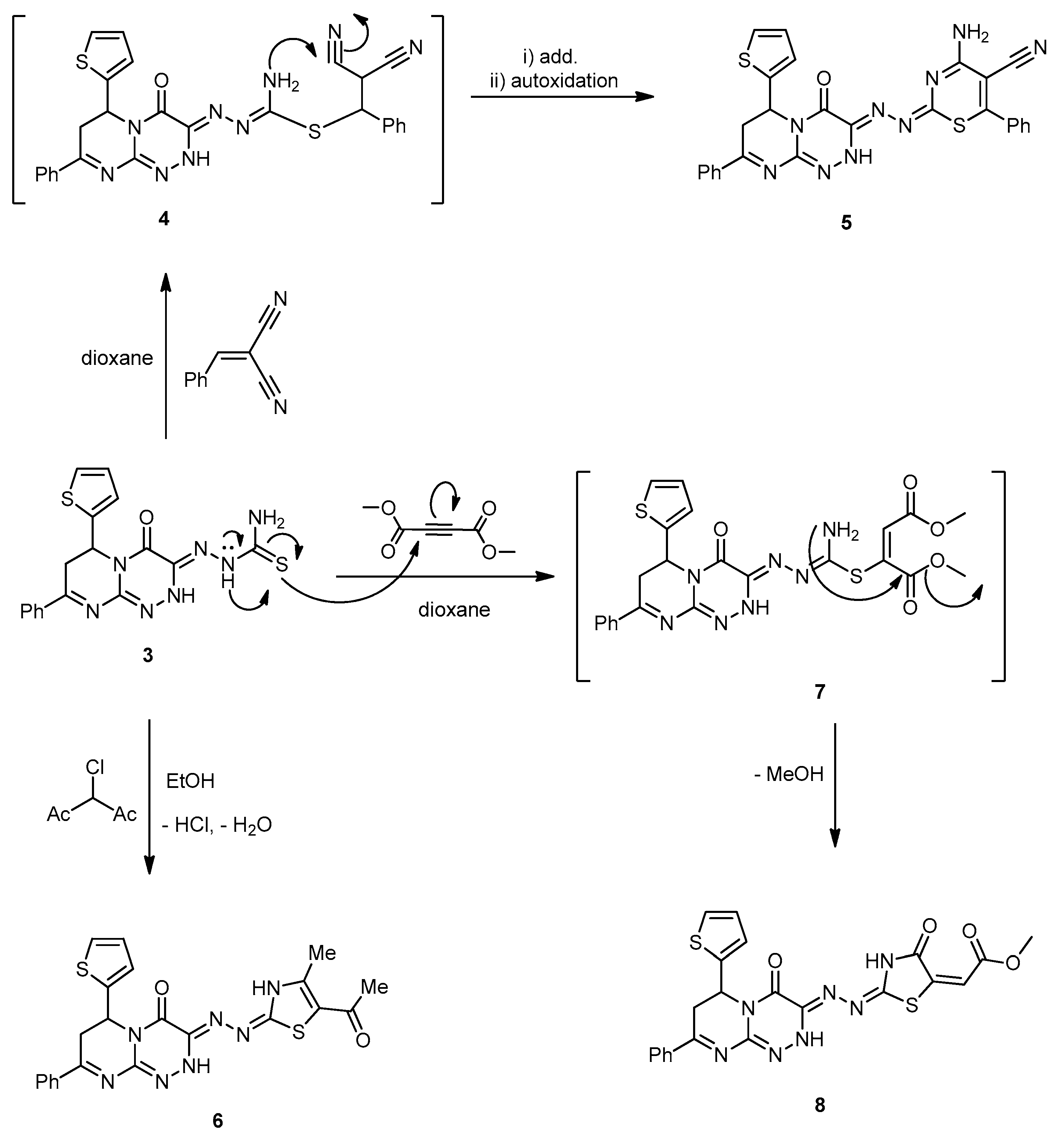
2-(4-Oxo-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazin-3(4H)-ylidene) hydrazinecarbothioamide (3). A mixture of compound 2 (0.32 g, 1 mmol) and thiosemicarbazide (0.09 g, 1 mmol) was refluxed in HCl (0.5 mL) and EtOH (30 mL) for 8 h. The mixture was filtered while hot, and the cooled filtrate was poured onto H2O. The formed solid was filtered, washed with H2O, and thoroughly dried and recrystallized from EtOH to afford 3 as yellowish-brown powder with an 80% yield; mp 271–273 °C; IR (KBr): (cm−1) 685 (C–S–C), 1290 (C=S), 1610–1620 (3 C=N), 1646 (amidic C=O), 3410–3230 (2 NH and NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.81 (dd, J = 13.7 and 4.7 Hz, 1H), 2.90 (dd, J = 9.5 and 13.7 Hz, 1H), 5.10 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 8.56 (s, 2H, NH2 Deutr. Exch), 10.59 (s, 1H, NH Deutr. Exch); 13C-NMR (75 MHz, DMSO-d6): 41.0 (CH2), 51.9 (CH), 127.0, 127.6, 128.1, 128.,2, 128.8, 129.,3, 131.1, 140.5 (C–Ar), 144.6, 149.1, 164.5 (3 C=N–), 160.4 (C=O), 181.2 (C=S); MS (m/z, %): 397.08 (M+, 20). Anal. Calcd for C17H15N7OS2 (397.48): C, 51.37; H, 3.80; N, 24.67%. Found: C, 51.10; H, 3.59; N, 24.32%.
4-Amino-2-((4-oxo-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazin-3(4H)-ylidene)hydrazono)-6-phenyl-2H-1,3-thiazine-5-carbonitrile (5). A mixture of 3 (0.39 g, 1 mmol), and benzylidinemalononitrile (0.15 g, 1 mmol) in dioxane (20 mL) containing a few drops of piperidine (0.3 mL) was refluxed for 6 h. Then, the reaction mixture was left to cool to rt. The solid produced was collected, washed with MeOH, and recrystallized from DMF to produce 5 as yellowish-green solid with a 75% yield; mp 165–167 °C; IR (KBr): (cm−1) 685 (C–S–C), 1610–1620 (4 C=N), 1672 (amidic C=O), 2219 (C≡N), 3186–3325 (NH, NH2); MS (m/z, %): 549.12 (M+, 18). Anal. Calcd for C27H19N9OS2 (549.63): C, 59.00; H, 3.48; N, 22.94%. Found: C, 58.85; H, 3.25; N, 22.67%.
