2. Results and Discussion
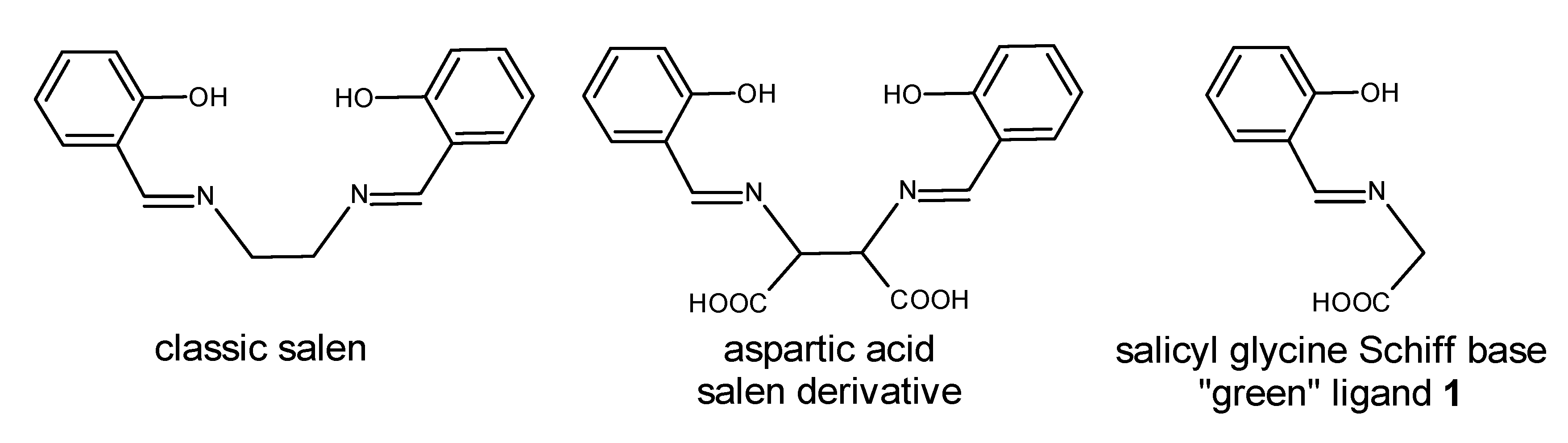
Our quest for
N,
O-ligands led us to literature references suggesting a simple condensation of salicylaldehyde with glycine in ethanol. The synthetic procedures for the desired imine described in the literature differed at several points, and a survey of these procedures is presented in
Table 1. To put our experimental findings into context, the details of the literature procedures are compared below. Only one reference described a procedure without the addition of a base [
15]. In this method, glycine was dissolved in 20 mL of water and salicylaldehyde in 10 mL ethanol. These solutions were mixed and refluxed for 3 h while turning orange. The workup was as follows: the volume was reduced to half, and the desired product crystallized upon cooling; the product was filtered off, washed with ethanol and ether, and finally dried. The crude product was recrystallized with an ethanol-propanol mixture (60:40) to give the desired product with a 66% yield. In the Zhu synthesis [
16], similar reactant solutions were used (methanol was used instead of ethanol), but the aqueous solution was alkalized with KOH. In addition, shorter reaction time and room temperature were used. The product remained in solution and was not separated, so the yield was not specified. Gürkan performed the reaction [
17] at low temperature (10 °C) under an N
2 atmosphere. Contrary to Zhu, NaOH was used as the base. After 2 h, the solid product was formed and recrystallized with methanol-ether (1:1) to give, after filtration, the ligand with a 58% yield. Panchal was the first to publish a synthetic method that avoided water [
18]. He carried out the reaction in ethanol using a stoichiometric addition of KOH at 50 °C for 1 h. The product was crystallized by diethyl ether diffusion giving a product with 63% yield. In a similar way, Zhao [
19] performed his synthesis at room temperature but did not specify a reaction time. The yield and separation methodology were the same as in the Panchal method.
