Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap–Deep Learning


 and
and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Optimization of Hyperparameters in the DeepSnap–DL Approach
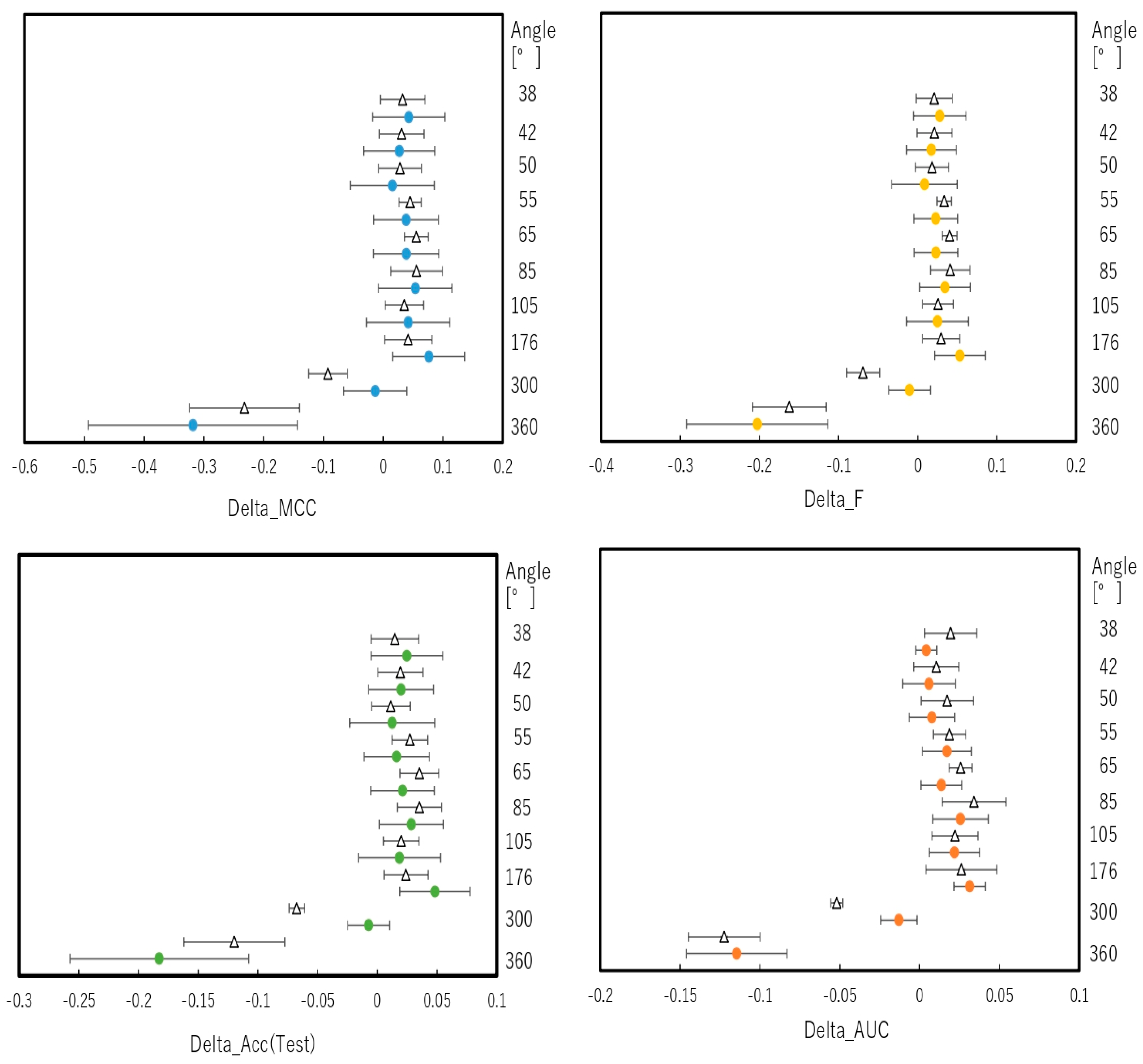
2.2. Snapshot Angles and Input Data Split of Chemical Compounds in the DeepSnap–DL Approach
2.3. Contribution of Threshold of AhR Activation in Prediction Performance of the DeepSnap–DL Approach
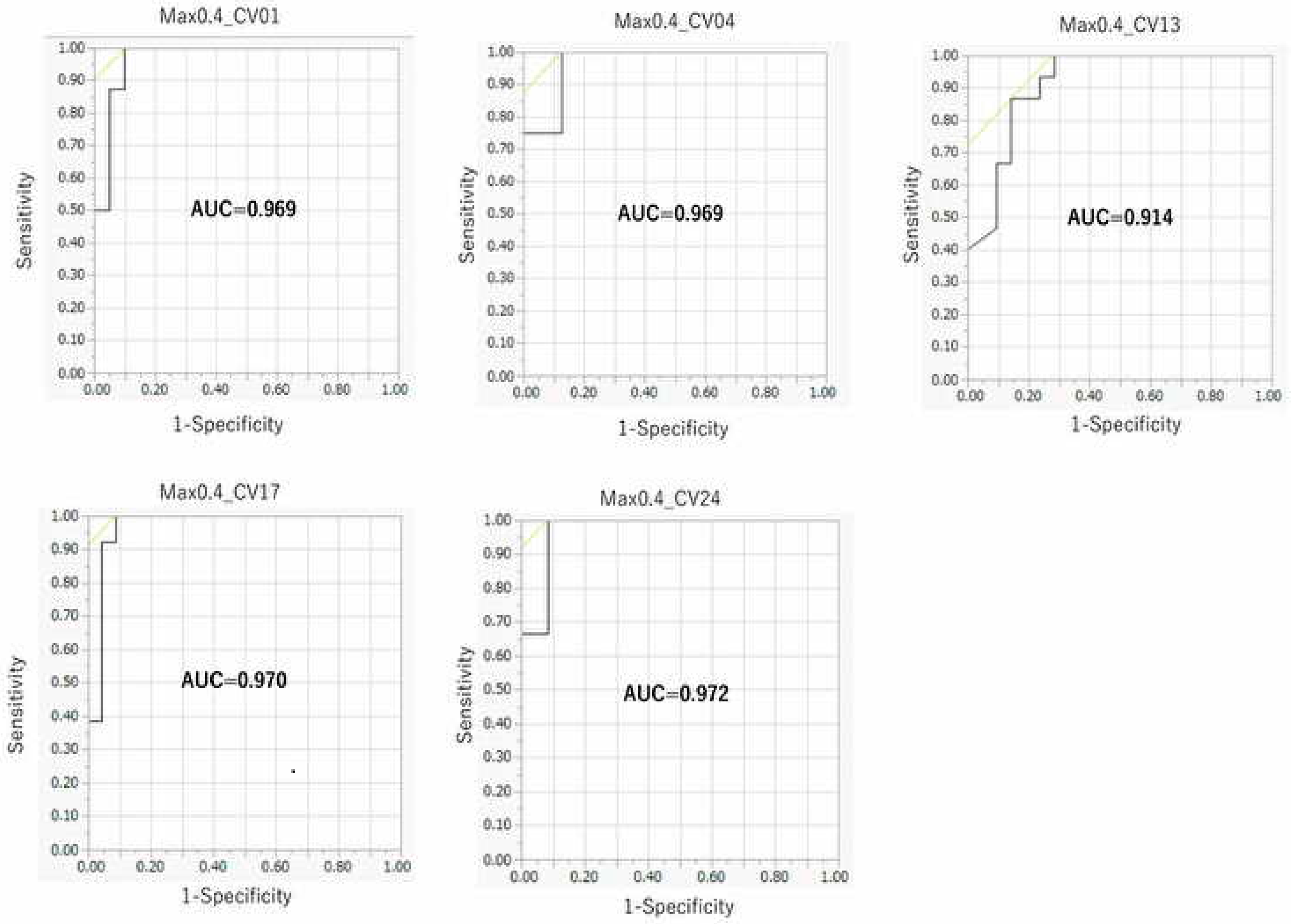
2.4. Comparison between the DeepSnap–DL Approach and Four Conventional MLs
3. Conclusions
4. Materials and Methods
4.1. AhR Activation Assay
4.2. Data Split by Endpoint
4.3. Preparation of Dataset
4.4. DeepSnap
4.5. ML Models
4.6. Evaluation of the Predictive Model
- Sensitivity = ΣTPs / (ΣTPs + ΣFNs)
- Specificity = ΣTNs / (ΣTNs + ΣFPs)
- BAC = (sensitivity + specificity)/2
- Accuracy = (TP + TN) / (TP + FP + TN + FN)
- Precision = TP / (TP + FP)
- Recall = TP / (TP + FN)
- F-measure = 2 × Recall × Precision / (Recall + Precision)
- MCC = (TP × TN − FP × FN)/
4.7. PCA and Cluster Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| 2D | two-dimensional |
| 3D | three-dimensional |
| Acc (Test) | accuracy in the test dataset |
| AhR | aryl hydrocarbon receptor |
| AO | adverse outcome |
| AOP | adverse outcome pathway |
| AUC | area under the curve |
| Acc (Val) | accuracy in the validation dataset |
| BAC | balanced accuracy |
| BSs | batch sizes |
| CB | CatBoost |
| CNN | convolutional neural network |
| DIGITS | deep learning GPU training system |
| DL | deep learning |
| DModX | distance to the model in X space |
| DNNs | deep neural networks |
| DILI | drug-induced liver injury |
| F | F value |
| FN | false negative |
| FP | false positive |
| LGBM | light gradient boosting machine |
| LR | learning rate |
| Loss (Val) | loss in the validation dataset |
| MAX value | maximum fold-change value of AhR reporter activity |
| MCC | Matthews correlation coefficient |
| MDCs | metabolism-disrupting chemicals |
| MIE | molecular initiating event |
| ML | machine learning |
| MOE | molecular operating environment |
| PCA | principle component analysis |
| RF | random forest |
| ROC | receiver operating characteristic |
| SMILES | simplified molecular input line entry system |
| TN | true negative |
| TP | true positive |
| XGB | eXtreme gradient boosting |
| XRE | xenobiotic-response element |
References
- Cave, M.; Falkner, K.C.; Ray, M.; Joshi-Barve, S.; Brock, G.; Khan, R.; Bon Homme, M.; McClain, C.J. Toxicant-associated steatohepatitis in vinyl chloride workers. Hepatology 2010, 51, 474–481. [Google Scholar] [CrossRef]
- Kaiser, J.P.; Lipscomb, J.C.; Wesselkamper, S.C. Putative mechanisms of environmental chemical-induced steatosis. Int. J. Toxicol. 2012, 31, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Al-Eryani, L.; Wahlang, B.; Falkner, K.C.; Guardiola, J.J.; Clair, H.B.; Prough, R.A.; Cave, M. Identification of environmental chemicals-associated with the development of toxicant-associated fatty liver disease in rodents. Toxicol. Pathol. 2015, 43, 482–497. [Google Scholar] [CrossRef] [PubMed]