3-((5-Acetyl-4-methylthiazol-2(3H)-ylidene)hydrazono)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazin-4(3H)-one (6). 3-Chloro-2,4-pentanedione (0.13 mL, 1 mmol) was added to a solution of compound 3 (0.39 g, 1 mmol) in EtOH (30 mL), and the mixture was refluxed for 6 h. The reaction mixture was cooled down, diluted with H2O (50 mL), and sodium acetate (0.49 g, 6 mmol) was added, and the mixture was stirred for 15 min at rt. The obtained product was filtered, washed with H2O, dried, and recrystallized from EtOH to produce 6 as a yellow powder with a 65% yield; mp 145–147 °C; IR (KBr): (cm−1) 681, 690 (2C–S–C), 1610–1620 (4C=N), 1653, 1710 (2 C=O), 3185–3215 (2 NH); 1H-NMR (300 MHz, DMSO-d6): δ 2.51 (s, 3H, Me), 2.72 (s, 3H, MeCO), 2.82 (dd, J = 12.4 and 4.6 Hz, 1H), 2.93 (dd, J = 8.4 and 12.3 Hz, 1H), 5.01 (dd, J = 8.6 and 3.8 Hz, 1H), 7.68–7.79 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 10.21, 10.83 (s, 2H, 2NHDeutr. Exch); 13C-NMR (75 MHz, DMSO-d6): 18.4, 25.7 (2Me), 41.0 (CH2), 52.3 (CH), 127.1, 127.4, 128.0, 128.2, 128.5, 129.4, 131.1, 140.5 (C–Ar), 144.6, 158.0, 158.3, 164.5 (4 C=N–), 160.3, 192.6 (C=O); MS (m/z, %): 477.09 (M+, 34%). Anal. Calcd for C22H19N7O2S2 (477.56): C, 55.33; H, 4.01; N, 20.53%. Found: C, 55.19; H, 3.81; N, 20.18%.
Methyl 2-(4-oxo-2-(-(4-oxo-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4] triazin-3(4H)-ylidene)hydrazono)thiazolidin-5-ylidene)acetate (8). To a well-stirred solution of 3 (0.39 g, 1 mmol) in dry dioxane (20 mL), a solution of dimethyl acetylenedicarboxylate (0.11 mL, 1 mmol) in dioxane (20 mL) was added dropwise. The mixture was stirred for further 2 h at rt, then the reaction mixture was refluxed for a further 8 h (the reaction was monitored by TLC analyses). The solvent was evaporated under a vacuum; the formed solid was filtered and then recrystallized from MeOH–H2O to produce 8 as yellowish-brown powder in an 80% yield; mp 228–230 °C; IR (KBr): (cm−1) 681, 694 (2C–S–C), 1610–1620 (4C=N), 1653, 1710, 1730 (3 C=O), 3185–3215 (2 NH); 1H-NMR (300 MHz, DMSO-d6): δ 2.72 (s, 3H, MeCO), 2.82 (dd, J = 12.4 and 4.6 Hz, 1H), 2.93 (dd, J = 8.4 and 12.3 Hz, 1H), 3.77 (s, 3H, Me), 5.01 (dd, J = 8.6 and 3.8 Hz, 1H), 6.20 ( s, 1H, =C–H), 7.68–7.79 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 10.21, 10.81 (s, 2H, 2NHDeutr. Exch); MS (m/z, %): 507.08 (M+, 16%). Anal. Calcd for C22H17N7O4S2 (507.54): C, 52.06; H, 3.38; N, 19.32%. Found: C, 51.98; H, 3.15; N, 19.18%.
3-Chloro-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-4H-pyrimido[2,1-c][1,2,4]triazin-4-one (
9) and
3-hydrazinyl-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-4H-pyrimido[2,1-c][1,2,4]triazin-4-one (
10). They were synthesized according to our reported works [
25,
26].
Compound 9. Off-white powder with a 75% yield; mp 176–178 °C; IR (KBr): (cm−1) 685 (C–Cl), 687 (C–S–C), 1610–1620 (3 C=N), 1645 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 13.7 and 4.7 Hz, 1H), 2.91 (dd, J = 9.5 and 13.7 Hz, 1H), 5.14 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H); 13C-NMR (75 MHz, DMSO-d6): 41.0 (CH2), 51.8 (CH), 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 131.1, 140.5 (C–Ar), 144.6, 164.5 (2 C=N–), 149.1, (=C–Cl), 160.3 (C=O), 181.2 (C=S); MS (m/z, %): 342.03 (M+, 55), 344.01 (M++2, 18). Anal. Calcd for C16H11ClN4OS (342.80): C, 56.06; H, 3.23; N, 16.34%. Found: C, 55.98; H, 3.01; N, 16.21%.
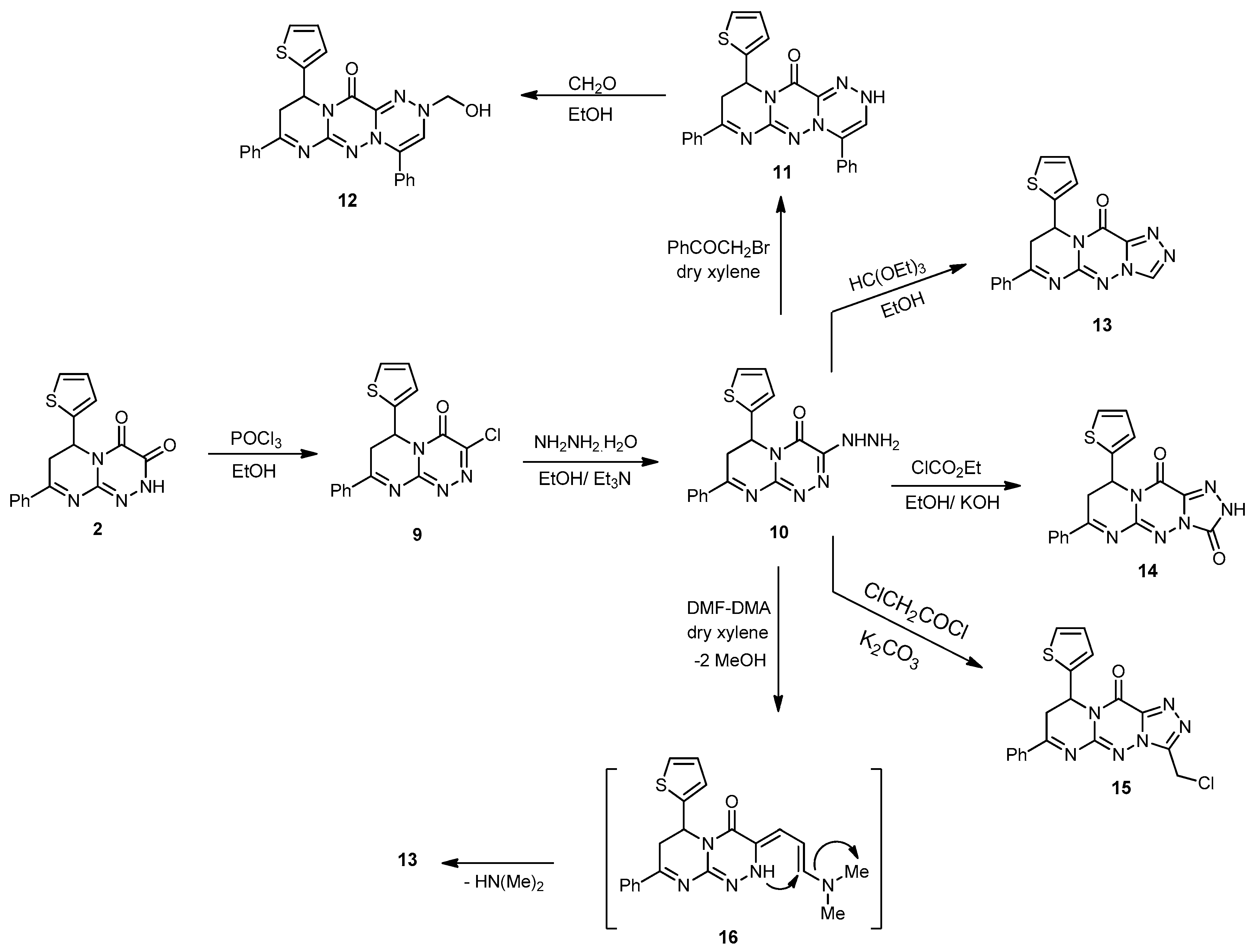
Compound 10. Yellowish-brown powder with an 80% yield; mp 220–222 °C; IR (KBr): (cm−1) 687 (C–S–C), 1610–1620 (3 C=N), 1645 (amidic C=O), 3211–3351 (NH, NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.85 (dd, J = 13.7 and 4.7 Hz, 1H), 2.91 (dd, J = 9.5 and 13.7 Hz, 1H), 4.82 (s, 2H, NH2Deutr. Exch), 5.14 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.25 (d, J = 4.6, 1H, Thioph.(C5)-H), 8.32 (s, 1H, NH Deutr. Exch); MS (m/z, %): 338.08 (M+, 30). Anal. Calcd for C16H14N6OS (338.39): C, 56.79; H, 4.17; N, 24.84%. Found: C, 56.51; H, 4.01; N, 24.63%.
