
Towards an Understanding of the Mode of Action of Human Aromatase Activity for Azoles through Quantum Chemical Descriptors-Based Regression and Structure Activity Relationship Modeling Analysis



Abstract
1. Introduction
2. Results and Discussion
2.1. Classification Modeling
2.2. Regression Modeling
2.2.1. Modeling of the Aromatase Antagonist Activity of Triazoles
2.2.2. Modeling of Aromatase Antagonist Activity of Diazoles
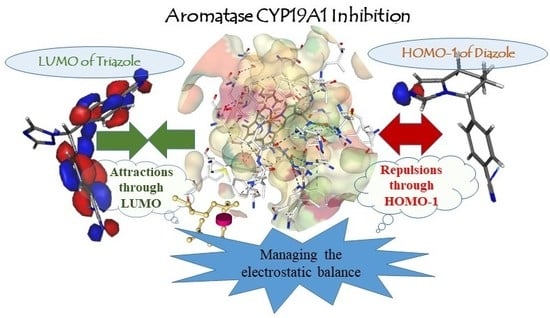
2.2.3. Electronic Interaction Aspects for Antagonist Activity of Diazoles and Triazoles
2.2.4. Modelling of Aromatase Agonist Activity for Diazole
2.2.5. Modeling of Agonist Activity for Thiazole/Oxazole
3. Materials and Methods
3.1. Data Collection
3.2. Data Curation
3.3. Dataset
3.4. Descriptor Calculation
3.5. Regression Model Development and Validation
3.6. Structure Activity Relationship Modeling and Evaluation
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Eicher, T.; Hauptmann, S.; Speicher, A. The Chemistry of Heterocycles: Structures, Reactions, Synthesis, and Applications; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Gonzalez, F.J.; Gelboin, H.V. Human cytochromes P450: evolution and cDNA-directed expression. Environ. Heal. Perspect. 1992, 98, 81–85. [Google Scholar] [CrossRef] [PubMed]
- Mermer, A.; Demirbas, N.; Demirbas, A.; Colak, N.; Ayaz, F.A.; Alagumuthu, M.; Arumugam, S. Synthesis, biological activity and structure activity relationship studies of novel conazole analogues via conventional, microwave and ultrasound mediated techniques. Bioorganic Chem. 2018, 81, 55–70. [Google Scholar] [CrossRef] [PubMed]
- Guerrero-Perilla, C.; Bernal, F.A.; Coy-Barrera, E. Insights into the interaction and binding mode of a set of antifungal azoles as inhibitors of potential fungal enzyme-based targets. Mol. Divers. 2018, 22, 929–942. [Google Scholar] [CrossRef] [PubMed]
- Brueggemeier, R.W.; Hackett, J.C.; Díaz-Cruz, E.S. Aromatase Inhibitors in the Treatment of Breast Cancer. Endocr. Rev. 2005, 26, 331–345. [Google Scholar] [CrossRef]
- Petkov, P.; Temelkov, S.; Villeneuve, D.; Ankley, G.; Mekenyan, O. Mechanism-based categorization of aromatase inhibitors: a potential discovery and screening tool. SAR QSAR Environ. Res. 2009, 20, 657–678. [Google Scholar] [CrossRef]
- Ghosh, D.; Griswold, J.; Erman, M.; Pangborn, W. Structural basis for androgen specificity and oestrogen synthesis in human aromatase. Nat. 2009, 457, 219–223. [Google Scholar] [CrossRef]
- Ghosh, D.; Lo, J.; Morton, D.; Valette, D.; Xi, J.; Griswold, J.; Hubbell, S.; Egbuta, C.; Jiang, W.; An, J.; et al. Novel Aromatase Inhibitors by Structure-Guided Design. J. Med. Chem. 2012, 55, 8464–8476. [Google Scholar] [CrossRef]
- Hamadeh, I.S.; Patel, J.N.; Rusin, S.; Tan, A.R. Personalizing aromatase inhibitor therapy in patients with breast cancer. Cancer Treat. Rev. 2018, 70, 47–55. [Google Scholar] [CrossRef]
- Kang, H.; Xiao, X.; Huang, C.; Yuan, Y.; Tang, D.; Dai, X.; Zeng, X. Potent aromatase inhibitors and molecular mechanism of inhibitory action. Eur. J. Med. Chem. 2018, 143, 426–437. [Google Scholar] [CrossRef]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular Docking of Aromatase Inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Saxena, A.; Devillers, J.; Bhunia, S.; Bro, E. Modelling inhibition of avian aromatase by azole pesticides. SAR QSAR Environ. Res. 2015, 26, 757–782. [Google Scholar] [CrossRef] [PubMed]