- AbdulHameed, M.D.M.; Pannala, V.R.; Wallqvist, A. Mining Public Toxicogenomic Data Reveals Insights and Challenges in Delineating Liver Steatosis Adverse Outcome Pathways. Front. Genet. 2019, 10, 1007. [Google Scholar] [CrossRef] [PubMed]
- Geng, N.; Ren, X.; Gong, Y.; Zhang, H.; Wang, F.; Xing, L.; Cao, R.; Xu, J.; Gao, Y.; Giesy, J.P.; et al. Integration of metabolomics and transcriptomics reveals short-chain chlorinated paraffin-induced hepatotoxicity in male Sprague-Dawley rat. Environ. Int. 2019, 133, 105231. [Google Scholar] [CrossRef] [PubMed]
- La Merrill, M.A.; Johnson, C.L.; Smith, M.T.; Kandula, N.R.; Macherone, A.; Pennell, K.D.; Kanaya, A.M. Exposure to Persistent Organic Pollutants (POPs) and Their Relationship to Hepatic Fat and Insulin Insensitivity among Asian Indian Immigrants in the United States. Environ. Sci. Technol. 2019, 53, 13906–13918. [Google Scholar] [CrossRef]
- Sargis, R.M.; Heindel, J.J.; Padmanabhan, V. Interventions to Address Environmental Metabolism-Disrupting Chemicals: Changing the Narrative to Empower Action to Restore Metabolic Health. Front. Endocrinol. 2019, 10, 33. [Google Scholar] [CrossRef]
- Ostapowicz, G.; Fontana, R.J.; Schiødt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Acute Liver Failure Study Group. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Weaver, R.J.; Blomme, E.A.; Chadwick, A.E.; Copple, I.M.; Gerets, H.H.J.; Goldring, C.E.; Guillouzo, A.; Hewitt, P.G.; Ingelman-Sundberg, M.; Jensen, K.G.; et al. Managing the challenge of drug-induced liver injury: A roadmap for the development and deployment of preclinical predictive models. Nat. Rev. Drug Discov. 2020, 19, 131–148. [Google Scholar] [CrossRef]
- Kaplowitz, N. Idiosyncratic drug hepatotoxicity. Nat. Rev. Drug Discov. 2005, 4, 489–499. [Google Scholar] [CrossRef]
- Thakkar, S.; Li, T.; Liu, Z.; Wu, L.; Roberts, R.; Tong, W. Drug-induced liver injury severity and toxicity (DILIst): Binary classification of 1279 drugs by human hepatotoxicity. Drug. Discov. Today 2019, 25, 201–208. [Google Scholar] [CrossRef] [PubMed]
- Vandenberg, L.N.; Colborn, T.; Hayes, T.B.; Heindel, J.J.; Jacobs, D.R., Jr.; Lee, D.H.; Shioda, T.; Soto, A.M.; vom Saal, F.S.; Welshons, W.V.; et al. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr. Rev. 2012, 33, 378–455. [Google Scholar] [CrossRef] [PubMed]
- Denison, M.S.; Nagy, S.R. Activation of the aryl hydrocarbon receptor by structurally diverse exogenous and endogenous chemicals. Annu. Rev. Pharmacol. Toxicol. 2003, 43, 309–334. [Google Scholar] [CrossRef]
- Moura-Alves, P.; Faé, K.; Houthuys, E.; Dorhoi, A.; Kreuchwig, A.; Furkert, J.; Barison, N.; Diehl, A.; Munder, A.; Constant, P.; et al. AhR sensing of bacterial pigments regulates antibacterial defence. Nature 2014, 512, 387–392. [Google Scholar] [CrossRef]
- Bock, K.W. Functions of aryl hydrocarbon receptor (AHR) and CD38 in NAD metabolism and nonalcoholic steatohepatitis (NASH). Biochem. Pharmacol. 2019, 169, 113620. [Google Scholar] [CrossRef]