4,8-Ciphenyl-10-(thiophen-2-yl)-9,10-dihydropyrimido[2,1-c][1,2,4]triazino[3,4-f][1,2,4]triazin-12(2H)-one (11). To a solution of 10 (0.33 g, 1 mmol) in absolute EtOH (25 mL), phenacyl bromide (0.19 g, 1 mmol) was added. The mixture was refluxed for 7 h (monitored by TLC), then left to cool. The precipitate formed was filtered, washed with MeOH, and recrystallized from EtOH to produce 11 as yellow crystals with a 78% yield; mp 242–244 °C; IR (KBr): (cm−1) 689 (C–S–C), 1611–1624 (3 C=N), 1648 (amidic C=O), 3231(NH); 1H-NMR (300 MHz, DMSO-d6): δ 2.81 (dd, J = 13.7 and 4.7 Hz, 1H), 2.90 (dd, J = 9.5 and 13.7 Hz, 1H), 4.94 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 13H, Ar–H, Triazin.(C6)-H and Thioph.(C3,4)-2H, 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H), 8.32 (s, 1H, NH Deutr. Exch); MS (m/z, %): 438.13 (M+, 21). Anal. Calcd for C16H12N4O2S (438.50): C, 65.74; H, 4.14; N, 19.17%. Found: C, 65.50; H, 4.07; N, 19.03%.
2-(Hydroxymethyl)-4,8-diphenyl-10-(thiophen-2-yl)-9,10-dihydropyrimido[2,1-c][1,2,4]triazino [3,4-f][1,2,4]triazin-12(2H)-one (12). A mixture of 11 (0.86 g, 2 mmol) and CH2O solution (0.12 mL, 4 mmol) in EtOH (30 mL) was refluxed for 8 h. The precipitate obtained on cooling was filtered, washed several times with H2O, and recrystallized from DMF to yield 12 as off-white crystals with a 75% yield; mp 182–184 °C; IR (KBr): (cm−1) 685 (C–S–C), 1609–1621(3 C=N), 1645 (amidic C=O), 3315 (OH); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 13.7 and 4.7 Hz, 1H), 2.92 (dd, J = 9.5 and 13.7 Hz, 1H), 4.94 (dd, J = 9.5 and 3.9 Hz, 1H), 4.32 (s, 2H, CH2-OH), 5.19 (s, 1H, OH Deutr. Exch), 7.67–7.78 (m, 13H, Ar–H, Triazin.(C6)-H and Thioph.(C3,4)-2H), 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 468.15 (M+, 60). Anal. Calcd for C25H20N6O2S (468.53): C, 64.09; H, 4.30; N, 17.94%. Found: C, 64.11; H, 4.20; N, 17.59%.
7-Phenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(9H)-one (13). Method A: A mixture of compound 10 (0.33 g, 1 mmol) and HC(OEt)3 (15 mL) was refluxed for 6 h. The obtained solid on cooling was filtered, dried, and recrystallized from EtOH to yield compound 13 (76%).
Method B: A solution of 10 (0.33 g, 1 mmol) and DMF-DMA (0.22 mL, 2 mmol) in dry xylene (25 mL) was refluxed for 8 h. The solvent was then removed in vacuo, and the solid so obtained was triturated with petroleum ether, collected by filtration, and recrystallized from EtOH to yield 13 as a brown powder with a 62% yield; mp 212–214 °C; IR (KBr): (cm−1) 687 (C–S–C), 1610–1620 (4 C=N), 1645 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.81 (dd, J = 13.5 and 4.5 Hz, 1H), 2.89 (dd, J = 9.2 and 13.5 Hz, 1H), 5.14 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H), 9.52 (s, 1H, Triazol.(C3)-H); 13C-NMR (75 MHz, DMSO-d6): 32.1, 40.1 (2CH2), 51.8 (CH), 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 131.1, 140.5 (C–Ar), 153.5, 157.6, 160.5, 164.5 (4 C=N–), 160.3 (C=O); MS (m/z, %): 348.08 (M+, 15). Anal. Calcd for C16H12N4O2S (348.38): C, 58.61; H, 3.47; N, 24.12%. Found: C, 58.32; H, 3.12; N, 24.01%.
7-Phenyl-9-(thiophen-2-yl)-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazine-3,11-dione (14). To an ice-cooled suspension of KOH (0.11 g, 2 mmol) in absolute EtOH (20 mL), compound 10 (0.66 g, 2 mmol) and ethyl chloroformate (2 mL) were added. The mixture was refluxed for 5 h, then allowed to cool and poured into H2O. The precipitate that obtained was filtered and recrystallized from MeOH to afford 14 as brown crystals in a 65% yield; mp 257–259 °C; IR (KBr): (cm−1) 687 (C–S–C), 1610–1620 (3 C=N), 1642–1655 (2 amidic C=O), 3210 (NH); 1H-NMR (300 MHz, DMSO-d6): δ 2.80 (dd, J = 13.7 and 4.7 Hz, 1H), 2.88 (dd, J = 9.5 and 13.7 Hz, 1H), 5.13 (dd, J = 9.5 and 3.9 Hz, 1H), 7.65–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H), 9.41 (s, 1H, NH Deutr. Exch); MS (m/z, %): 364.07 (M+, 45). Anal. Calcd for C17H12N6O2S (364.38): C, 56.04; H, 3.32; N, 23.06%. Found: C, 56.11; H, 3.03; N, 23.01%.
3-(Chloromethyl)-7-phenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4] triazin-11(9H)-one (15). A mixture of 10 (0.33 g, 1 mmol) and chloroacetyl chloride (0.11 mL, 1 mmol) in dry dioxane (25 mL) in the presence of anhydrous K2CO3 (0.13 g, 1 mmol) was refluxed in a water bath for 8 h. The reaction mixture was evaporated under reduced pressure, and the residue was triturated with H2O. The formed solid was filtered off, washed with H2O, dried, and recrystallized from EtOH to produce 15 as brown powder with a 76% yield; mp 260–262 °C; IR (KBr): (cm−1) 659 (C–Cl), 687 (C–S–C), 1610–1620 (4 C=N), 1645 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.82 (dd, J = 13.5 and 4.5 Hz, 1H), 2.90 (dd, J = 9.2 and 13.5 Hz, 1H), 4.23 (s, 2H, CH2), 5.16 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.20 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 398.05 (M++2, 15), 396.02 (42): Anal. Calcd for C18H13ClN6OS (396.85): C, 54.48; H, 3.30; N, 21.18%. Found: C, 54.37; H, 3.11; N, 21.04%.