- Hao, Y.; Sun, G.; Fan, T.; Sun, X.; Liu, Y.; Zhang, N.; Zhao, L.; Zhong, R.; Peng, Y. Prediction on the mutagenicity of nitroaromatic compounds using quantum chemistry descriptors based QSAR and machine learning derived classification methods. Ecotoxicol. Environ. Saf. 2019, 186, 109822. [Google Scholar] [CrossRef] [PubMed]
- Nantasenamat, C.; Worachartcheewan, A.; Prachayasittikul, S.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. QSAR modeling of aromatase inhibitory activity of 1-substituted 1,2,3-triazole analogs of letrozole. Eur. J. Med. Chem. 2013, 69, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Xia, X.-R.; Monteiro-Riviere, N.A.; Riviere, J.E. An index for characterization of nanomaterials in biological systems. Nat. Nanotechnol. 2010, 5, 671–675. [Google Scholar] [CrossRef]
- Worachartcheewan, A.; Mandi, P.; Prachayasittikul, V.; Toropova, A.P.; Toropov, A.A.; Nantasenamat, C. Large-scale QSAR study of aromatase inhibitors using SMILES-based descriptors. Chemom. Intell. Lab. Syst. 2014, 138, 120–126. [Google Scholar] [CrossRef]
- Song, Z.; Liu, Y.; Dai, Z.; Liu, W.; Zhao, K.; Zhang, T.; Hu, Y.; Zhang, X.; Dai, Y. Synthesis and aromatase inhibitory evaluation of 4- N -nitrophenyl substituted amino-4 H -1,2,4-triazole derivatives. Bioorganic Med. Chem. 2016, 24, 4723–4730. [Google Scholar] [CrossRef]
- Lee, S.; Barron, M.G. 3D-QSAR study of steroidal and azaheterocyclic human aromatase inhibitors using quantitative profile of protein–ligand interactions. J. Chem. 2018, 10, 2. [Google Scholar] [CrossRef]
- Xie, H.; Qiu, K.; Xie, X. Pharmacophore modeling, virtual screening, and 3D-QSAR studies on a series of non-steroidal aromatase inhibitors. Med. Chem. Res. 2015, 24, 1901–1915. [Google Scholar] [CrossRef]
- Roy, P.P.; Roy, K. Docking and 3D-QSAR studies of diverse classes of human aromatase (CYP19) inhibitors. J. Mol. Model. 2010, 16, 1597–1616. [Google Scholar] [CrossRef]
- Shoombuatong, W.; Schaduangrat, N.; Nantasenamat, C. Towards understanding aromatase inhibitory activity via QSAR modeling. EXCLI J. 2018, 17, 688–708. [Google Scholar]
- Ghodsi, R.; Hemmateenejad, B. QSAR study of diarylalkylimidazole and diarylalkyltriazole aromatase inhibitors. Med. Chem. Res. 2016, 25, 834–842. [Google Scholar] [CrossRef]
- Nagar, S.; Saha, A. Modeling of diarylalkyl-imidazole and diarylalkyl-triazole derivatives as potent aromatase inhibitors for treatment of hormone-dependent cancer. J. Comput. Chem. 2010, 31, 2342–2353. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, T.; Gini, G.; Bakhtyari, N.G.; Benfenati, E. Mining toxicity structural alerts from SMILES: A new way to derive Structure Activity Relationships. In Proceedings of the 2011 IEEE Symposium on Computational Intelligence and Data Mining (CIDM), Paris, France, 11–15 April 2011; pp. 120–127. [Google Scholar]
- Chattaraj, P.K.; Sarkar, U.; Roy, D.R. Electrophilicity index. Chem. Rev. 2006, 106, 2065–2091. [Google Scholar] [CrossRef] [PubMed]
- Levine, I. Quantum Chemistry Vol. 6; Pearson Prentice Hall: Upper Saddle River, NJ, USA, 2009; Available online: http://www.dll.cheminfinity.com/arks/wp-file/books/Ira-N.Levine-Quantum-Chemistry-Pearson-(2013)-cheminfinity.com.pdf (accessed on 22 October 2014).