However, despite many attempts, we were unable to repeat any of the five procedures listed in
Table 1, although these approaches consist of classical methods of Schiff base synthesis with precipitation of the desired product often in a fine crystal form. Finally, after modifying the conditions, we obtained the desired product
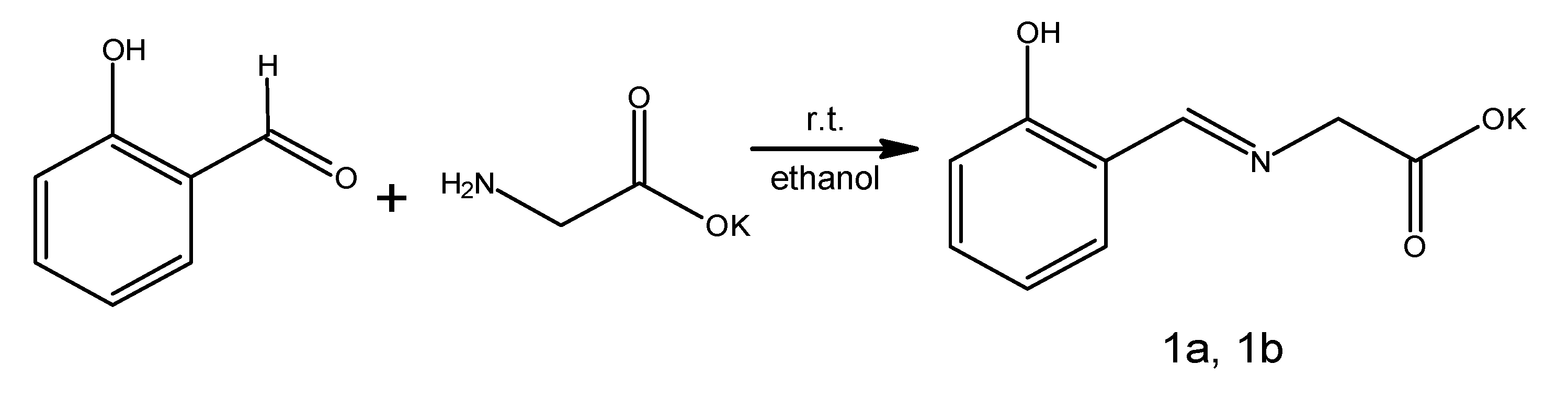
1—potassium
N-[(2-hydroxyphenyl)methylene]glycinate. It is worth noting that we identified the factor determining success. Initial efforts focused on obtaining the non-salt product. Chohan’s conditions [
15] were adopted; however, the results achieved differed from the published report. Separation of an orange, oily, residue on the bottom of the flask was observed. After reducing the volume and cooling, a solid product was not obtained. After 24 h, a few single crystals appeared, which, according to NMR, corresponded to the expected product but with a large excess of glycine. Because the attempt was unsuccessful, we modified the reaction conditions. Changing the solvent to ethanol resulted in the incomplete dissolution of glycine, as expected. During the reaction, the mixture changed to yellow, and glycine gathered at the bottom of the flask. After 3 h of heating, the remaining glycine was filtered, and the filtrate evaporated. A solid product was not obtained, and further analysis did not confirm the expected product. The reaction was carried out using a water:ethanol mixture (1:2), but results similar to those using an excess of water were obtained. Another solvent mixture (5 parts water to 1 part ethanol) was used just to dissolve the aldehyde. The mixture again turned yellow with significant solid formation; though, regrettably, NMR analysis showed that it was only glycine. After performing the above-described approaches no satisfying experimental results were obtained. Our observations led to the conclusion that, on the one hand, water disables separation of the expected product, while, on the other, dissolution of glycine is difficult in alcohol only. Conversion of glycine into a salt allows its dissolution in alcohol. Thus, we turned our efforts to obtaining the product in salt form by adopting the Zhao conditions [
19]. Equimolar alkalization of glycine with KOH enabled the formation of a salt that was soluble in ethanol, allowing us to obtain a homogenous mixture with salicylaldehyde. Upon its addition, the mixture changed color immediately to an intense orange with the immediate formation of an orange precipitate removing the need for diethyl ether addition for product separation. The solid was filtered off and air-dried. During this time, we observed in the bulk solid an exceptionally interesting process of color transformation from an intense orange to a bright yellow. The initial yield of the reaction was 84%; however, NMR analysis indicated contamination with the substrates. The reaction was optimized, and after a few trials (using methanol as a solvent), the pure product was produced with a final yield of 74% (
Scheme 2).
Spectroscopic data were initially consistent with our expectations and unambiguously confirmed by X-ray crystallography; though, curiously, the spectroscopic data obtained did not agree with the literature references and the overall observations during the synthesis.
However, it was observed that the freshly formed orange crystals (1a) rearranged within 1–2 days in the air into a new, yellow form (1b). This observation was independent of the scale. Crystals of both forms were collected and analyzed by X-ray diffraction. Freshly crystallized crude product 1a formed an orthorhombic Pcca lattice. On the other hand, the yellow crystals 1b crystallized in the monoclinic P21/c space group.
The chemical composition of both crystalline forms is similar—
1a represents pure compound
1 without additional solvent molecules, while
1b contains additional disordered water molecules. It is worth noting that the oxygen atom O3 from the main residue (carboxyl group) and the potassium ion are also disordered (
Figure 1). However, the structural differences between the two crystals are more significant. In order to determine the conformation of the two forms, the torsion angles C7-N1-C8-C9 were analyzed. For form
1a, its value of 172.39(18)° corresponds to the antiperiplanar conformation along the N1-C8 bond. In the case of form
1b, the angle mentioned is equal to 124.46(44)°, corresponding to the anticlinal conformation. This conformation is present in nickel complexes with substituted glycine of the same Schiff base skeleton; however, the torsions fall between 106.36–112.09° [
20,
21]. On the other hand, copper complexes with similar ligand derivatives form synclinal conformations with a range from 58.85–72.27° for their torsion angles [
22]. Nevertheless, none of the reported structures of the glycine-salicylaldehyde imine derivatives forms the antiperiplanar conformation as found in the metastable structure
1a.