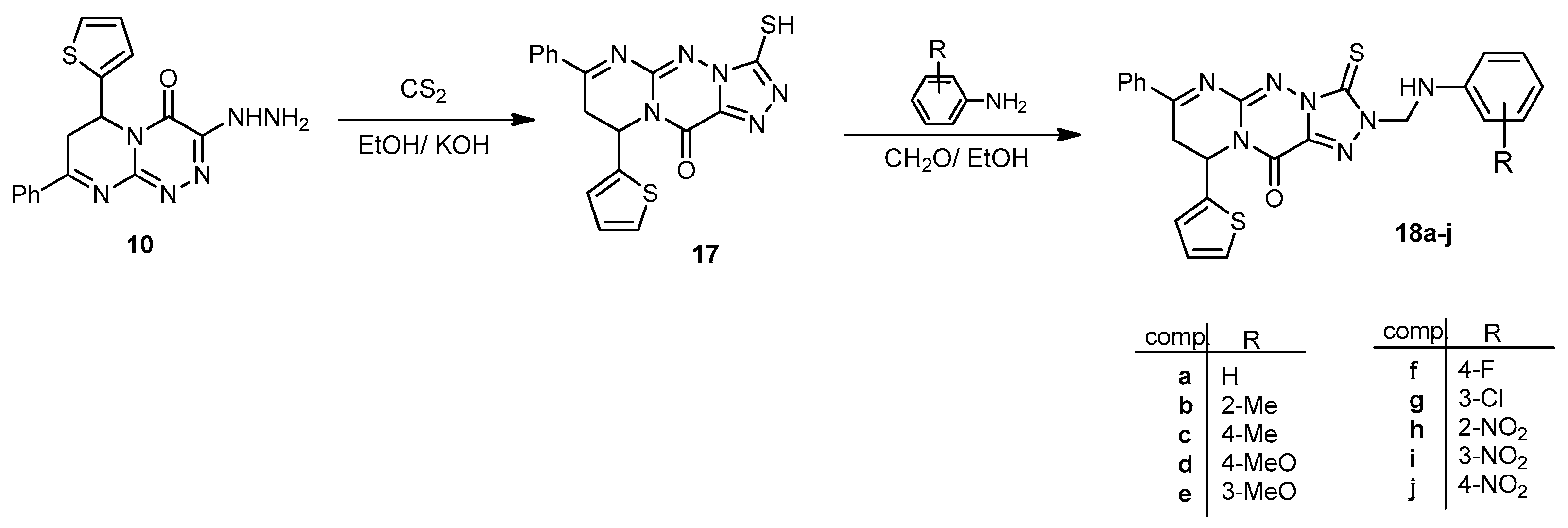
3-Mercapto-7-phenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(9H)-one (17). To a stirred suspension of compound 10 (0.66 g, 2 mmol) in EtOH (20 mL), ethanolic potassium hydroxide (20 mL, 0.01 mol) and CS2 (5 mL) were added dropwise. The reaction mixture was then refluxed for 7 h. After cooling and evaporation of the solvent under reduced pressure, the potassium salt obtained was dissolved in H2O and acidified with 2 N aqueous HCl. The precipitate obtained was filtered and recrystallized from EtOH to yield 17 as a colorless powder in a 78% yield; mp 232–234 °C; IR (KBr): (cm−1) 687 (C–S–C), 1610–1620 (4 C=N), 1641 (amidic C=O), 2580 (S–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.81 (dd, J = 13.5 and 4.3 Hz, 1H), 2.90 (dd, J = 9.3 and 13.6 Hz, 1H), 5.14 (dd, J = 9.5 and 3.9 Hz, 1H), 7.62–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.18 (d, J = 4.5, 1H, Thioph.(C5)-H), 11.52 (s, 1H, SH Deutr. Exch); 13C-NMR (75 MHz, DMSO-d6): 32.1, 40.1 (2CH2), 51.8 (CH), 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 131.1, 140.5 (C–Ar), 153.5, 157.6, 164.5 (3 C=N–), 160.3 (C=O), 167.9 (N=C–SH); MS (m/z, %): 380.05 (M+, 20). Anal. Calcd for C17H12N6OS2 (380.45): C, 53.67; H, 3.18; N, 22.09%. Found: C, 53.38; H, 3.11; N, 22.01%.
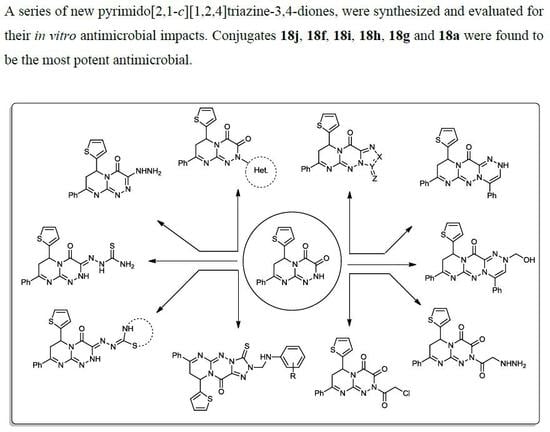
General procedure for the synthesis of 2-(((substitutedphenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-ones (18a–j). A mixture of the thiol compound 17 (0.38 g, 1 mmol); 37% HCHO solution (2 mL); and the selected substituted anilines (1 mmol) in EtOH (30 mL) containing a few drops of HCl was stirred at rt for 7–10 h (monitored by TLC). The obtained solid was collected and recrystallized from the proper solvent to yield the respective products (18a–j).
7-Phenyl-2-((phenylamino)methyl)-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c] [1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18a). A yellowish powder from MeOH with a 72% yield; mp 218–220 °C; IR (KBr): (cm−1) 687 (C–S–C), 1609–1624 (3 C=N), 1643 (amidic C=O), 1281 C=S), 3219 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.79 (dd, J = 12.9 and 4.2 Hz, 1H), 2.86 (dd, J = 8.8 and 12.9 Hz, 1H), 4.82 (s, 1H, N–H Deutr. Exch), 5.09 (dd, J = 8.9 and 3.8 Hz, 1H), 5.42 (s, 2H, CH2), 7.68–7.78 (m, 12H, Ar–H and Thioph.(C3,4)-2H), 8.18 (d, J = 4.5, 1H, Thioph.(C5)-H); 13C-NMR (75 MHz, DMSO-d6): 40.1, 76.1 (2CH2), 52.2 (CH), 113.4, 120.8, 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 129.5, 131.1, 140.6 (C–Ar), 142.6, 144.6, 164.5 (3 C=N–), 160.2 (C=O), 178.9 (C=S); MS (m/z, %): 485.10 (M+, 23). Anal. Calcd for C24H19N7OS2 (485.11): C, 59.36; H, 3.94; N, 20.19%. Found: C, 59.14; H, 3.78; N, 20.06%.
