
Mesoscale Assembly of Bisteroidal Esters from Terephthalic Acid

 , , ,
, , ,
Abstract
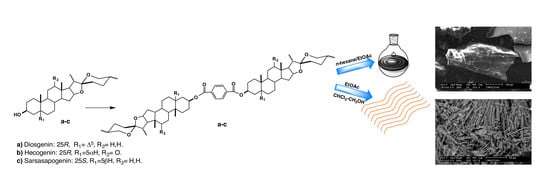
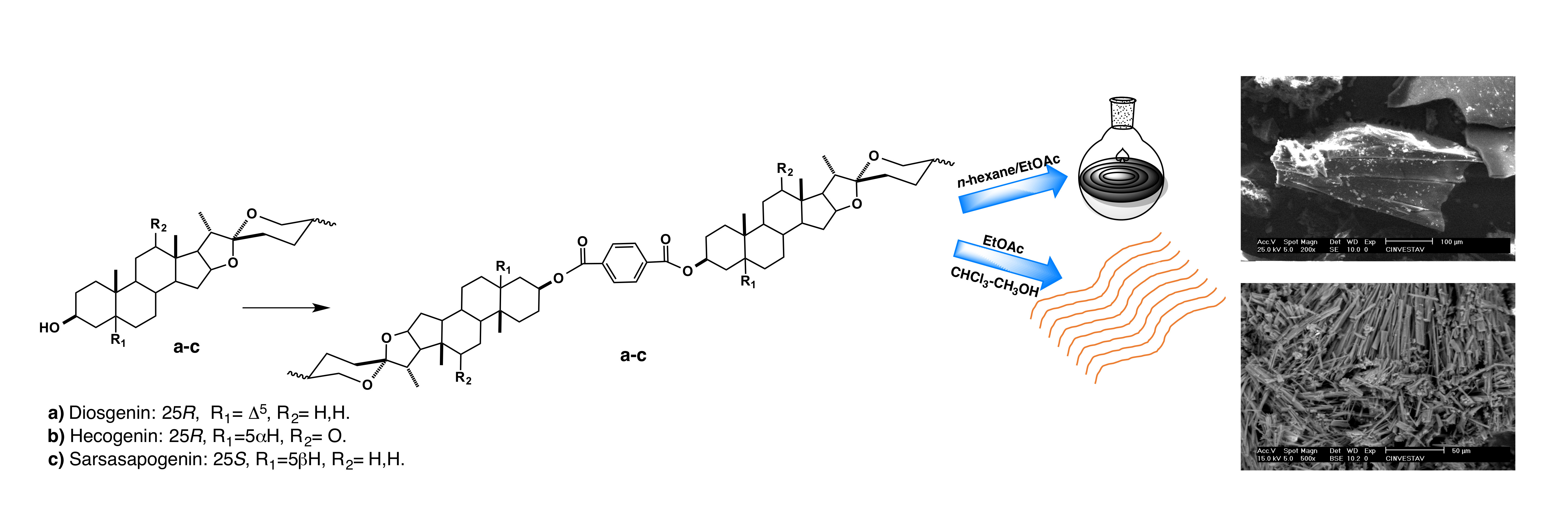
1. Introduction
2. Results