Protonation of the nitrogen atom N1 in both structures results in an N1-C7 bond length of 1.287(2) in
1a and 1.301(5) Å in
1b, similar to the standard value [
23].
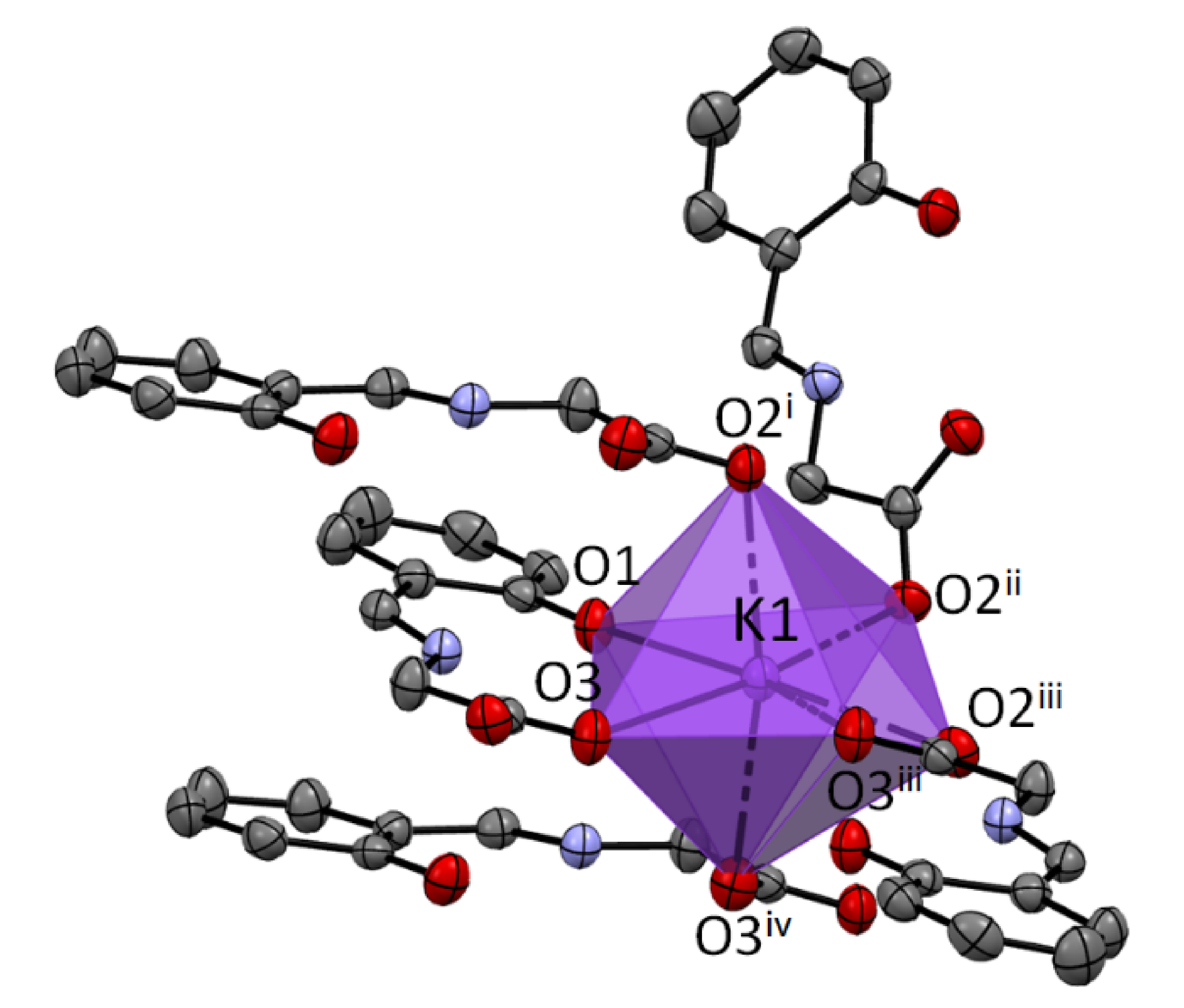
The coordination number of potassium is 7 for the structure
1a. The K ion is bonded to one O1, three O2, and three O3 atoms. It creates a very regular 1:4:2 7-coordinate geometry with only the O1 atom and one O3 atom in a unique position (
Figure 2).
In 1a, the parallel arrangements of the neighboring aromatic rings with the common potassium cations are most probably a consequence of their π-stacking. Intermolecular interactions in 1b do not reveal π-stacking of the aromatic systems, but consist of bonding to the carboxyl groups.