7-Phenyl-9-(thiophen-2-yl)-3-thioxo-2-((o-tolylamino)methyl)-8,9-dihydro-2H-pyrimido[2,1-c] [1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18b). A yellowish powder from acetone with a 79% yield; mp 202–204 °C; IR (KBr): (cm−1) 686 (C–S–C), 1610–1625 (3 C=N), 1646 (amidic C=O), 1285 C=S), 3221 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.43 (s, 3H, Me), 2.82 (dd, J = 13.2 and 4.4 Hz, 1H), 2.87 (dd, J = 8.9 and 12.8 Hz, 1H), 4.81 (s, 1H, N–H Deutr. Exch), 5.10 (dd, J = 8.7 and 3.6 Hz, 1H), 5.41 (s, 1H, CH2), 7.65–7.79 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.20 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 499.01 (M+, 36). Anal. Calcd for C25H21N7OS2 (499.12): C, 60.10; H, 4.24; N, 19.62%. Found: C, 60.01; H, 4.17; N, 19.51%.
7-Phenyl-9-(thiophen-2-yl)-3-thioxo-4-((o-tolylamino)methyl)-8,9-dihydro-2H-pyrimido[2,1-c] [1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18c). A yellowish powder from MeOH with a 75% yield; mp 165–167 °C; IR (KBr): (cm−1) 686 (C–S–C), 1610–1625 (3 C=N), 1646 (amidic C=O), 1285 C=S), 3221 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.33 (s, 3H, Me), 2.81 (dd, J = 13.2 and 4.4 Hz, 1H), 2.89 (dd, J = 8.9 and 12.8 Hz, 1H), 4.80 (s, 1H, N–H Deutr. Exch), 5.09 (dd, J = 8.7 and 3.6 Hz, 1H), 5.38 (s, 1H, CH2), 7.65–7.79 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.22 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 499.02 (M+, 51). Anal. Calcd for C25H21N7OS2 (499.12): C, 60.10; H, 4.24; N, 19.62%. Found: C, 60.02; H, 4.15; N, 19.49%.
2-(((4-Methoxyphenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18d). A yellowish-brown solid EtOH in a 70% yield; mp 196–198 °C; IR (KBr): (cm−1) 685 (C–S–C), 1610–1620 (3 C=N), 1643 (amidic C=O), 1291 C=S), 3281 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 12.8 and 4.1 Hz, 1H), 2.90 (dd, J = 8.8 and 12.7 Hz, 1H), 3.81 (s, 3H, OMe), 4.81 (s, 1H, N–H Deutr. Exch), 5.10 (dd, J = 8.7 and 3.6 Hz, 1H), 5.40 (s, 1H, CH2), 7.60–7.79 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.4, 1H, Thioph.(C5)-H); MS (m/z, %): 515.19 (M+, 19). Anal. Calcd for C25H21N7O2S2 (515.61): C, 58.24; H, 4.11; N, 19.02%. Found: C, 58.11; H, 3.98; N, 18.95%.
2-(((3-Methoxyphenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18e). Yellow crystals from EtOH with an 80% yield; mp 210–212 °C; IR (KBr): (cm−1) 680 (C–S–C), 1613–1624 (3 C=N), 1641 (amidic C=O), 1290 C=S), 3283 (N–H); MS (m/z, %): 515.13 (M+, 23). Anal. Calcd for C25H21N7O2S2 (515.61): C, 58.24; H, 4.11; N, 19.02%. Found: C, 58.15; H, 3.85; N, 18.98%.
2-(((3-Fluorophenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18f). A yellowish powder from acetone in a 75% yield; mp 267–269 °C; IR (KBr): (cm−1) 681 (C–S–C), 1610–1620 (3 C=N), 1643 (amidic C=O), 1291 C=S), 3163 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.84 (dd, J = 12.8 and 4.1 Hz, 1H), 2.94 (dd, J = 8.8 and 12.7 Hz, 1H), 4.83 (s, 1H, N–H Deutr. Exch), 4.09 (dd, J = 8.7 and 3.6 Hz, 1H), 5.43 (s, 1H, CH2), 7.10–8.09 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.20 (d, J = 4.4, 1H, Thioph.(C5)-H); MS (m/z, %): 503.10 (M+, 23). Anal. Calcd for C24H18FN7OS2 (503.57): C, 57.24; H, 3.60; N, 19.47%. Found: C, 57.13; H, 3.36; N, 19.25%.
2-(((3-Chlorophenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18g). A pale brown powder from DMF–MeOH (1:3) in a 77% yield; mp 276–278 °C; IR (KBr): (cm−1) 715 (C–Cl), 681 (C–S–C), 1610–1620 (3 C=N), 1643 (amidic C=O), 1291 C=S), 3163 (N–H); MS (m/z, %): 522.02 (M+ +2, 9), 520.01 (30). Anal. Calcd for C24H18ClN7OS2 (520.03): C, 55.43; H, 3.49; N, 18.85%. Found: C, 55.19; H, 3.24; N, 18.63%.
2-(((2-Nitrophenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18h). A yellow powder from EtOH in a 71% yield; mp 203–205 °C; IR (KBr): (cm−1) 681 (C–S–C), 1610–1620 (3 C=N), 1390, 1520 (NO2), 1643 (amidic C=O), 1291 C=S), 3253 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 12.8 and 4.1 Hz, 1H), 2.90 (dd, J = 8.8 and 12.7 Hz, 1H), 4.81 (s, 1H, N–H Deutr. Exch), 5.10 (dd, J = 8.7 and 3.6 Hz, 1H), 5.45 (s, 1H, CH2), 7.60–8.09 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.4, 1H, Thioph.(C5)-H); MS (m/z, %): 530.09 (M+, 35). Anal. Calcd for C24H18ClN7OS2 (530.58): C, 54.33; H, 3.42; N, 21.12%. Found: C, 54.24; H, 3.19; N, 21.01%.
2-(((3-Nitrophenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c] [1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18i). A yellow powder from EtOH in a 71% yield; mp 154–456 °C; IR (KBr): (cm−1) 681 (C–S–C), 1610–1620 (3 C=N), 1390, 1520 (NO2), 1643 (amidic C=O), 1291 C=S), 3253 (N–H); MS (m/z, %): 530.09 (M+, 55). Anal. Calcd for C24H18ClN7OS2 (530.58): C, 54.33; H, 3.42; N, 21.12%. Found: C, 54.24; H, 3.19; N, 21.01%.
2-(((4-Nitrophenyl)amino)methyl)-7-phenyl-9-(thiophen-2-yl)-3-thioxo-8,9-dihydro-2H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(3H)-one (18j). A yellow powder from MeOH–H2O in a 78% yield; mp 281–283 °C; IR (KBr): (cm−1) 681 (C–S–C), 1610–1620 (3 C=N), 1390, 1520 (NO2), 1643 (amidic C=O), 1291 (C=S), 3253 (N–H); 1H-NMR (300 MHz, DMSO-d6): δ 22.83 (dd, J = 12.8 and 4.1 Hz, 1H), 2.90 (dd, J = 8.8 and 12.7 Hz, 1H), 3.81 (s, 3H, OMe), 4.81 (s, 1H, N–H Deutr. Exch), 5.10 (dd, J = 8.7 and 3.6 Hz, 1H), 5.40 (s, 1H, CH2), 7.60–7.79 (m, 11H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.4, 1H, Thioph.(C5)-H); 40.1, 76.1 (2CH2), 52.2 (CH), 113.4, 120.8, 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 129.5, 131.1, 140.6 (C–Ar), 142.6, 144.6, 164.5 (3 C=N–), 160.2 (C=O), 178.9 (C=S); MS (m/z, %): 530.09 (M+, 40). Anal. Calcd for C24H18ClN7OS2 (530.58): C, 54.33; H, 3.42; N, 21.12%. Found: C, 54.18; H, 3.25; N, 20.98%.