- Klimenko, K.; Rosenberg, S.A.; Dybdahl, M.; Wedebye, E.B.; Nikolov, N.G. QSAR modelling of a large imbalanced aryl hydrocarbon activation dataset by rational and random sampling and screening of 80,086 REACH pre-registered and/or registered substances. PLoS ONE 2019, 14, e0213848. [Google Scholar] [CrossRef]
- Vogeley, C.; Esser, C.; Tüting, T.; Krutmann, J.; Haarmann-Stemmann, T. Role of the Aryl Hydrocarbon Receptor in Environmentally Induced Skin Aging and Skin Carcinogenesis. Int. J. Mol. Sci. 2019, 20, 6005. [Google Scholar] [CrossRef]
- Lawal, A.O. Air particulate matter induced oxidative stress and inflammation in cardiovascular disease and atherosclerosis: The role of Nrf2 and AhR-mediated pathways. Toxicol. Lett. 2017, 270, 88–95. [Google Scholar] [CrossRef]
- Seok, S.H.; Lee, W.; Jiang, L.; Molugu, K.; Zheng, A.; Li, Y.; Park, S.; Bradfield, C.A.; Xing, Y. Structural hierarchy controlling dimerization and target DNA recognition in the AHR transcriptional complex. Proc. Natl. Acad. Sci. USA 2017, 114, 5431–5436. [Google Scholar] [CrossRef]
- Vogel, C.F.A.; Haarmann-Stemmann, T. The aryl hydrocarbon receptor repressor—More than a simple feedback inhibitor of AhR signaling: Clues for its role in inflammation and cancer. Curr. Opin. Toxicol. 2017, 2, 109–119. [Google Scholar] [CrossRef]
- Larigot, L.; Juricek, L.; Dairou, J.; Coumoul, X. AhR signaling pathways and regulatory functions. Biochim. Open. 2018, 7, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Labadie, B.W.; Bao, R.; Luke, J.J. Reimagining IDO Pathway Inhibition in Cancer Immunotherapy via Downstream Focus on the Tryptophan-Kynurenine-Aryl Hydrocarbon Axis. Clin. Cancer Res. 2019, 25, 1462–1471. [Google Scholar] [CrossRef] [PubMed]
- Tarnow, P.; Tralau, T.; Luch, A. Chemical activation of estrogen and aryl hydrocarbon receptor signaling pathways and their interaction in toxicology and metabolism. Expert. Opin. Drug Metab. Toxicol. 2019, 15, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Fader, K.A.; Zacharewski, T.R. Beyond the Aryl Hydrocarbon Receptor: Pathway Interactions in the Hepatotoxicity of 2,3,7,8-Tetrachlorodibenzo-p-dioxin and Related Compounds. Curr. Opin. Toxicol. 2017, 2, 36–41. [Google Scholar] [CrossRef] [PubMed]
- Roman, Á.C.; Carvajal-Gonzalez, J.M.; Merino, J.M.; Mulero-Navarro, S.; Fernández-Salguero, P.M. The aryl hydrocarbon receptor in the crossroad of signalling networks with therapeutic value. Pharmacol. Ther. 2018, 185, 50–63. [Google Scholar] [CrossRef]
- Lu, P.; Cai, X.; Guo, Y.; Xu, M.; Tian, J.; Locker, J.; Xie, W. Constitutive Activation of the Human Aryl Hydrocarbon Receptor in Mice Promotes Hepatocarcinogenesis Independent of Its Coactivator Gadd45b. Toxicol. Sci. 2019, 167, 581–592. [Google Scholar] [CrossRef]
- Kennedy, G.D.; Nukaya, M.; Moran, S.M.; Glover, E.; Weinberg, S.; Balbo, S.; Hecht, S.S.; Pitot, H.C.; Drinkwater, N.R.; Bradfield, C.A. Liver tumor promotion by 2,3,7,8-tetrachlorodibenzo-p-dioxin is dependent on the aryl hydrocarbon receptor and TNF/IL-1 receptors. Toxicol. Sci. 2014, 140, 135–143. [Google Scholar] [CrossRef]