In both structures
1a and
1b, intramolecular hydrogen bonds are present (
Table 2). The structure
1a is stabilized by two hydrogen bonds between the N1 atom and the oxygen atoms O1 and O3. According to the characteristics of hydrogen bonds proposed by Desiraju and Steiner [
24], they can be classified as strong, conventional bonds. Structure
1b is also stabilized by an intramolecular hydrogen bond with parameters corresponding to strong, conventional bonds. It is worth noting that no hydrogen bond between N1 and the oxygen atoms from the disordered carboxylic group is observed, although a weak intermolecular hydrogen bond between C8 and the O3A atom from the carboxylic group occurs. There are also weak hydrogen bonds involving the water molecule with the O2W atom.
Table S1 contains detailed geometric parameters of the hydrogen bonds present in both structures. The characteristics of the fingerprint plots and the Hirshfeld surface are given in the
Supplementary Material.
Synthesis of the Schiff base
1 in the presence of water was not successful; however, encouraged by reports on the formation of metal complexes with ligand
1 using a variety of transition metal ions (Cu, Co, and Mn), we thought that it was still possible to synthesize them, as metal complexes tend to be more stable structures [
15]. Thus, complexation of Cu, Zn, Fe, Mn, and Mo ions with ligand
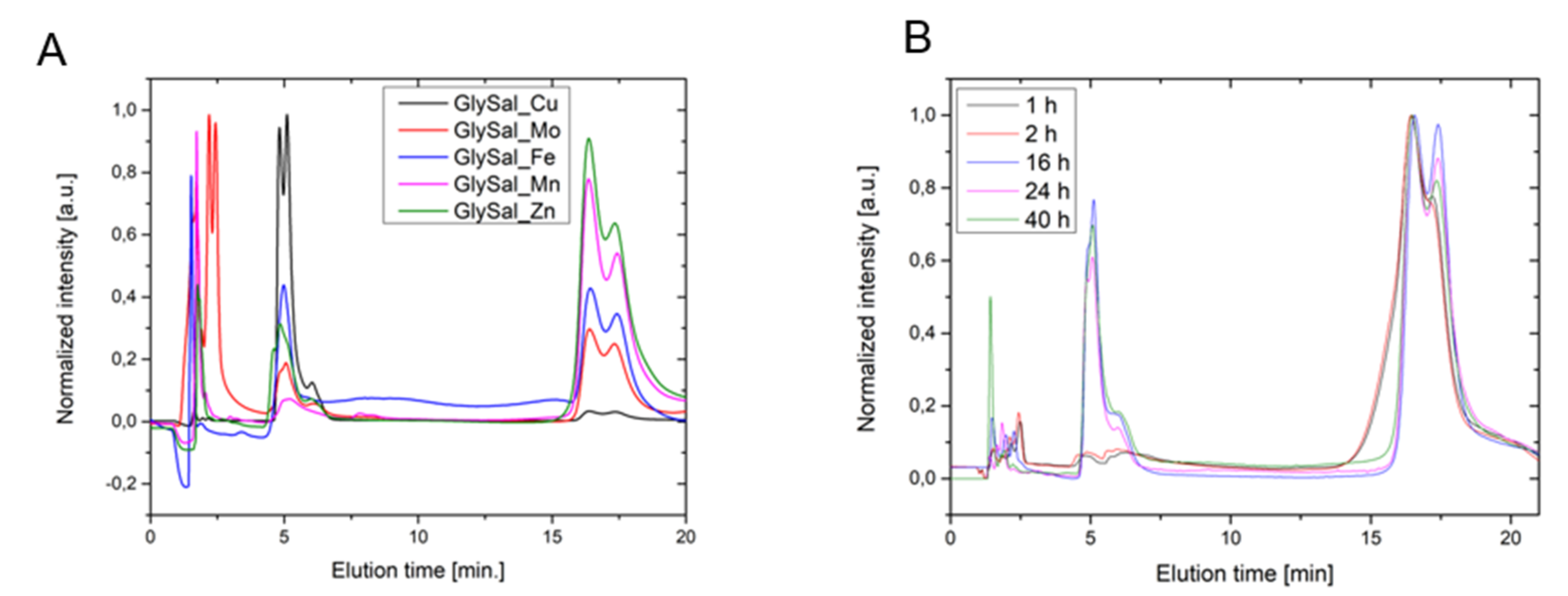
1 in an equimolar ratio (see experimental section for details) in water was performed. During these reactions, the formation of colored mixtures was observed, potentially indicating successful complexation. To confirm the formation of these new products, the reaction mixtures were analyzed by HPLC using MeOH:H
2O (30:70) as the eluent. All the chromatograms showed evidence of polar products between 0–4 min (metal sulfates, glycine, and salicylic acid) and a peak at 5 min, the area of which was proportionally reversible to a broad peak between 15–20 min corresponding to salicylaldehyde (
Figure 3). The peak appearing around 5 min was presumed to be the polar metal complex of
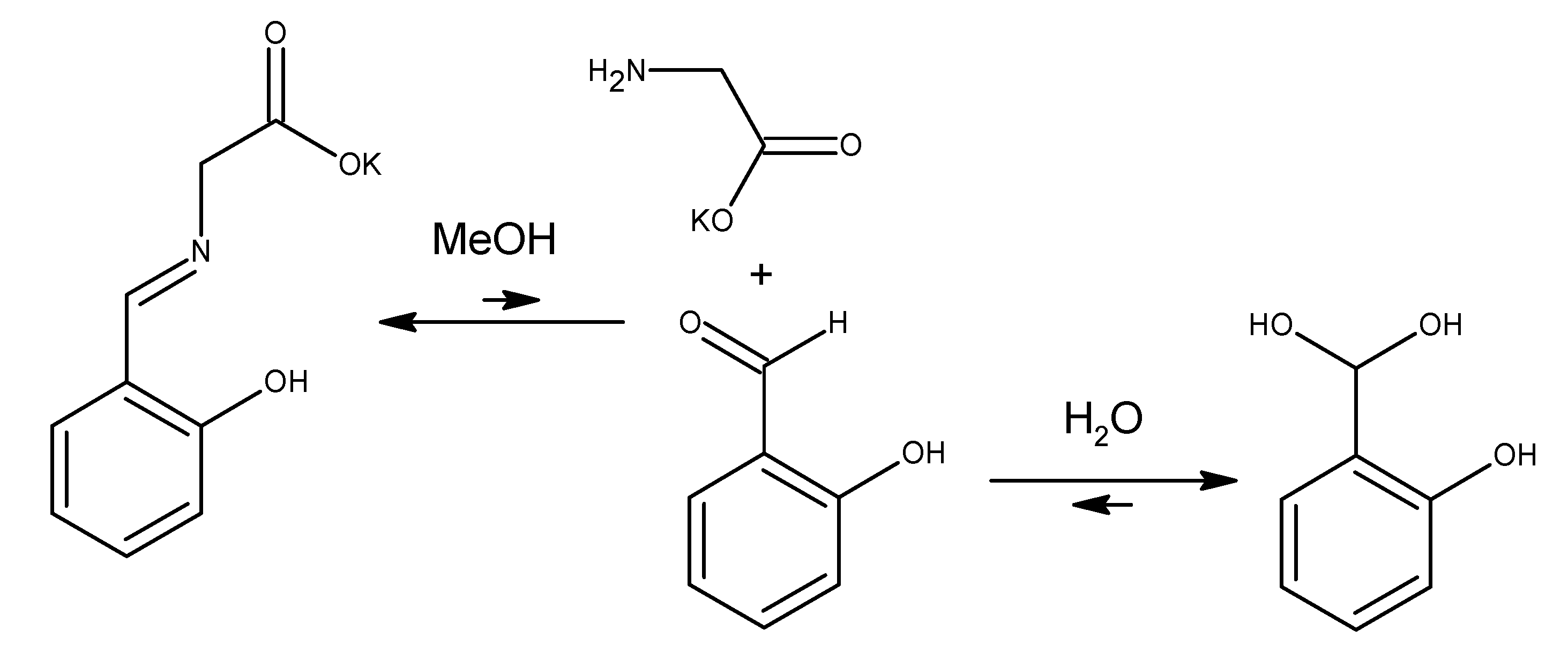
1, but the presence of salicylaldehyde in different amounts indicated hydrolysis. These observations led us to assess the behavior of
1 in water over time. In this experiment, we observed that ligand