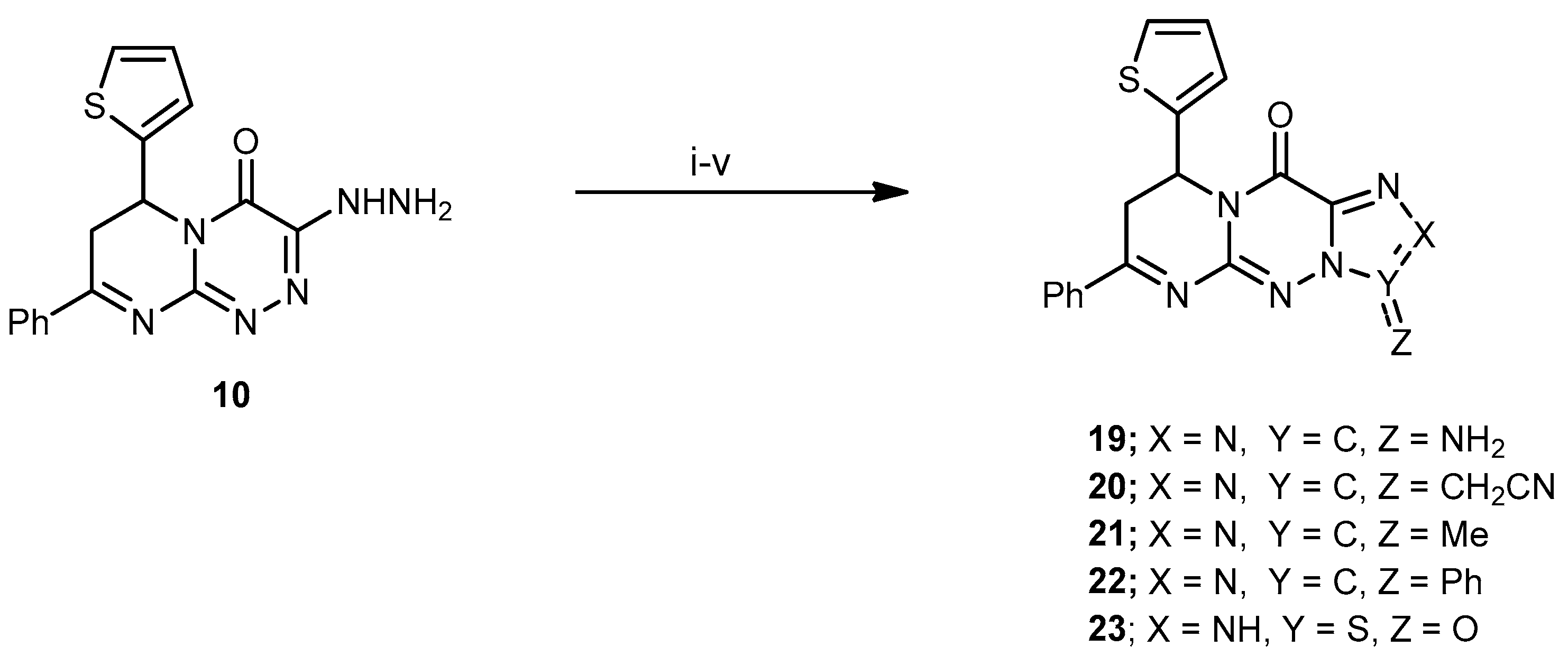
3-Amino-7-phenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(9H)-one (19). A mixture of compound 10 (0.33 g, 1 mmol) and KSCN (0.48 g, 5 mmol) was refluxed for 7 h in glacial AcOH (25 mL), and the reaction mixture was allowed to cool to rt, then poured into H2O. The solid formed was collected by filtration, dried, and recrystallized from EtOH to produce 19 as a light yellow solid in a 76% yield; mp 205–207 °C; IR (KBr): (cm−1) 683 (C–S–C), 1613–1625 (4 C=N), 1650 (amidic C=O), 3340 (NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 13.7 and 4.7 Hz, 1H), 2.91 (dd, J = 9.5 and 13.7 Hz, 1H), 5.10 (dd, J = 9.5 and 3.9 Hz, 1H), 6.86 (s, 2H, NH2 Deutr. Exch), 7.69–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.21 (d, J = 4.4, 1H, Thioph.(C5)-H); MS (m/z, %): 363.09 (M+, 50). Anal. Calcd for C17H13N7OS (363.40): C, 56.19; H, 3.61; N, 26.98%. Found: C, 56.01; H, 3.51; N, 26.78%.
2-(11-Oxo-7-phenyl-9-(thiophen-2-yl)-9,11-dihydro-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f] [1,2,4]triazin-3-yl)acetonitrile (20). A mixture of 10 (0.33 g, 1 mmol) and ethyl 2-cyanoacetate (0.11 mL, 1 mmol) in EtOH (30 mL) containing piperidine (0.3 mL) was refluxed for 8 h, then left to cool at rt. The solid product was filtered off, washed with cold EtOH, and recrystallized from EtOH to yield 20 as pale-yellow crystals at a 65% yield; mp 178–180 °C; IR (KBr): (cm−1) 687 (C–S–C), 1610–1620 (4 C=N), 1686 (amidic C=O), 2218 (C≡N); 1H-NMR (300 MHz, DMSO-d6): δ 2.81 (dd, J = 13.5 and 4.5 Hz, 1H), 2.90 (dd, J = 9.2 and 13.5 Hz, 1H), 3.83 (s, 2H, CH2), 5.16 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H); 13C-NMR (75 MHz, DMSO-d6): 13.1, 40.1 (2CH2), 52.5 (CH), 127.0, 127.6, 128.1, 128.2, 128.8, 129.3, 131.1, 140.5 (C–Ar), 153.5, 157.6, 160.5, 164.5 (4 C=N–), 160.3 (C=O); MS (m/z, %): 387.08 (M+, 75). Anal. Calcd for C19H13N7OS (387.42): C, 58.90; H, 3.38; N, 25.31%. Found: C, 58.71; H, 3.15; N, 25.14%.
3-Methyl-7-phenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(9H)-one (21). Compound 10 (0.33 g, 1 mmol) was dissolved in Ac2O (15 mL) and refluxed for 6 h. The reaction mixture was allowed to cool to rt and poured onto H2O (50 mL). The resulted solid was filtered off, washed with H2O several times, dried, and recrystallized using CHCl3 to produce 21 as a brown powder in a 60% yield; mp 156–158 °C; IR (KBr): (cm−1) 685 (C–S–C), 1611–1625 (4 C=N), 1645 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.43 (s, 3H, Me), 2.81 (dd, J = 13.5 and 4.5 Hz, 1H), 2.89 (dd, J = 9.2 and 13.5 Hz, 1H), 5.14 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.25 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 362.09 (M+, 42). Anal. Calcd for C18H14N6OS (362.41): C, 59.65; H, 3.89; N, 23.19%. Found: C, 59.47; H, 3.62; N, 23.07%.