- Lata, S. Vikas Externally predictive quantum-mechanical models for the adsorption of aromatic organic compounds by graphene-oxide nanomaterials. SAR QSAR Environ. Res. 2019, 30, 847–863. [Google Scholar] [CrossRef] [PubMed]
- Roy, K. Quantitative Structure-Activity Relationships in Drug Design, Predictive Toxicology, and Risk Assessment; IGI Global: Hershey, PA, USA, 2015. [Google Scholar]
- Feher, M.; Schmidt, J.M. Property Distributions: Differences Between Drugs, Natural Products, and Molecules from Combinatorial Chemistry. Cheminstry 2003, 34, 218–227. [Google Scholar]
- Kassimi, N.E.-B.; Doerksen, R.J.; Thakkar, A.J. Polarizabilities of Aromatic Five-Membered Rings: Azoles. J. Phys. Chem. 1995, 99, 12790–12796. [Google Scholar] [CrossRef]
- Guégan, F.; Mignon, P.; Tognetti, V.; Joubert, L.; Morell, C. Dual descriptor and molecular electrostatic potential: complementary tools for the study of the coordination chemistry of ambiphilic ligands. Phys. Chem. Chem. Phys. 2014, 16, 15558–15569. [Google Scholar] [CrossRef]
- Şahin, Z.; Ertaş, M.; Berk, B.; Biltekin, S.N.; Yurttaş, L.; Demirayak, S. Studies on non-steroidal inhibitors of aromatase enzyme; 4-(aryl/heteroaryl)-2-(pyrimidin-2-yl)thiazole derivatives. Bioorg. Med. Chem. 2018, 26, 1986–1995. [Google Scholar] [CrossRef]
- Ertas, M.; Şahin, Z.; Berk, B.; Yurttas, L.; Biltekin, S.N.; Demirayak, S.; Berk, B. Pyridine-substituted thiazolylphenol derivatives: Synthesis, modeling studies, aromatase inhibition, and antiproliferative activity evaluation. Arch. Pharm. 2018, 351, 1700272. [Google Scholar] [CrossRef]
- Seth, A. Thiazolidinediones (TZDs) affect osteoblast viability and biomarkers independently of the TZD effects on aromatase. Horm. Metab. Res. 2013, 45, 1–8. [Google Scholar]
- Vosooghi, M.; Firoozpour, L.; Rodaki, A.; Pordeli, M.; Safavi, M.; Ardestani, S.K.; Dadgar, A.; Asadipour, A.; Moshafi, M.H.; Foroumadi, A. Design, synthesis, docking study and cytotoxic activity evaluation of some novel letrozole analogs. DARU J. Pharm. Sci. 2014, 22, 83. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Shi, H.-F.; Zhao, J.-F.; He, Y.-P.; Zhang, H.-B.; Liu, J.-P. Design, synthesis and aromatase inhibitory activities of novel indole-imidazole derivatives. Bioorg. Med. Chem. Lett. 2013, 23, 1760–1762. [Google Scholar] [CrossRef] [PubMed]
- Di Matteo, M.; Ammazzalorso, A.; Andreoli, F.; Caffa, I.; De Filippis, B.; Fantacuzzi, M.; Giampietro, L.; Maccallini, C.; Nencioni, A.; Parenti, M.D.; et al. Synthesis and biological characterization of 3-(imidazol-1-ylmethyl)piperidine sulfonamides as aromatase inhibitors. Bioorg. Med. Chem. Lett. 2016, 26, 3192–3194. [Google Scholar] [CrossRef] [PubMed]
- Jeong, S.; Woo, M.M.; Flockhart, D.A.; Desta, Z. Inhibition of drug metabolizing cytochrome P450s by the aromatase inhibitor drug letrozole and its major oxidative metabolite 4,4′-methanol-bisbenzonitrile in vitro. Cancer Chemother. Pharmacol. 2009, 64, 867–875. [Google Scholar] [CrossRef]
- Carhart, R.E.; Smith, D.H.; Venkataraghavan, R. Atom pairs as molecular features in structure-activity studies: definition and applications. J. Chem. Inf. Model. 1985, 25, 64–73. [Google Scholar] [CrossRef]
- Galvez, J.; Garcia, R.; Salabert, M.T.; Soler, R. Charge Indexes. New Topological Descriptors. J. Chem. Inf. Model. 1994, 34, 520–525. [Google Scholar] [CrossRef]
- EPA. “Tox21Assay”. Available online: ftp://newftp.epa.gov/comptox/High_Throughput_Screening_Data/Standard_Lab_Protocol_Tox21_Assays/Tox21Assay_SLPs%20and%20Descriptions_2016.zip (accessed on 31 January 2018).
