Effect of Initial Conformation on the Starch Biopolymer Film Formation Studied by NMR



Abstract
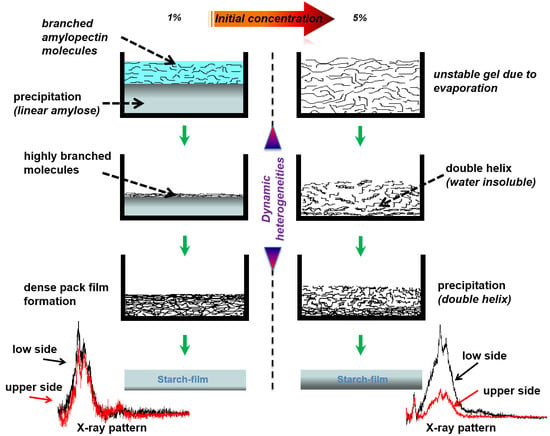
1. Introduction
2. Results and Discussion
2.1. Film Formation Study Using a Micro Camera
2.2. Real-Time Film Formation Study Using Single-Sided NMR Scanner
2.2.1. Profiling of the Samples
2.2.2. Determination of Sample Shrinking Rate
2.2.3. Determination of T2 at Different Heights
2.2.4. Determination of T2 of Starch Samples at an Early Stage
2.3. Dried Film Characterizations
2.3.1. Single-Sided NMR Study
2.3.2. X-Ray Diffraction (XRD) Study
3. Proposed Mechanism of Starch Film Formation
4. Materials and Methods
4.1. Materials
4.2. Methods
4.2.1. Sample Preparation
4.2.2. Samples
4.2.3. Film Formation Study Using a Micro Camera
4.2.4. Film Formation Study Using the Single-Sided NMR Scanner
4.2.5. Dried Film Characterizations
5. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Belgacem, M.N.; Gandini, A. Monomers, Polymers and Composites from Renewable Resources; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Barsby, T.L.; Donald, A.M.; Frazier, P.J. Starch: Advances in Structure and Function; Royal Society of Chemistry: London, UK, 2001. [Google Scholar]
- BeMiller, J.N.; Whistler, R.L. Starch: Chemistry and Technology; Academic Press: Cambridge, MA, USA, 2009. [Google Scholar]
- Jenkins, P.J.; Donald, A.M. The influence of amylose on starch granule structure. Int. J. Biol. Macromol. 1995, 17, 315–321. [Google Scholar] [CrossRef]
- Jobling, S. Improving starch for food and industrial applications. Curr. Opin. Plant Biol. 2004, 7, 210–218. [Google Scholar] [CrossRef] [PubMed]
- Krogars, K.; Heinämäki, J.; Karjalainen, M.; Rantanen, J.; Luukkonen, P.; Yliruusi, J. Development and characterization of aqueous amylose-rich maize starch dispersion for film formation. Eur. J. Pharm. Biopharm. 2003, 56, 215–221. [Google Scholar] [CrossRef]
- Robyt, J.F. Essentials of Carbohydrate Chemistry; Springer: New York, NY, USA, 1998. [Google Scholar]
- Ring, S.G. Some studies on starch gelation. Starch 1985, 37, 80–83. [Google Scholar] [CrossRef]
- Jenkins, P.J.; Donald, A.M. Gelatinisation of starch: A combined SAXS/WAXS/DSC and SANS study. Carbohydr. Res. 1998, 308, 133–147. [Google Scholar] [CrossRef]
- Tananuwong, K.; Reid, D.S. Differential scanning calorimetry study of glass transition in frozen starch gels. J. Agric. Food Chem. 2004, 52, 4308–4317. [Google Scholar] [CrossRef] [PubMed]
- Chatakanonda, P.; Varavinit, S.; Chinachoti, P. Relationship of gelatinization and recrystallization of cross-linked rice to glass transition temperature. Cereal Chem. J. 2000, 77, 315–319. [Google Scholar] [CrossRef]
- Svoboda, V.; Liaw, B.Y. In situ transient study of polymer film growth via simultaneous correlation of charge, mass, and ellipsometric measurements. Pure Appl. Chem. 2008, 80, 2439–2449. [Google Scholar] [CrossRef]
- Peura, M.; Karppinen, T.; Soovre, A.; Salmi, A.; Tenkanen, M.; Hæggström, E.; Serimaa, R. Crystallization and shear modulus of a forming biopolymer film determined by in situ x-ray diffraction and ultrasound reflection methods. J. Appl. Phys. 2008, 104, 23513. [Google Scholar] [CrossRef]