3,7-Diphenyl-9-(thiophen-2-yl)-8H-pyrimido[2,1-c][1,2,4]triazolo[3,4-f][1,2,4]triazin-11(9H)-one (22). A mixture of compound 10 (0.33 g, 1 mmol, and benzoyl chloride (15 mL) was refluxed for 4 h. The reaction mixture was allowed to cool to rt; the resulting solid was filtered off, washed with cold MeOH, dried, and recrystallized using MeOH to yield 22 as orange crystals at a 76% yield; mp 275–277 °C; IR (KBr): (cm−1) 680 (C–S–C), 1614–1630 (4 C=N), 1641 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.45 (s, 3H, Me), 2.83 (dd, J = 13.5 and 4.5 Hz, 1H), 2.92 (dd, J = 9.2 and 13.5 Hz, 1H), 5.16 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 12H, Ar–H and Thioph.(C3,4)-2H), 8.22 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 424.11 (M+, 50). Anal. Calcd for C16H12N4O2S (424.48): C, 65.08; H, 3.80; N, 19.80%. Found: C, 65.01; H, 3.71; N, 19.51%.
7-Phenyl-9-(thiophen-2-yl)-8,9-dihydropyrimido[2,1-c][1,2,3,5]thiatriazolo[4,5-f][1,2,4]triazin-11 (2H)-one 3-oxide (23). A solution of compound 10 (0.33 g, 1 mmol) in EtOH (25 mL) treated with EtONa (0.23 g, 10 mmol sodium metal in 5 mL absolute EtOH) and thionyl chloride was heated in a water bath for 8 h. The mixture was diluted with H2O and extracted with ethyl acetate; the organic layers were dried over anhydrous Na2SO4; the ethyl acetate was removed under reduced pressure and recrystallized by EtOH to produce 23 as a yellow solid in a 65% yield; mp 231–233 °C; IR (KBr): (cm−1) 685 (C–S–C), 1611–1625 (4 C=N), 1331 (S=O), 1645 (amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.80 (dd, J = 13.5 and 4.5 Hz, 1H), 2.90 (dd, J = 9.2 and 13.5 Hz, 1H), 5.11 (dd, J = 9.5 and 3.9 Hz, 1H), 7.65–7.79 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 10.01 (s, 1H, NH Deutr. Exch); MS (m/z, %): 384.05 (M+, 10). Anal. Calcd for C16H12N6O2S2 (384.44): C, 49.99; H, 3.15; N, 21.86%. Found: C, 49.78; H, 3.02; N, 21.75%.
2-(2-Chloroacetyl)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazine-3,4-dione (24). A mixture of chloroacetyl chloride (0.02 mol) in absolute EtOH (20 mL) was added dropwise during 30 min to a well-stirred solution of 2 (0.32 g, 1 mmol) in absolute EtOH (30 mL) containing Et3N (0.5 mL). The mixture was stirred for a further 1 h at 80 °C; the reaction mixture was allowed to cool to rt, and then poured onto ice-cooled H2O. The produced solid was filtered off, dried, and recrystallized from EtOH/dioxane (2:1) to produce 24 as a yellowish-green powder in an 80% yield; mp 128–130 °C; IR (KBr): (cm−1) 688 (C–S–C), 715 (C–Cl), 1610–1626 (2 C=N), 1641–1682 (3 amidic C=O); 1H-NMR (300 MHz, DMSO-d6): δ 2.82 (dd, J = 13.7 and 4.7 Hz, 1H), 2.91 (dd, J = 9.5 and 13.8 Hz, 1H), 4.21 (s, 2H, CH2), 5.10 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.04 (d, J = 4.5, 1H, Thioph.(C5)-H); 13C-NMR (75 MHz, DMSO-d6): 41.0, 42.4 (2CH2), 51.8 (CH), 127.1, 127.4, 128.0, 128.2, 128.5, 129.4, 131.1, 140.5 (C–Ar), 144.6, 164.5 (2 C=N–), 157.1, 159.0, 165.7 (3C=O); MS (m/z, %): 402.05 (M++2, 20), 400.04 (58). Anal. Calcd for C18H13ClN4O3S (400.84): C, 53.94; H, 3.27; N, 13.98%. Found: C, 53.76; H, 3.11; N, 13.75%.
2-(2-Hydrazinylacetyl)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazine-3,4-dione (25). It was prepared according to the general procedure described for compound 10 as a reddish-brown powder from EtOH in a 76% yield; mp 227–229 °C; IR (KBr): (cm−1) 686 (C–S–C), 1610–1626 (2 C=N), 1641–1682 (3 amidic C=O), 3186–3350 (NH and NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.79 (dd, J = 13.7 and 4.7 Hz, 1H), 2.86 (dd, J = 9.5 and 13.8 Hz, 1H), 3.12 (s, 2H, CH2), 4.50 (s, 2H, NH2Deutr. Exch), 5.10 (dd, J = 9.5 and 3.9 Hz, 1H), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H, 8.04 (d, J = 4.5, 1H, Thioph.(C5)-H), 8.31 (s, 1H, NHDeutr. Exch); MS (m/z, %): 396.10 (M+, 15). Anal. Calcd for C18H16N6O3S (396.42): C, 54.54; H, 4.07; N, 21.20%. Found: C, 54.36; H, 3.87; N, 21.11%.
3-Amino-5-(3,4-dioxo-8-phenyl-6-(thiophen-2-yl)-3,4,6,7-tetrahydro-2H-pyrimido[2,1-c][1,2,4] triazin-2-yl)–1,6-dihydropyridazine-4-carbonitrile (26). To a solution of 25 (0.78 g, 1 mmol) in pyridine (15 mL), malononitrile (0.12 g, 2 mmol) was added, and the mixture was refluxed for 6 h. The reaction mixture was left to cool at rt. The precipitate that formed was collected and recrystallized from CHCl3 to yield 26 as yellow-orange powder in a 75% yield; mp 221–223 °C; IR (KBr): (cm−1) 685 (C–S–C), 1610–1620 (3 C=N), 1665–1686 (2amidic C=O), 2218 (C≡N), 3186–3350 (NH and NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.83 (dd, J = 13.5 and 4.5 Hz, 1H), 2.94 (dd, J = 9.2 and 13.5 Hz, 1H), 3.34 (s, 2H, Pyridaz.(C6)-2H), 5.12 (dd, J = 9.5 and 3.9 Hz, 1H), 6.81 (s, H, NHDeutr. Exch), 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 8.50 (s, 2H, NH2Deutr. Exch); MS (m/z, %): 444.10 (M+, 62). Anal. Calcd for C21H16N8O2S (444.47): C, 56.75; H, 3.63; N, 25.21%. Found: C, 56.54; H, 3.41; N, 25.12%.
2-(5-Hydroxy-7-methyl-2,3-dihydropyrimido[4,5-c]pyridazin-4-yl)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazine-3,4-dione (27). A solution of 26 (0.44 g, 1 mmol) in an Ac2O/pyridine mixture (30 mL, (2:1)) was heated in a water bath for 8 h, then cooled and poured into an ice/H2O mixture. The precipitate thus obtained was filtered off, washed several times with H2O, dried, and recrystallized from EtOH to yield 27 as yellowish crystals in a 76% yield; mp 240–242 °C; IR (KBr): (cm−1) 685 (C–S–C), 1615–1630 (4 C=N), 1662, 1679 (2amidic C=O), 3251 (NH), 3453 (br OH); 1H-NMR (300 MHz, DMSO-d6): δ 1.04 (s, 3H, Me), 2.79 (dd, J = 13.5 and 4.5 Hz, 1H), 2.87 (dd, J = 9.2 and 13.5 Hz, 1H), 3.32 (s, 2H, Pyridaz.(C6)-2H), 5.10 (dd, J = 9.5 and 3.9 Hz, 1H), 6.92 (s, H, NHDeutr. Exch); 7.67–7.78 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 9.31 (s, 1H, OHDeutr. Exch); MS (m/z, %): 486.12 (M+, 55%). Anal. Calcd for C23H18N8O3S (486.51): C, 56.78; H, 3.73; N, 23.03%. Found: C, 56.50; H, 3.61; N, 23.01%.