1 in water decomposed mostly to salicylaldehyde, glycine, and the substance, with a retention time of around 5 min. As the concentration of free amino acid increased, the intensity of the peak at 5 min also increased, allowing us to recognize this signal as most probably salicylaldehyde hydrate, which forms under the basic conditions present in the aqueous solution (
Scheme 3). The peak position can shift and depends on the amount of the base added.
We performed UV-Vis experiments to confirm our observation on the hydrolysis of compound
1 during HPLC and determined the rate of this phenomenon. We tested the behavior of compound
1 in methanol (HPLC grade) with the addition of water in various ratios. We also added separately Fe
3+ or Cu
2+ to assess whether these metals would form complexes or catalyze hydrolysis. Additionally, we took UV-Vis spectra of salicylaldehyde and glycine in methanol with the addition of KOH and the abovementioned salts in order to study the stability of the reactants and the formation of the complexes potentially formed after the eventual hydrolysis. The addition of 0.1 equivalent of Fe
3+ or Cu
2+ to the methanolic solution of
1 (
Figures S5 and S6) caused the absorbance at 400 nm to decrease immediately (in less than 5 min). This band, which is present in the spectrum of compound
1, is specific to the C = N imine bond. It did not disappear completely because in this region there is also a band of salicylaldehyde hydrate, which was identified by its spectra with the addition of KOH (
Figure S7). For this reason, during the experiment with the addition of one equivalent of water to compound
1, we did not observe any significant decrease in the absorbance (
Figure S12) because of the equilibrium between these compounds. Only 10 times more water made a change to this band (
Figure S13). The comparison of the spectra of the Fe-salicylaldehyde and Fe-glycine complexes to the mixture of
1 with Fe
3+ (
Figure S10) may indicate that after hydrolysis we also observed the formation of a complex of the aldehyde, which can confirm the hydrolysis. These results are supported by the HPLC observation that imine
1 hydrolyzes rapidly (within 5 min) in the presence of a metal, and therefore the complex of
1 cannot be obtained in water.