- Wang, Y.; Juhué, D.; Winnik, M.A.; Leung, O.; Goh, M.C. Atomic force microscopy study of latex film formation. Langmuir 1992, 8, 760–762. [Google Scholar] [CrossRef]
- Fissel, A.; Dabrowski, J.; Osten, H.J. Photoemission and ab initio theoretical study of interface and film formation during epitaxial growth and annealing of praseodymium oxide on Si(001). J. Appl. Phys. 2002, 91, 8986–8991. [Google Scholar] [CrossRef]
- Ghoshal, S.; Mattea, C.; Stapf, S. Inhomogeneity in the drying process of gelatin film formation: NMR microscopy and relaxation study. Chem. Phys. Lett. 2010, 485, 343–347. [Google Scholar] [CrossRef]
- Ghoshal, S.; Mattea, C.; Denner, P.; Stapf, S. Heterogeneities in Gelatin Film Formation Using Single-Sided NMR. J. Phys. Chem. B 2010, 114, 16356–16363. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, S.; Denner, P.; Stapf, S.; Mattea, C. Structural and dynamical heterogeneities in PVA films induced by evaporation during the formation process. Chem. Phys. Lett. 2011, 515, 231–234. [Google Scholar] [CrossRef]
- Ghoshal, S.; Denner, P.; Stapf, S.; Mattea, C. Study of the formation of poly(vinyl alcohol) films. Macromolecules 2012, 45, 1913–1923. [Google Scholar] [CrossRef]
- Mattea, C.; Ghoshal, S.; Pabjańczyk, E.; Stapf, S. Poly(vinyl alcohol)film formation process using single-sided low-field NMR relaxometry: effect of initial concentration. Micropor. Mesopor. Mat. 2013, 178, 27–30. [Google Scholar] [CrossRef]
- Ghoshal, S.; Mattea, C.; Du, L.; Stapf, S. Concentration and humidity effect on gelatin films studied by NMR. Zeitschrift für Physikalische Chemie 2012, 226, 1259–1270. [Google Scholar] [CrossRef]
- Ghoshal, S.; Stapf, S.; Mattea, C. Protein renaturation in the gelatin film formation process. Appl. Magn. Reson. 2014, 45, 145–154. [Google Scholar] [CrossRef]
- Ghoshal, S. Study of Polymer Film Formation and Their Characterization Using NMR, XRD and DSC. Ph.D. Thesis, Technische Universität Ilmenau, Ilmenau, Germany, 2012. [Google Scholar]
- Meiboom, S.; Gill, D. Modified Spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29, 688–691. [Google Scholar] [CrossRef]
- Kimmich, R.K. NMR: Tomography, Diffusion, Relaxometry; Springer: New York, NY, USA, 1997. [Google Scholar]
- Tizzotti, M.J.; Sweedman, M.C.; Tang, D.; Schaefer, C.; Gilbert, R.G. New 1 H NMR procedure for the characterization of native and modified food-grade starches. J. Agric. Food Chem. 2011, 59, 6913–6919. [Google Scholar] [CrossRef]
- Park, S.; Baker, J.O.; Himmel, M.E.; Parilla, P.A.; Johnson, D.K. Cellulose crystallinity index: measurement techniques and their impact on interpreting cellulase performance. Biotechnol. Biofuels 2010, 3, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Widjonarko, N.E. Introduction to Advanced X-ray Diffraction Techniques for Polymeric Thin Films. Coatings 2016, 6, 54. [Google Scholar] [CrossRef]