- Moreno-Marín, N.; Merino, J.M.; Alvarez-Barrientos, A.; Patel, D.P.; Takahashi, S.; González-Sancho, J.M.; Gandolfo, P.; Rios, R.M.; Muñoz, A.; Gonzalez, F.J.; et al. Aryl Hydrocarbon Receptor Promotes Liver Polyploidization and Inhibits PI3K, ERK, and Wnt/β-Catenin Signaling. iScience 2018, 4, 44–63. [Google Scholar] [CrossRef]
- Faidallah, H.M.; Girgis, A.S.; Tiwari, A.D.; Honkanadavar, H.H.; Thomas, S.J.; Samir, A.; Kalmouch, A.; Alamry, K.A.; Khan, K.A.; Ibrahim, T.S.; et al. Synthesis, antibacterial properties and 2D-QSAR studies of quinolone-triazole conjugates. Eur. J. Med. Chem. 2018, 143, 1524–1534. [Google Scholar] [CrossRef]
- El-Zahabi, H.S.A.; Khalifa, M.M.A.; Gado, Y.M.H.; Farrag, A.M.; Elaasser, M.M.; Safwat, N.A.; AbdelRaouf, R.R.; Arafa, R.K. New thiobarbituric acid scaffold-based small molecules: Synthesis, cytotoxicity, 2D-QSAR, pharmacophore modelling and in-silico ADME screening. Eur. J. Pharm. Sci. 2019, 130, 124–136. [Google Scholar] [CrossRef]
- Khan, K.; Khan, P.M.; Lavado, G.; Valsecchi, C.; Pasqualini, J.; Baderna, D.; Marzo, M.; Lombardo, A.; Roy, K.; Benfenati, E. QSAR modeling of Daphnia magna and fish toxicities of biocides using 2D descriptors. Chemosphere 2019, 229, 8–17. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Du, Z.; Lv, W.J.; Zhang, X.Y.; Zhai, H.L. In silico toxicity evaluation of dioxins using structure-activity relationship (SAR) and two-dimensional quantitative structure-activity relationship (2D-QSAR). Arch. Toxicol. 2019, 93, 3207–3218. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Fang, Y.; Cao, W.; Liang, F.; Pan, S.; Xu, X. Quantitative Structure-Activity Relationships for the Flavonoid-Mediated Inhibition of P-Glycoprotein in KB/MDR1 Cells. Molecules 2019, 24, 1661. [Google Scholar] [CrossRef]
- Danishuddin Khan, A.U. Descriptors and their selection methods in QSAR analysis: Paradigm for drug design. Drug Discov. Today 2016, 21, 1291–1302. [Google Scholar] [CrossRef]
- Zhao, L.; Wang, W.; Sedykh, A.; Zhu, H. Experimental Errors in QSAR Modeling Sets: What We Can Do and What We Cannot Do. ACS Omega 2017, 2, 2805–2812. [Google Scholar] [CrossRef]
- Luque Ruiz, I.; Gómez-Nieto, M.Á. Building of Robust and Interpretable QSAR Classification Models by Means of the Rivality Index. J. Chem. Inf. Model. 2019, 59, 2785–2804. [Google Scholar] [CrossRef]
- Plante, A.; Shore, D.M.; Morra, G.; Khelashvili, G.; Weinstein, H. A Machine Learning Approach for the Discovery of Ligand-Specific Functional Mechanisms of GPCRs. Molecules 2019, 24, 2097. [Google Scholar] [CrossRef]
- Koutsoukas, A.; Monaghan, K.J.; Li, X.; Huan, J.J. Deep-learning: Investigating deep neural networks hyper-parameters and comparison of performance to shallow methods for modeling bioactivity data. Cheminform 2017, 9, 42. [Google Scholar] [CrossRef]
- Lenselink, E.B.; Ten Dijke, N.; Bongers, B.; Papadatos, G.; van Vlijmen, H.W.T.; Kowalczyk, W.; IJzerman, A.P.; van Westen, G.J.P. Beyond the hype: Deep neural networks outperform established methods using a ChEMBL bioactivity benchmark set. J. Cheminform. 2017, 9, 45. [Google Scholar] [CrossRef]