Despite the general high stability of Schiff bases, it was found that this particular base undergoes hydrolysis in aqueous solutions at room temperature. Moreover, this reaction is followed by consecutive condensations, which most likely lead to polymeric, dark products within several days. Additionally, it is important to note that any attempts to synthesize transition metal complexes (such as Fe, Cu, and Mn) of
1 in aqueous media failed due to the accelerated hydrolysis of the imine. However, stable complexes were obtained from a non-aqueous, aprotic environment such as DMF (see
Supplementary Material for synthetic details ), but their contact with water still led to hydrolysis products. Based on the above facts, it can be concluded that the presence of water has a destructive effect on the ligand and leads to hydrolysis. There were two crystallographic structures of compound
1, which differed in the content of water in the crystallographic unit cell. Therefore, to get a better idea of the chemical nature of this compound, additional experiments were performed. Literature reports indicated that metastable Schiff bases were obtained by the solvent-free method, thus limiting the adverse effect of water on the reaction balance [
25,
26,
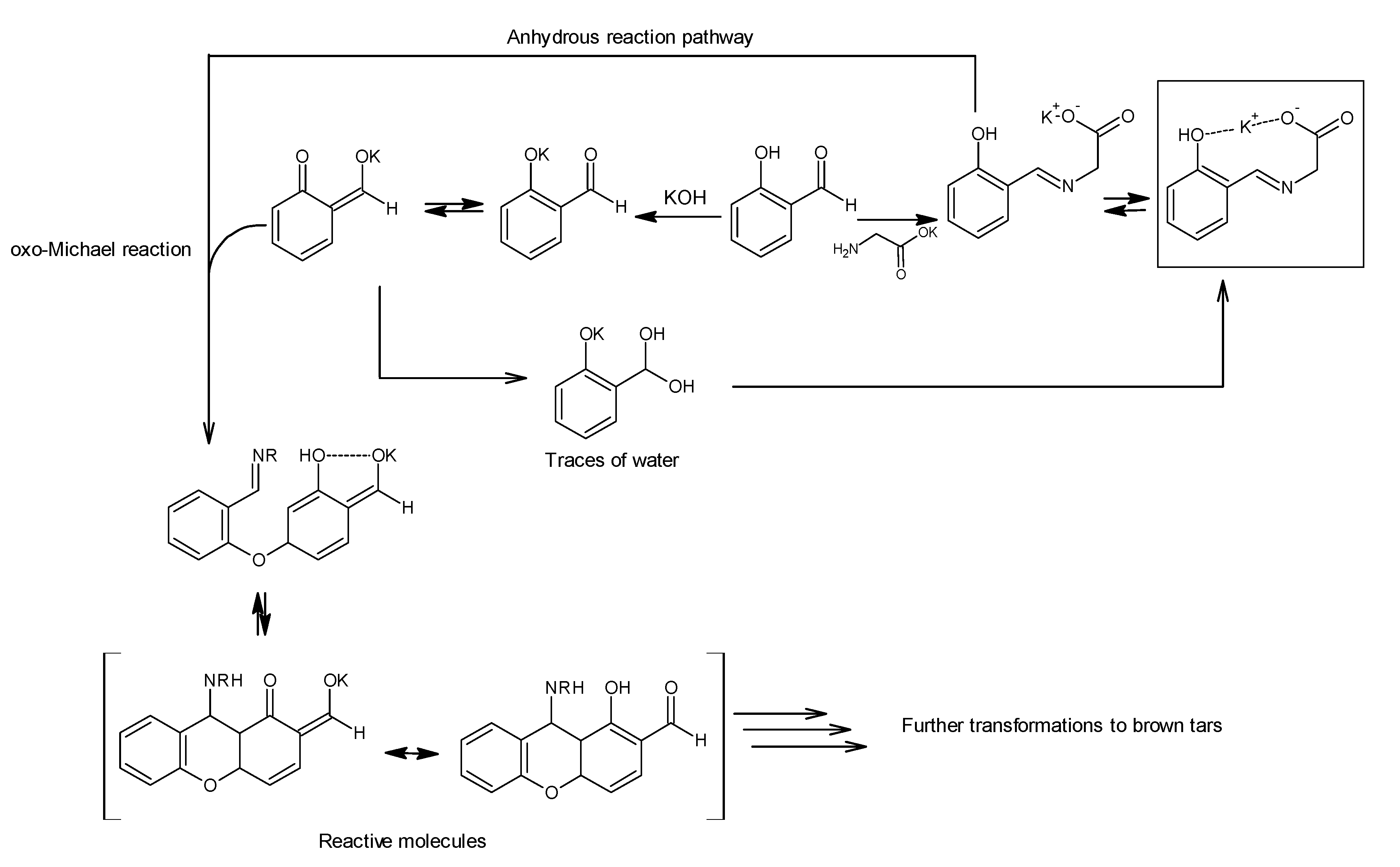
27]. Similar experiments were undertaken “without water” as follows: glycine and potassium hydroxide were stirred, and freshly distilled salicylaldehyde was added slowly. An emulsion formed which, while stirring, began to intensively heat up and change color. This reaction was carried out at 70 °C. After 30 min, we still observed large amounts of salicylaldehyde (conversion monitored by TLC). After 1 h, we quenched the reaction, and the reaction mixture was worked up to give a metastable oil-solid system. In addition, TLC analysis showed the presence of many new, inseparable products. A
1H-NMR of this mixture confirmed our assumptions that we had obtained a very complicated mixture in which many signals were observed in the aromatic range (see
Figure S7). To clarify the above observations, we hypothesized that trace amounts of water present in methanol led to the desired product, whereas an excess of water led to hydrolysis and, in its absence, other, different side reactions occurred. It is probable that under these anhydrous conditions, as the next step of condensation, the resulting ligand reacts with salicylaldehyde in an oxo-Michael reaction to form a chromanone derivative which can further react with compounds present in the mixture (
Scheme 4) [
28,
29,
30,
31,
32].