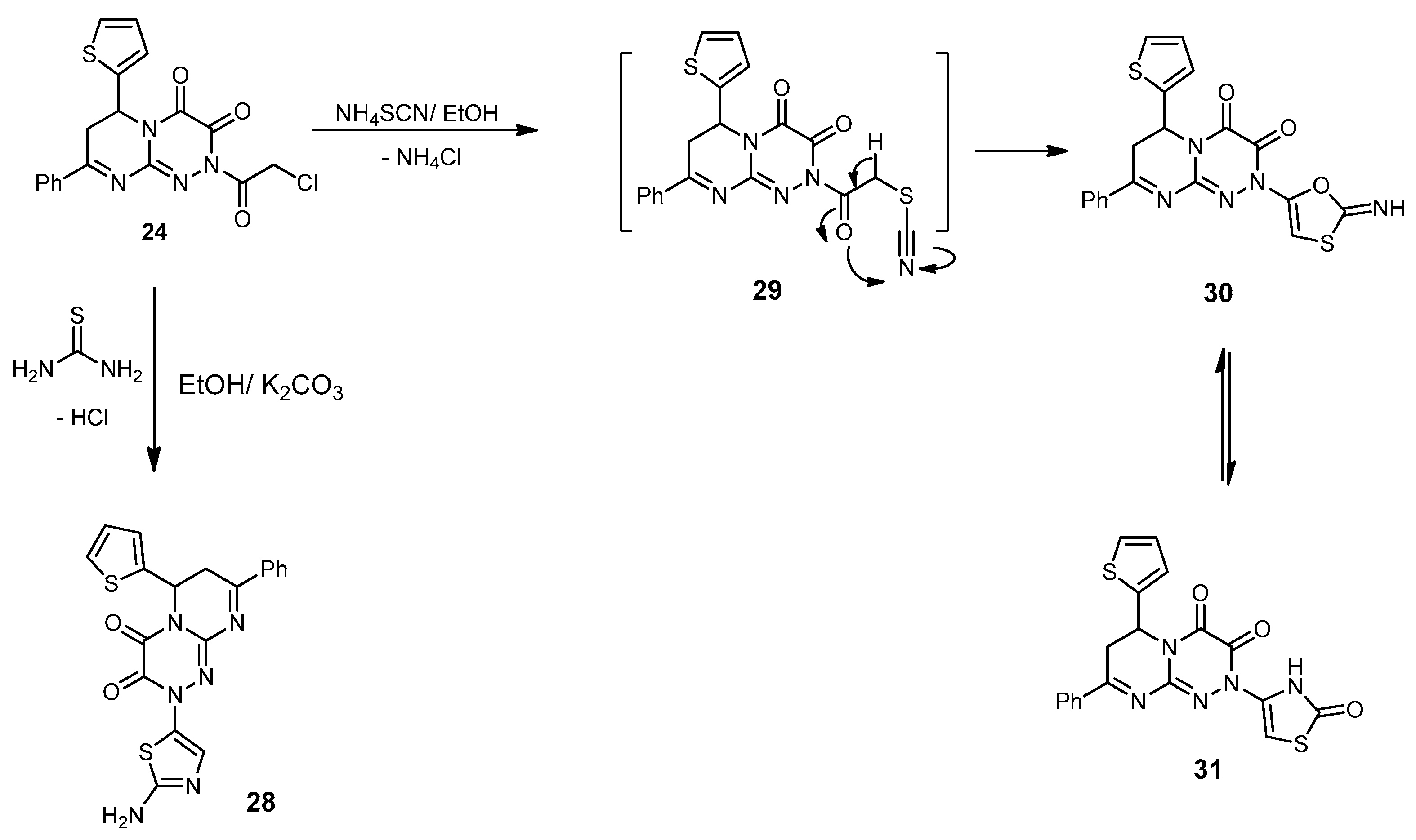
2-(2-Aminothiazol-5-yl)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4] triazine-3,4-dione (28). A mixture of 24 (0.40 g, 1 mmol) and an equimolar amount (1 mmol) of thiourea in dry EtOH (30 mL) containing anhydrous K2CO3 (0.13 g, 1 mmol) was refluxed for 8 h. The excess solvent was removed under reduced pressure, and the residue was triturated with MeOH. The formed product was filtered, washed with cold MeOH, dried, and recrystallized from the DMF–MeOH (1:3) to yield 28 as a brown solid in an 80% yield; mp 231–233 °C; IR (KBr): (cm−1) 679, 685 (2C–S–C), 1610–1620 (3 C=N), 1665–1686 (2amidic C=O), 3215 (NH2); 1H-NMR (300 MHz, DMSO-d6): δ 2.78 (dd, J = 12.4 and 4.6 Hz, 1H), 2.91 (dd, J = 8.1 and 12.1 Hz, 1H), 5.01 (dd, J = 8.6 and 3.8 Hz, 1H), 6.21 (s, 1H, Thiaz.(C4)-H), 6.51 (s, 2H, NH2Deutr. Exch), 7.68–7.79 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H); MS (m/z, %): 422.06 (M+, 20). Anal. Calcd for C19H14N6O2S2 (422.48): C, 54.01; H, 3.34; N, 19.89%. Found: C, 54.12; H, 3.11; N, 19.57%.
2-(2-Oxo-2,3-dihydrothiazol-4-yl)-8-phenyl-6-(thiophen-2-yl)-6,7-dihydro-2H-pyrimido[2,1-c][1,2,4]triazine-3,4-dione (31). A mixture of 24 (0.40 g, 1 mmol) and NH4SCN (0.15 g, 2 mmol) in absolute EtOH (30 mL) was refluxed for 8 h. The reaction mixture was allowed to stand overnight; the solid produced was filtered and then recrystallized from EtOH to produce 31 as yellow crystals in a 76% yield; mp 189–191 °C; IR (KBr): (cm−1) 679, 689 (2C–S–C), 1610–1620 (C=N), 1651–1686 (3 amidic C=O), 3185 (NH); 1H-NMR (300 MHz, DMSO-d6): δ 2.78 (dd, J = 12.4 and 4.6 Hz, 1H), 2.92 (dd, J = 8.4 and 12.3 Hz, 1H), 5.01 (dd, J = 8.6 and 3.8 Hz, 1H), 6.83 (s, 1H, Thiaz.(C5)-H), 7.68–7.79 (m, 7H, Ar–H and Thioph.(C3,4)-2H), 8.21 (d, J = 4.5, 1H, Thioph.(C5)-H), 10.13 (s, 1H, NHDeutr. Exch); MS (m/z, %): 423.03 (M+, 75). Anal. Calcd for C19H13N5O3S2 (423.47): C, 53.89; H, 3.09; N, 16.54%. Found: C, 53.68; H, 3.21; N, 16.28%.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}