- Le Bail, P.; Bizot, H.; Buléon, A. ‘B’ to ‘A’ type phase transition in short amylose chains. Carbohydr. Polymers 1993, 21, 99–104. [Google Scholar] [CrossRef]
- Zhai, M.; Zhao, L.; Yoshii, F.; Kume, T. Study on antibacterial starch/chitosan blend film formed under the action of irradiation. Carbohydr. Polymers 2004, 57, 83–88. [Google Scholar] [CrossRef]
- Green, M.M.; Blankenhorn, G.; Hart, H. Which starch fraction is water-soluble, amylose or amylopectin? J. Chem. Educ. 1975, 52, 729–730. [Google Scholar] [CrossRef]
- Kabalnov, A.; Wennerström, H. Diffusion in evaporating solutions. Soft Matter 2009, 5, 4712. [Google Scholar] [CrossRef]
- Talja, R.A.; Helén, H.; Roos, Y.H.; Jouppila, K. Effect of various polyols and polyol contents on physical and mechanical properties of potato starch-based films. Carbohydr. Polymers 2007, 67, 288–295. [Google Scholar] [CrossRef]
- Zhang, Y.; Han, J.H. Mechanical and thermal characteristics of pea starch films plasticized with monosaccharides and polyols. J. Food Sci. 2006, 71, E109–E118. [Google Scholar] [CrossRef]
- Jansson, A.; Thuvander, F. Influence of thickness on the mechanical properties for starch films. Carbohydr. Polymers 2004, 56, 499–503. [Google Scholar] [CrossRef]
- Casanova, F.; Perlo, J.; Blümich, B. Single-Sided NMR; Springer: New York, NY, USA, 2011. [Google Scholar]
- Perlo, J.; Casanova, F.; Blümich, B. Profiles with microscopic resolution by single-sided NMR. J. Magnetic Reason. 2005, 176, 64–70. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds are not available from the authors. |











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Initial Concentration (%) of Starch in the Solvent (w/v) | Solvent | Initial Physical Status of the Sample | Study | Thickness of the Final 3 Film (µm) |
|---|---|---|---|---|---|
| a | 1 | H2O | suspension | Using micro camera | - |
| b | 5 | H2O | gel | Using micro camera | - |
| c | 1 | H2O | suspension | Profiling 1 at 11.7 MHz (1H) and XRD | 130 ± 15 |
| d | 5 | H2O | gel | Profiling 2 at 11.7 MHz (1H) and XRD | 170 ± 10 |
| e | 1 | H2O | suspension | T2 study at 11.7 MHz (1H) | - |
| f | 1 | D2O | suspension | T2 study at 11.7 MHz (1H) | - |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghoshal, S.; Mattea, C.; Denner, P.; Stapf, S. Effect of Initial Conformation on the Starch Biopolymer Film Formation Studied by NMR. Molecules 2020, 25, 1227. https://doi.org/10.3390/molecules25051227
Ghoshal S, Mattea C, Denner P, Stapf S. Effect of Initial Conformation on the Starch Biopolymer Film Formation Studied by NMR. Molecules. 2020; 25(5):1227. https://doi.org/10.3390/molecules25051227
Chicago/Turabian StyleGhoshal, Sushanta, Carlos Mattea, Paul Denner, and Siegfried Stapf. 2020. "Effect of Initial Conformation on the Starch Biopolymer Film Formation Studied by NMR" Molecules 25, no. 5: 1227. https://doi.org/10.3390/molecules25051227
APA StyleGhoshal, S., Mattea, C., Denner, P., & Stapf, S. (2020). Effect of Initial Conformation on the Starch Biopolymer Film Formation Studied by NMR. Molecules, 25(5), 1227. https://doi.org/10.3390/molecules25051227