- Baskin, I.I. Machine Learning Methods in Computational Toxicology. Methods Mol. Biol. 2018, 1800, 119–139. [Google Scholar] [CrossRef]
- Russo, D.P.; Zorn, K.M.; Clark, A.M.; Zhu, H.; Ekins, S. Comparing Multiple Machine Learning Algorithms and Metrics for Estrogen Receptor Binding Prediction. Mol. Pharm. 2018, 15, 4361–4370. [Google Scholar] [CrossRef] [PubMed]
- Kato, Y.; Hamada, S.; Goto, H. Validation study of QSAR/DNN models using the competition datasets. Mol. Inform. 2020, 39, e1900154. [Google Scholar] [CrossRef] [PubMed]
- Beltran, J.A.; Aguilera-Mendoza, L.; Brizuela, C.A. Optimal selection of molecular descriptors for antimicrobial peptides classification: An evolutionary feature weighting approach. BMC Genom. 2018, 19, 672. [Google Scholar] [CrossRef] [PubMed]
- Kausar, S.; Falcao, A.O. Analysis and Comparison of Vector Space and Metric Space Representations in QSAR Modeling. Molecules 2019, 24, 1698. [Google Scholar] [CrossRef]
- Veltri, D.; Kamath, U.; Shehu, A. Improving Recognition of Antimicrobial Peptides and Target Selectivity through Machine Learning and Genetic Programming. IEEE/ACM Trans. Comput. Biol. Bioinform. 2017, 14, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Uesawa, Y. Quantitative structure-activity relationship analysis using deep learning based on a novel molecular image input technique. Bioorg. Med. Chem. Lett. 2018, 28, 3400–3403. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Optimization of a Deep-Learning Method Based on the Classification of Images Generated by Parameterized Deep Snap a Novel Molecular-Image-Input Technique for Quantitative Structure-Activity Relationship (QSAR) Analysis. Front. Bioeng. Biotechnol. 2019, 7, 65. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. Prediction Model with High-Performance Constitutive Androstane Receptor (CAR) Using DeepSnap-Deep Learning Approach from the Tox21 10K Compound Library. Int. J. Mol. Sci. 2019, 20, 4855. [Google Scholar] [CrossRef]
- Matsuzaka, Y.; Uesawa, Y. DeepSnap-Deep Learning Approach Predicts Progesterone Receptor Antagonist Activity with High Performance. Front. Bioeng. Biotechnol. 2020, 7, 485. [Google Scholar] [CrossRef]
- Buda, M.; Maki, A.; Mazurowski, M.A. A systematic study of the class imbalance problem in convolutional neural networks. Neural Netw. 2018, 106, 249–259. [Google Scholar] [CrossRef]
- Saddala, M.S.; Lennikov, A.; Huang, H. Discovery of Small-Molecule Activators for Glucose-6-Phosphate Dehydrogenase (G6PD) Using Machine Learning Approaches. Int. J. Mol. Sci. 2020, 21, 1523. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Tao, W.; Gao, T.; Li, H.; Lu, Y.H.; Su, Z.M. Improving the accuracy of Density Functional Theory (DFT) calculation for homolysis bond dissociation energies of Y-NO bond: Generalized regression neural network based on grey relational analysis and principal component analysis. Int. J. Mol. Sci. 2011, 12, 2242–2261. [Google Scholar] [CrossRef] [PubMed]