- Gadaleta, D.; Lombardo, A.; Toma, C.; Benfenati, E. A new semi-automated workflow for chemical data retrieval and quality checking for modeling applications. J. Chem. 2018, 10, 60. [Google Scholar] [CrossRef]
- Achar, P.N.; Aubert, A.-M. Springer correspondences for dihedral groups 2007. Transform. Groups 2008, 13, 1–24. [Google Scholar] [CrossRef]
- Bajusz, D.; Racz, A.; Héberger, K. Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J. Cheminform. 2015, 7, 20. [Google Scholar] [CrossRef]
- ChemAxon, Marvin 5.1.10. Available online: http://www.chemaxon.com (accessed on 25 June 2013).
- MOPAC2012. Stewart Computational Chemistry. Available online: http://OpenMOPAC.net (accessed on 11 May 2018).
- Stewart, J.J.P. Optimization of parameters for semiempirical methods VI: more modifications to the NDDO approximations and re-optimization of parameters. J. Mol. Model. 2013, 19, 1–32. [Google Scholar] [CrossRef]
- Allouche, A.R. Gabedit—A graphical user interface for computational chemistry softwares. J. Comput. Chem. 2011, 32, 174–182. [Google Scholar] [CrossRef]
- Szabo, A.; Ostlund, N.S. Quantum Chemistry: Introduction to Advanced Electronic Structure Theory; MacMillan: New York, NY, USA, 1982. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Yap, C.W. PaDEL-descriptor: An open source software to calculate molecular descriptors and fingerprints. J. Comput. Chem. 2011, 32, 1466–1474. [Google Scholar] [CrossRef] [PubMed]
- Neese, F. The ORCA program system. Wiley Interdiscip. Rev. 2012, 2, 73–78. [Google Scholar] [CrossRef]
- Parr, R.G. Density Functional Theory of Atoms and Molecules. In Horizons of Quantum Chemistry; Springer Science and Business Media LLC: Berlin, Germany, 1980; pp. 5–15. [Google Scholar]
- Reenu, V.; Chayawan, C. Does electron-correlation has any role in the quantitative structure–activity relationships? J. Mol. Graph. Model. 2013, 42, 7–16. [Google Scholar]
- Dragon (Software for Molecular Descriptor Calculation). 2017. Available online: https://chm.kode-solutions.net. (accessed on 25 June 2013).
- Gramatica, P.; Chirico, N.; Papa, E.; Cassani, S.; Kovarich, S. QSARINS: A new software for the development, analysis, and validation of QSAR MLR models. J. Comput. Chem. 2013, 34, 2121–2132. [Google Scholar] [CrossRef]
- Todeschini, R.; Consonni, V.; Maiocchi, A. The K correlation index: theory development and its application in chemometrics. Chemom. Intell. Lab. Syst. 1999, 46, 13–29. [Google Scholar] [CrossRef]
- Vian, M.; Raitano, G.; Roncaglioni, A.; Benfenati, E. In silico model for mutagenicity (Ames test), taking into account metabolism. Mutagenesis 2019, 34, 41–48. [Google Scholar] [CrossRef]
- Lučić, B.; Batista, J.; Lovrić, M.; Vikić-Topić, D.; Nadramija, D.; Bešlo, D.; Kržić, A.S.; Bojović, V. Estimation of Random Accuracy and its Use in Validation of Predictive Quality of Classification Models within Predictive Challenges. Croat. Chem. Acta 2019, 92, 379–391. [Google Scholar] [CrossRef]
- Batista, J.; Vikić-Topić, D.; Lučić, B. The Difference Between the Accuracy of Real and the Corresponding Random Model is a Useful Parameter for Validation of Two-State Classification Model Quality. Croat. Chem. Acta 2016, 89, 527–534. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
| SA_ID | Name | SMARTS 1 | Structure | Activity Related | LR Value Using Eq. 7 | Accuracy (Acc) of SA Using Eq. 6 | Statistical Reliability | Literature Reliability [17,32,33,34,35,36,37,38] | Relevant Information of Selected SA and Distributions |