Additionally, we performed experimental studies on the thermal stability of both forms
1a and
1b in their solid states and the reversibility of their transformation to each other. Thus, the obtained product from the reaction containing both forms
1a and
1b was dried on a moisture analyzer to remove water (
Figure S16), leading to an intense orange compound, which was further analyzed by TGA under an argon atmosphere (
Figure S15). No weight loss that could be assigned to water was detected up to 200 °C. At this temperature, decomposition of the compound started; thus, we assume that it was the pure
1a form of the Schiff base. Furthermore, the
1a form was exposed to moisture in a phytotron chamber (see
SI for the details) until the whole volume of the compound turned light yellow, suggesting the exclusive formation of the
1b form. Measurement of the moisture content for this form of compound
1 with a moisture analyzer showed that 7.69% of the mass was water, which almost corresponds to its stoichiometric content of 7.65%. Additionally, TGA analysis of the
1b form showed a 7% weight loss in the temperature range from 25 °C to 100 °C, which can be assigned to the removal of water, while above this temperature the course of
1b’s decomposition was identical with
1a, confirming that both compounds are chemically identical. The difference in water content between those two analyses may be connected with the transformation of
1b into the more stable
1a form, as confirmed by crystallographic structures, during the few hours that it took us to transport the sample for TGA analysis, despite our efforts to preserve the
1b compound unchanged. Finally, we dried the
1b form to
1a, and the course of the TGA decomposition was identical with the original
1a form, confirming the full reversibility of the hydration process and the lack of any negative influence on both structures.
4. Materials and Methods
Glycine (5.0 g, 67 mmol) and one equiv. of KOH (3.7 g, 66 mmol) were dissolved in methanol (50 mL). A solution of salicylaldehyde (8.1 g; 66 mmol) in methanol (50 mL) was added. The color of the solution rapidly turned yellow with the formation of an orange precipitate. The mixture was stirred for 1 h at room temperature. The resulting solid product was filtered and air-dried. During drying, the color of the product changed from orange to yellow. Yield 74%, (10.7 g, 49 mmol) m.p. = 219–221 °C.
NMR spectra were recorded on an Agilent 400 MHz spectrometer at room temperature. Aromatic signals were assigned by simulations using ChemDraw® software (license No. 2013781467484).
1H-NMR (400 MHz, CD3OD) δ 8.34 (s, 1H, CAr-CH=N), 7.30 (d, J. = 7.6 Hz, 1H o-CAr-H), 7.27 (dd, J. = 8.2, 7.4 Hz, 1H, p-CAr-H), 6.79 (dd, J. = 8.2, 0.8 Hz, 1H, m-CAr-H-CAr-OH), 6.73 (ddd, J. = 7.6, 7.4, 0.8 Hz, 1H, m-CAr-H-CAr-H), 4.23 (m, 2H, =N-CH2). 13C-NMR (400 MHz, CD3OD) δ 175.80 (COO), 168.00 (CH=N), 167.92 (CAr-OH), 134.85 (p-CAr-H), 133.61 (o-CAr-H), 119.78 (m-CAr-H), 119.17 (CAr-CH=N), 117.88 (CAr-H-CAr-OH), 60.89 (=N-CH2). MS for C9H9NO3 [M+H]+: calculated = 180.1; found: 180.2.
4.1. Attempted Syntheses of Cu, Zn, Fe, Mn, and Mo. Metal. Complexes with Ligand 1
HPLC analyses were carried out using a Dionex UltiMate 3000 HPLC system equipped with an autosampler. Chromatographic separation was achieved with Thermo Scientific Hypersil GOLD HPLC (125 × 3 mm; 5 μm particle size) column, using water (solvent A, 70%) and methanol (solvent B, 30%). Sample injection volumes were 10 μL, the flow rate was 1 mL/min, and the column temperature was maintained using an oven at T = 23 °C.