- Okamura, M.; Shizu, R.; Hosaka, T.; Sasaki, T.; Yoshinari, K. Possible involvement of the competition for the transcriptional coactivator glucocorticoid receptor-interacting protein 1 in the inflammatory signal-dependent suppression of PXR-mediated CYP3A induction in vitro. Drug Metab. Pharmacokinet. 2019, 34, 272–279. [Google Scholar] [CrossRef] [PubMed]
- CORINA Classic—High-Quality 3D Molecular Models. Available online: https://www.mn-am.com/products/corina/ (accessed on 3 March 2020).
- Ambe, K.; Ishihara, K.; Ochibe, T.; Ohya, K.; Tamura, S.; Inoue, K.; Yoshida, M.; Tohkin, M. In Silico Prediction of Chemical-Induced Hepatocellular Hypertrophy Using Molecular Descriptors. Toxicol. Sci. 2018, 162, 667–675. [Google Scholar] [CrossRef] [PubMed]
- IMAGENET Large Scale Visual Recognition Challenge 2012 (ILSVRC2012). Available online: http://image-net.org/challenges/LSVRC/2012/browse-synsets/ (accessed on 3 March 2020).
- IMAGENET. Available online: http://www.image-net.org/ (accessed on 3 March 2020).
- Shukla, R.; Lipasti, M.; Van Essen, B.; Moody, A.; Maruyama, N. REMODEL: Rethinking Deep CNN Models to Detect and Count on a NeuroSynaptic System. Front. Neurosci. 2019, 13, 4. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.T.; Lee, E.H.; Lee, S. Study on the Classification Performance of Underwater Sonar Image Classification Based on Convolutional Neural Networks for Detecting a Submerged Human Body. Sensors 2019, 20, 94. [Google Scholar] [CrossRef]
- Toğaçar, M.; Ergen, B.; Cömert, Z. BrainMRNet: Brain tumor detection using magnetic resonance images with a novel convolutional neural network model. Med. Hypotheses 2020, 134, 109531. [Google Scholar] [CrossRef]
- Park, S.J.; Palvanov, A.; Lee, C.H.; Jeong, N.; Cho, Y.I.; Lee, H.J. The development of food image detection and recognition model of Korean food for mobile dietary management. Nutr. Res. Pract. 2019, 13, 521–528. [Google Scholar] [CrossRef]
- Motta, D.; Santos, A.Á.B.; Winkler, I.; Machado, B.A.S.; Pereira, D.A.D.I.; Cavalcanti, A.M.; Fonseca, E.O.L.; Kirchner, F.; Badaró, R. Application of convolutional neural networks for classification of adult mosquitoes in the field. PLoS ONE 2019, 14, e0210829. [Google Scholar] [CrossRef]
- Yang, Y.; Yan, L.F.; Zhang, X.; Han, Y.; Nan, H.Y.; Hu, Y.C.; Hu, B.; Yan, S.L.; Zhang, J.; Cheng, D.L.; et al. Glioma Grading on Conventional MR Images: A Deep Learning Study With Transfer Learning. Front. Neurosci. 2018, 12, 804. [Google Scholar] [CrossRef]
- Szegedy, C.; Liu, W.; Jia, Y.; Sermanet, Y.; Reed, S.; Anguelov, D.; Erhan, D.; Vanhoucke, V.; Rabinovich, A. Going Deeper with Convolutions. arXiv 2014, arXiv:1409.4842v1. [Google Scholar]
- Kim, J.Y.; Lee, H.E.; Choi, Y.H.; Lee, S.J.; Jeon, J.S. CNN-based diagnosis models for canine ulcerative keratitis. Sci. Rep. 2019, 9, 14209. [Google Scholar] [CrossRef] [PubMed]
- Moriwaki, H.; Tian, Y.S.; Kawashita, N.; Takagi, T. Mordred: A molecular descriptor calculator. J. Cheminform. 2018, 10, 4. [Google Scholar] [CrossRef] [PubMed]
- Mordred-Descriptor/Mordred. Available online: https://github.com/mordred-descriptor/mordred/ (accessed on 3 March 2020).