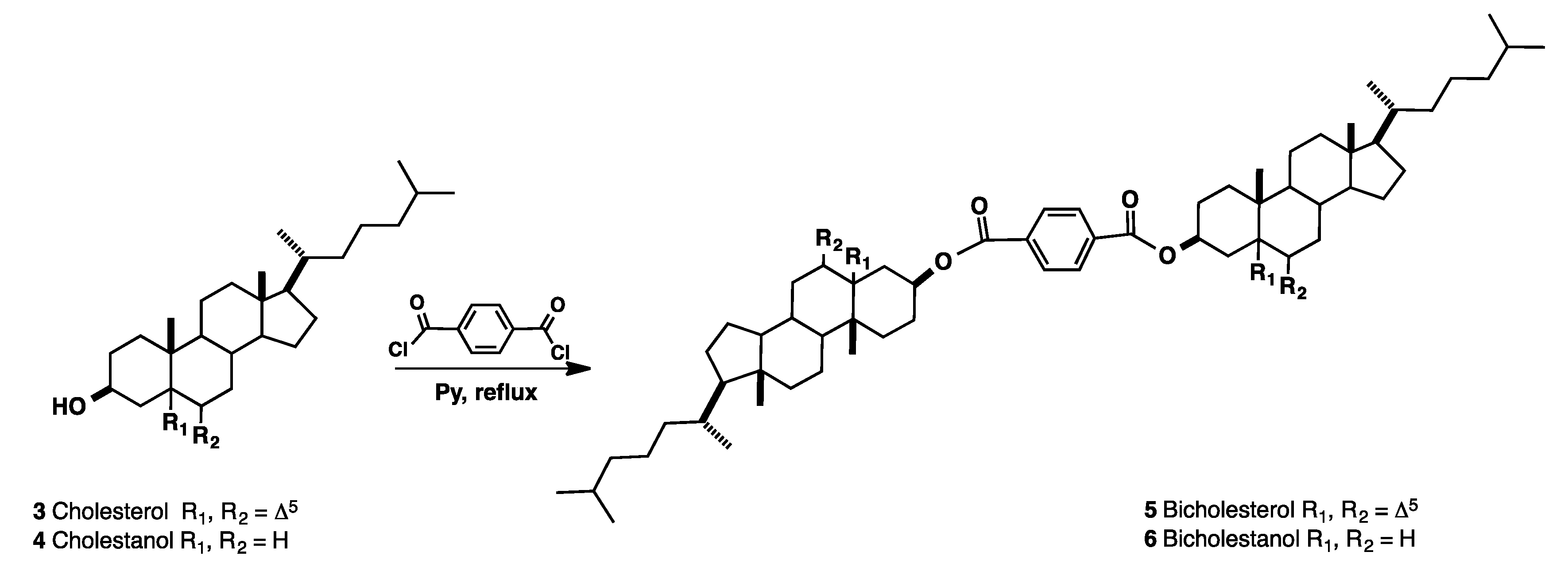
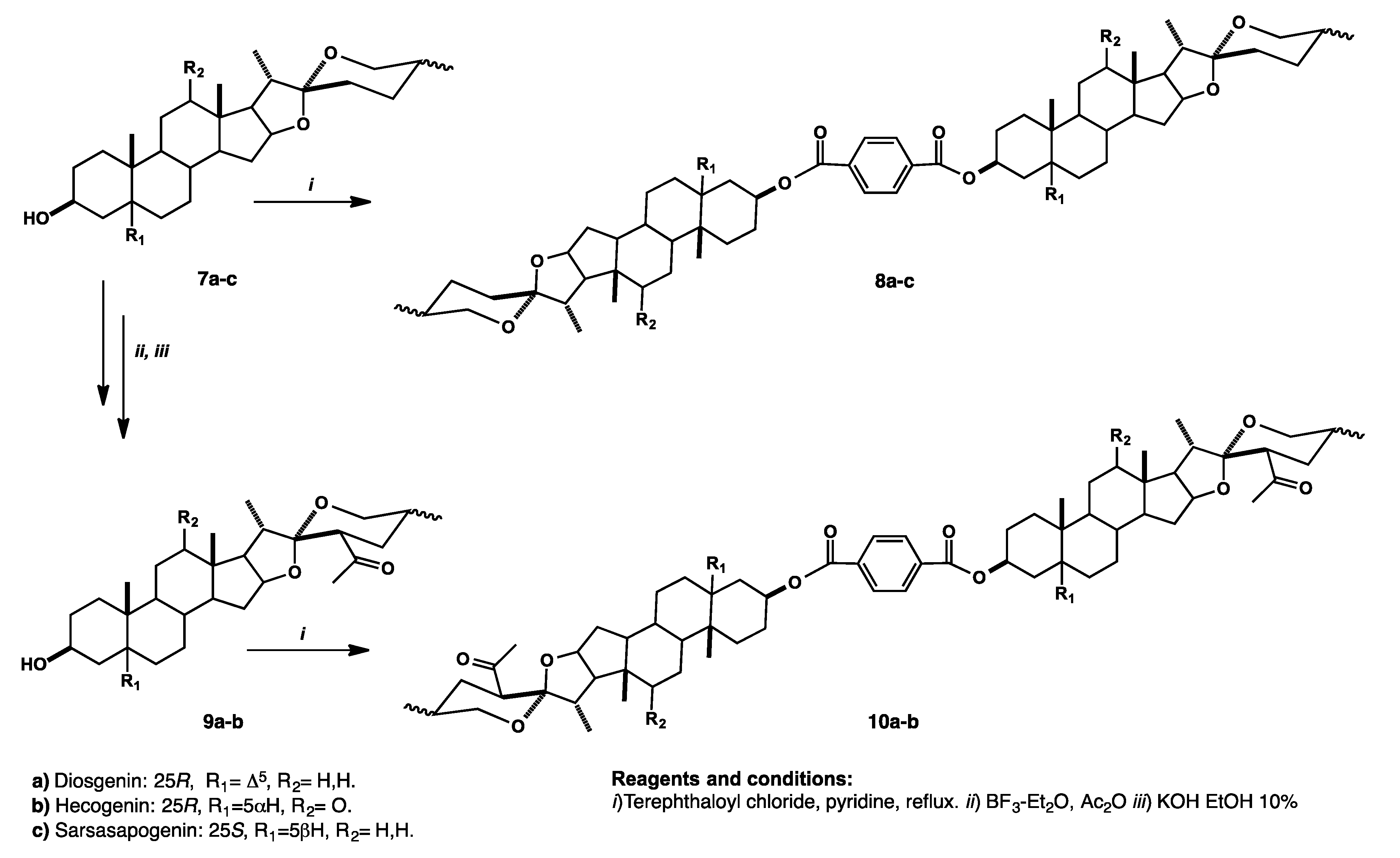
Synthesis and Purification of Bisteroidal Esters