|---|---|---|---|---|---|---|---|---|---|
| SA1 | 1,3-thiazoles | c2cscn2 |  | Active agonist | inf | 1.0 | High 0.9 < Acc ≤ 1 | Medium | - |
| SA2 | para substituted chlorobenzenes | Clc1ccc(CC)cc1 |  | Active antagonist | inf | 1.0 | High 0.9 < Acc ≤ 1 | High | Antagonist Diazoles (n = 9; Average pIC50 = 8.40) Antagonist Triazole (n = 7; Average pIC50 = 7.89) |
| SA3 | 1,2,4-triazoles | n1cncn1 |  | Active antagonist | inf | 1.0 | High 0.9 < Acc ≤ 1 | High | - |
| SA4 | carboxylic acids | C(=O)O |  | Active agonist | 12.83 | 0.92 | High 0.9 < Acc ≤ 1 | High | Agonist Diazoles (n = 7; AveragepEC50 = 8.88) Agonist Monozoles(n = 4; AveragepEC50 = 9.09; Incorrect prediction = 1 triazoles) |
| SA5 | ethyl(propyl)amine | CCCN(CC) |  | Active antagonist | 1.71 | 0.67 | Low to medium 0.6 < Acc ≤ 0.7 | Low | - |
| SA6 | benzylimidazoles | c1cn(cn1)C(c1ccccc1) |  | Active antagonist | inf | 1.0 | High 0.9 < Acc ≤ 1 | High | Antagonist Diazoles (n = 3; Average pIC50=9.79) |
| SA7 | 1-phenyl-1H-Imidazoles | c1c(cccc1)n1ccnc1 |  | Active antagonist | inf | 1.0 | High 0.9 < Acc ≤ 1 | High | Antagonist Diazoles (n = 3; Average pIC50 = 7.87) |
| SA8 | Formamides | C(=O)N |  | Active agonist | 4.96 | 0.81 | Medium to high 0.8 < Acc ≤ 0.9 | Medium | Agonist Diazoles (n = 9; Average pEC50 = 8.53) Agonist Monozoles (n = 8; AveragepEC50 = 8.47; Incorrect prediction = 4 diazoles) |
| SA9 | chlorobenzenes | Clc1cccc(c1) |  | Active antagonist | 11.57 | 0.93 | High 0.9 < Acc ≤ 1 | High | Antagonist Diazoles (n = 16; Average pIC50 = 8.45) Antagonist Triazoles (n = 11; Average pIC50 = 7.72; Incorrect prediction = 2 diazoles) |
| SA10 | Carbon chain | CC |  | Active antagonist | 1.67 | 0.66 | Low to medium 0.6 < Acc ≤ 0.7 | Low | - |
| SA11 | purines | c1ncnc2c1ncn2 |  | Active agonist | 2.33 | 0.67 | Low to medium 0.6 < Acc ≤ 0.7 | Low to medium | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chayawan, C.; Toma, C.; Benfenati, E.; Caballero Alfonso, A.Y. Towards an Understanding of the Mode of Action of Human Aromatase Activity for Azoles through Quantum Chemical Descriptors-Based Regression and Structure Activity Relationship Modeling Analysis. Molecules 2020, 25, 739. https://doi.org/10.3390/molecules25030739
Chayawan C, Toma C, Benfenati E, Caballero Alfonso AY. Towards an Understanding of the Mode of Action of Human Aromatase Activity for Azoles through Quantum Chemical Descriptors-Based Regression and Structure Activity Relationship Modeling Analysis. Molecules. 2020; 25(3):739. https://doi.org/10.3390/molecules25030739
Chicago/Turabian StyleChayawan, Chayawan, Cosimo Toma, Emilio Benfenati, and Ana Y. Caballero Alfonso. 2020. "Towards an Understanding of the Mode of Action of Human Aromatase Activity for Azoles through Quantum Chemical Descriptors-Based Regression and Structure Activity Relationship Modeling Analysis" Molecules 25, no. 3: 739. https://doi.org/10.3390/molecules25030739
APA StyleChayawan, C., Toma, C., Benfenati, E., & Caballero Alfonso, A. Y. (2020). Towards an Understanding of the Mode of Action of Human Aromatase Activity for Azoles through Quantum Chemical Descriptors-Based Regression and Structure Activity Relationship Modeling Analysis. Molecules, 25(3), 739. https://doi.org/10.3390/molecules25030739