- Random-Forest-Classifier. Available online: https://github.com/topics/random-forest-classifier/ (accessed on 3 March 2020).
- Xgboost/Python-Package. Available online: https://github.com/dmlc/xgboost/tree/master/python-package/ (accessed on 3 March 2020).
- Microsoft/Lightgbm. Available online: https://github.com/microsoft/LightGBM/ (accessed on 3 March 2020).
- Catboost/Catboost. Available online: https://github.com/catboost/catboost/ (accessed on 3 March 2020).
- Ivanov, M.V.; Levitsky, L.I.; Bubis, J.A.; Gorshkov, M.V. Scavager: A Versatile Postsearch Validation Algorithm for Shotgun Proteomics Based on Gradient Boosting. Proteomics 2019, 19, e1800280. [Google Scholar] [CrossRef]
- Yun, J.H.; Chun, S.M.; Kim, J.C.; Shin, H.I. Obesity cutoff values in Korean men with motor complete spinal cord injury: Body mass index and waist circumference. Spinal Cord 2019, 57, 110–116. [Google Scholar] [CrossRef]
- Liang, K.; Wang, C.; Yan, F.; Wang, L.; He, T.; Zhang, X.; Li, C.; Yang, W.; Ma, Z.; Ma, A.; et al. HbA1c Cutoff Point of 5.9% Better Identifies High Risk of Progression to Diabetes among Chinese Adults: Results from a Retrospective Cohort Study. J. Diabetes Res. 2018, 2018, 7486493. [Google Scholar] [CrossRef]
- Kona, R.; Qu, H.; Mattes, R.; Jancsik, B.; Fahmy, R.M.; Hoag, S.W. Application of in-line near infrared spectroscopy and multivariate batch modeling for process monitoring in fluid bed granulation. Int. J. Pharm. 2013, 452, 63–72. [Google Scholar] [CrossRef]
- Xiong, H.; Yu, L.X.; Qu, H. Batch-to-batch quality consistency evaluation of botanical drug products using multivariate statistical analysis of the chromatographic fingerprint. AAPS PharmSciTech 2013, 14, 802–810. [Google Scholar] [CrossRef]
- Zeng, S.; Chen, T.; Wang, L.; Qu, H. Monitoring batch-to-batch reproducibility using direct analysis in real time mass spectrometry and multivariate analysis: A case study on precipitation. J. Pharm. Biomed. Anal. 2013, 76, 87–95. [Google Scholar] [CrossRef]
- Stockdale, G.; Murphy, B.M.; D’Antonio, J.; Manning, M.C.; Al-Azzam, W. Comparability of higher order structure in proteins: Chemometric analysis of second-derivative amide I Fourier transform infrared spectra. J. Pharm. Sci. 2015, 104, 25–33. [Google Scholar] [CrossRef]
- Chakraborty, A.; Chaudhuri, P. A Wilcoxon-Mann-Whitney type test for infinite dimensional data. arXiv 2014, arXiv:1403.0201v1. [Google Scholar] [CrossRef]
- Dehling, H.; Fried, R.; Wendler, M. A Robust Method for Shift Detection in Time Series. arXiv 2015, arXiv:1506.03345v1. [Google Scholar]
- Dedecker, J.; Saulière, G. The Mann-Whitney U-statistic for α-dependent sequences. arXiv 2016, arXiv:1611.06828v1. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds that is a total of 201 of SMILES and MAX values, are available from the authors. |





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| MAX Scores | Average AUC ± SD | Average Acc (Test) ± SD | Average MCC ± SD | ||||||
|---|---|---|---|---|---|---|---|---|---|
| 0.10 | 0.863 | ± | 0.121 | 0.867 | ± | 0.099 | 0.624 | ± | 0.171 |
| 0.15 | 0.793 | ± | 0.111 | 0.750 | ± | 0.094 | 0.481 | ± | 0.139 |
| 0.20 | 0.853 | ± | 0.072 | 0.828 | ± | 0.036 | 0.622 | ± | 0.083 |
| 0.30 | 0.920 | ± | 0.029 | 0.940 | ± | 0.032 | 0.814 | ± | 0.024 |
| 0.35 | 0.936 | ± | 0.025 | 0.889 | ± | 0.039 | 0.774 | ± | 0.079 |
| 0.40 | 0.959 | ± | 0.025 | 0.922 | ± | 0.036 | 0.845 | ± | 0.075 |