3. Discussion
3.1. Structural Characterization of Bisteroidal Esters
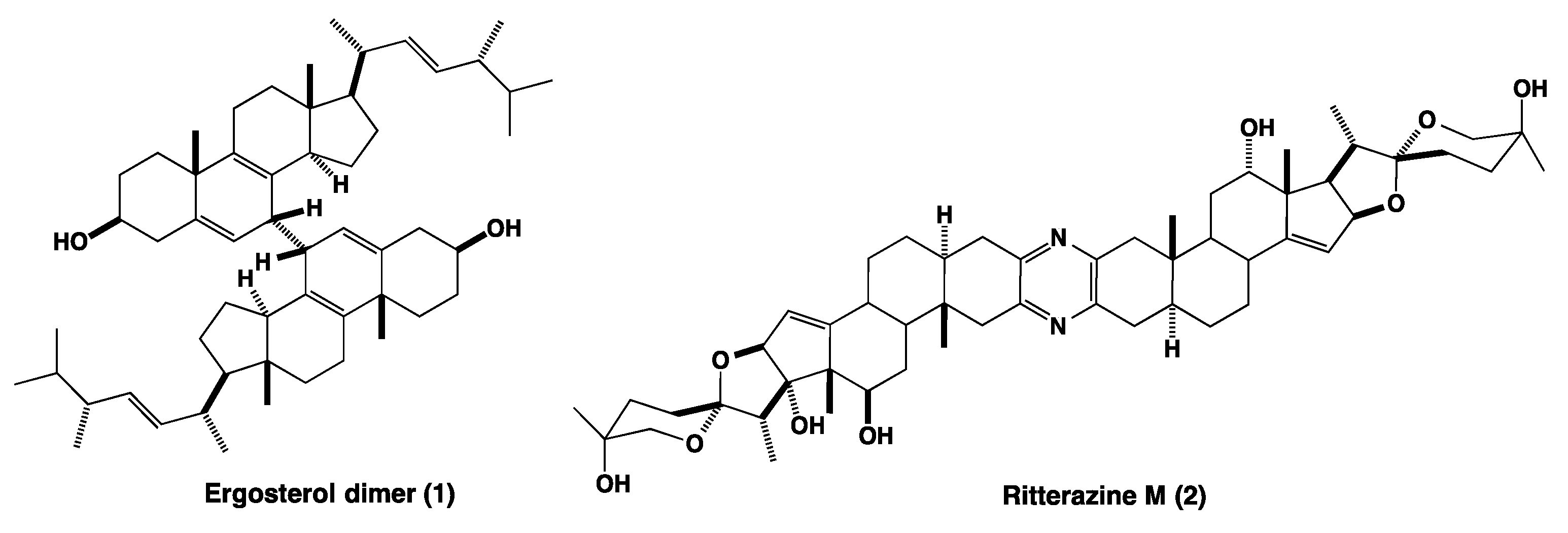