In a 50 mL round bottom flask, 10 mL of a 1 mM solution of ligand 1 was mixed with 10 mL of a 1 mM solution of metal sulfate and stirred for 1 h. 20 μL was then taken and diluted with distilled water to 2 mL, filtered through a 0.22 μm pore size syringe filter, and analyzed by HPLC.
4.2. Hydrolysis Experiment
In a 25 mL round bottom flask, 1 mmol of ligand 1 was dissolved in 10 mL of water and stirred for 40 h. After 1, 2, 16, 24, and 40 h, a 20 μL solution aliquot was diluted with distilled water to 2 mL, filtered through a 0.22 μm pore size syringe filter, and analyzed by HPLC.
4.3. UV-Vis. Measurements
UV-Vis analyses were carried out using a Hitachi U-2910 in the wavelength range 190–900 nm. 10 mm quartz cuvettes were used. Sample preparation: 10 mL of a 1 mM solution of ligand
1 in methanol (HPLC grade) was prepared, and then one equivalent or 10 equivalents of water was added, respectively. These solutions were transferred sequentially to the cuvette, capped, and measured for at least 6 h. 10 mL of a 1 mM solution of ligand
1 in methanol (HPLC grade) was then measured as a blank, followed by the addition of 0.1 and 1 equivalent FeCl
3 or CuCl
2, respectively, which were measured for at least 3 h. Additionally, the following solutions were prepared and measured: 1 mmol solution of salicylaldehyde + 1 equiv. KOH in methanol followed by the addition of 1 equiv. of FeCl
3; and 1 mmol solution of glycine + 1 equiv. KOH + 1 equiv. FeCl
3. All UV-Vis spectra are attached to the
supplementary material.
4.4. X-ray Diffraction Measurements
X-ray diffraction data were collected using an Xcalibur diffractometer with Mo Kα radiation (λ = 0.71073 Å) at room temperature for crystal
1a and using a SuperNova diffractometer with Cu Kα radiation (λ = 1.54184 Å) at 120 K for crystal
1b. Data were integrated with CrysAlis
Pro software (version 1.171.39.15e for 1a, 1.171.38.41q for 1b) [
33]. The phase problem was solved by direct methods with SHELXS-97 [
34]. The model parameters were refined by full-matrix least-squares on F
2 using SHELXL-2018/3 [
34]. All programs were included in the WINGX package (version 2018.2) [
35].
1a represents a pure compound without any additional solvent molecules. Compound 1b is a hydrate and is also a pure compound. The presence of the additional atoms results from the disordering of the carboxyl group and the K+ ions. The number of water molecules can be defined based on an analysis of the occupancies of the oxygen atoms in the water molecules. The O1W atom from the first water molecule has an occupancy equal to 0.5. This atom is placed in the special position (the coordination is 0.5, 0.5, 0.5), and thus the real occupancy is 1. The O2W atom from the second water molecule has an occupancy equal to 0.46(1). In the final refinement, it was fixed at 0.5. The hydrogen atoms that are present at the O2W atom were found on the difference Fourier map. In order to preserve the correct geometry of the water molecules, the hydrogen atoms were placed using a suitable HFIX. In the case of the O1W atoms, the hydrogen atoms were not visible on the difference Fourier map.
Crystal Data for 1a (C9H8NO3K; M = 217.26 g/mol): orthorhombic, space group Pcca (no. 54), a = 13.9484(12) Å, b = 18.5280(16) Å, c = 7.0215(8) Å, V = 1814.6(3) Å3, Z = 8, μ(MoKα) = 0.562 mm−1, Dcalc = 1.591 g/cm3, 12,574 reflections measured (6.8° ≤ 2Θ ≤ 52.6°), 1842 unique (Rint = 0.0496, Rsigma = 0.0281) which were used in all calculations. The final R1 was 0.0387 (I > 2σ(I)), and wR2 was 0.0900 (all data).
Crystal Data for 1b (C9H9O4NK; M = 234.27 g/mol): monoclinic, space group P21/c (no. 14), a = 18.879(3) Å, b = 6.2395(6) Å, c = 8.7555(9) Å, β = 99.765(12)°, V = 1016.4(2) Å3, Z = 4, μ(CuKα) = 4.567 mm−1, Dcalc = 1.531 g/cm3, 3619 reflections measured (9.5° ≤ 2Θ ≤ 117.6°), 1350 unique (Rint = 0.0320, Rsigma = 0.0421 which were used in all calculations. The final R1 was 0.0615 (I > 2σ(I)) and wR2 was 0.1854 (all data).
4.5. Moisture and Thermal Analysis
TGA curves were recorded using a LINSEIS STA PT1600 at a heating rate of 10 °C/min under an argon atmosphere over a 25 °C to 800 °C temperature range. Moisture analysis was performed on a Radwag MA 50/1.R.WH.
4.6. Mass Spectrometry
Mass spectrometry employed an API 4000 QTRAP tandem mass spectrometer detector equipped with an electrospray ionization (ESI) source (Applied Biosystems/MDS SCIEX, Foster City, CA, USA).

 ,
, 








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}