| 0.45 | 0.919 | ± | 0.045 | 0.883 | ± | 0.046 | 0.771 | ± | 0.092 |
| 0.50 | 0.953 | ± | 0.016 | 0.917 | ± | 0.020 | 0.842 | ± | 0.031 |
| 0.55 | 0.892 | ± | 0.073 | 0.861 | ± | 0.071 | 0.733 | ± | 0.127 |
| 0.40PMT | 0.539 | ± | 0.064 | 0.615 | ± | 0.093 | 0.216 | ± | 0.254 |
| MAX Scores | MLs | Average AUC ± SD | Average Acc (Test) ± SD | ||||
|---|---|---|---|---|---|---|---|
| 0.10 | RF | 0.642 | ± | 0.151 | 0.878 | ± | 0.037 |
| XGB | 0.720 | ± | 0.113 | 0.894 | ± | 0.053 | |
| LGBM | 0.746 | ± | 0.176 | 0.872 | ± | 0.046 | |
| CB | 0.702 | ± | 0.099 | 0.878 | ± | 0.037 | |
| 0.15 | RF | 0.660 | ± | 0.101 | 0.822 | ± | 0.032 |
| XGB | 0.736 | ± | 0.083 | 0.833 | ± | 0.062 | |
| LGBM | 0.795 | ± | 0.108 | 0.817 | ± | 0.072 | |
| CB | 0.736 | ± | 0.050 | 0.817 | ± | 0.046 | |
| 0.20 | RF | 0.652 | ± | 0.100 | 0.767 | ± | 0.032 |
| XGB | 0.687 | ± | 0.118 | 0.744 | ± | 0.050 | |
| LGBM | 0.710 | ± | 0.075 | 0.744 | ± | 0.066 | |
| CB | 0.744 | ± | 0.089 | 0.767 | ± | 0.042 | |
| 0.30 | RF | 0.682 | ± | 0.115 | 0.733 | ± | 0.050 |
| XGB | 0.770 | ± | 0.104 | 0.756 | ± | 0.099 | |
| LGBM | 0.751 | ± | 0.147 | 0.772 | ± | 0.050 | |
| CB | 0.802 | ± | 0.075 | 0.767 | ± | 0.075 | |
| 0.35 | RF | 0.737 | ± | 0.062 | 0.744 | ± | 0.087 |
| XGB | 0.743 | ± | 0.057 | 0.711 | ± | 0.085 | |
| LGBM | 0.732 | ± | 0.079 | 0.733 | ± | 0.093 | |
| CB | 0.770 | ± | 0.049 | 0.744 | ± | 0.046 | |
| 0.40 | RF | 0.716 | ± | 0.059 | 0.700 | ± | 0.057 |
| XGB | 0.724 | ± | 0.026 | 0.711 | ± | 0.015 | |
| LGBM | 0.715 | ± | 0.049 | 0.728 | ± | 0.050 | |
| CB | 0.719 | ± | 0.048 | 0.678 | ± | 0.050 | |
| 0.45 | RF | 0.724 | ± | 0.132 | 0.711 | ± | 0.119 |
| XGB | 0.733 | ± | 0.094 | 0.672 | ± | 0.069 | |
| LGBM | 0.709 | ± | 0.111 | 0.678 | ± | 0.085 | |
| CB | 0.754 | ± | 0.097 | 0.650 | ± | 0.061 | |
| 0.50 | RF | 0.739 | ± | 0.083 | 0.650 | ± | 0.075 |
| XGB | 0.702 | ± | 0.059 | 0.583 | ± | 0.059 | |
| LGBM | 0.702 | ± | 0.067 | 0.622 | ± | 0.082 | |
| CB | 0.744 | ± | 0.067 | 0.633 | ± | 0.080 | |
| 0.55 | RF | 0.713 | ± | 0.059 | 0.644 | ± | 0.057 |
| XGB | 0.737 | ± | 0.063 | 0.656 | ± | 0.025 | |
| LGBM | 0.728 | ± | 0.071 | 0.656 | ± | 0.064 | |
| CB | 0.769 | ± | 0.080 | 0.683 | ± | 0.097 | |
| 0.40PMT | RF | 0.452 | ± | 0.105 | 0.522 | ± | 0.077 |
| XGB | 0.531 | ± | 0.066 | 0.578 | ± | 0.060 | |
| LGBM | 0.467 | ± | 0.031 | 0.489 | ± | 0.032 | |
| CB | 0.467 | ± | 0.048 | 0.533 | ± | 0.063 | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsuzaka, Y.; Hosaka, T.; Ogaito, A.; Yoshinari, K.; Uesawa, Y. Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap–Deep Learning. Molecules 2020, 25, 1317. https://doi.org/10.3390/molecules25061317
Matsuzaka Y, Hosaka T, Ogaito A, Yoshinari K, Uesawa Y. Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap–Deep Learning. Molecules. 2020; 25(6):1317. https://doi.org/10.3390/molecules25061317
Chicago/Turabian StyleMatsuzaka, Yasunari, Takuomi Hosaka, Anna Ogaito, Kouichi Yoshinari, and Yoshihiro Uesawa. 2020. "Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap–Deep Learning" Molecules 25, no. 6: 1317. https://doi.org/10.3390/molecules25061317
APA StyleMatsuzaka, Y., Hosaka, T., Ogaito, A., Yoshinari, K., & Uesawa, Y. (2020). Prediction Model of Aryl Hydrocarbon Receptor Activation by a Novel QSAR Approach, DeepSnap–Deep Learning. Molecules, 25(6), 1317. https://doi.org/10.3390/molecules25061317