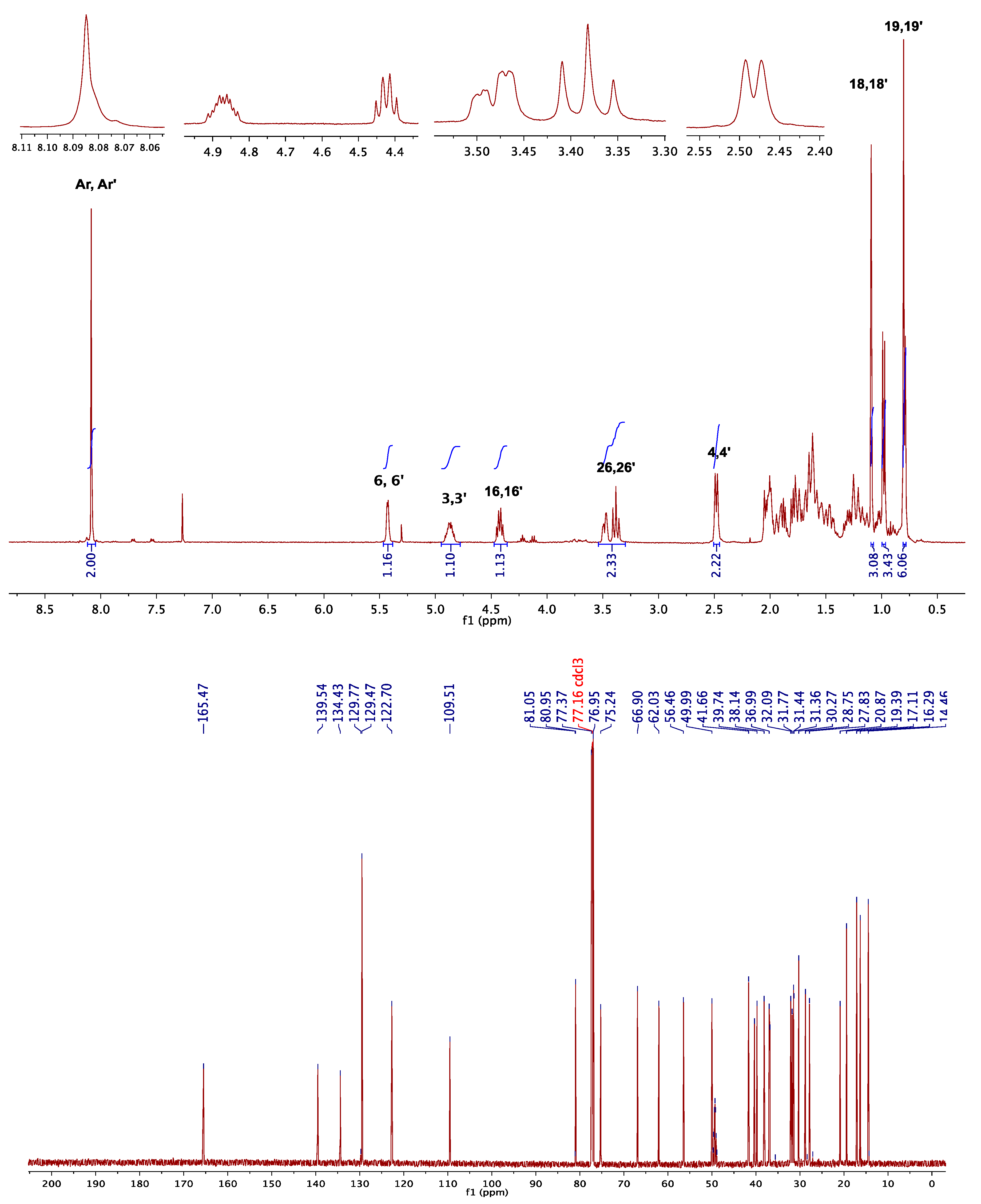
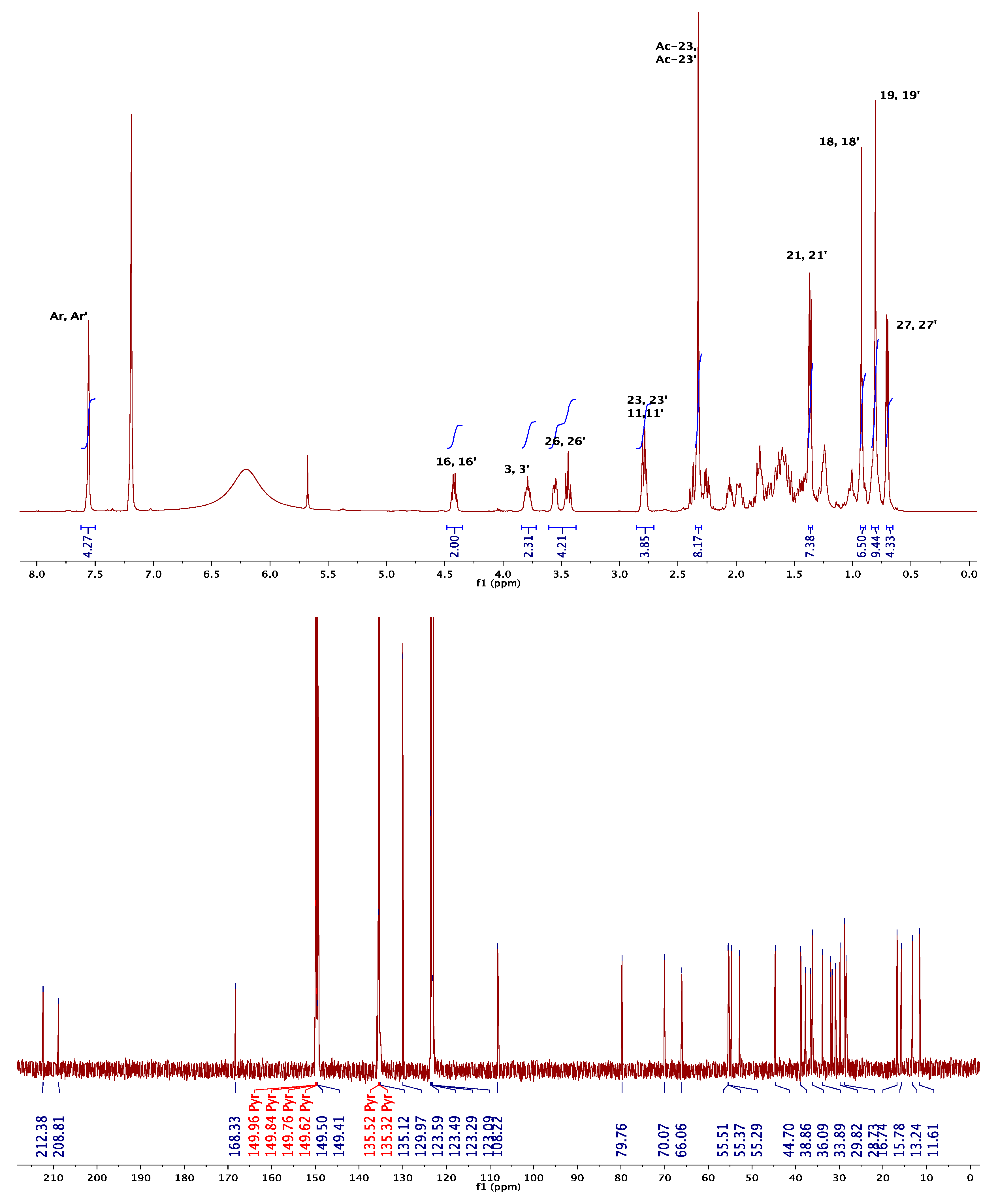
3.1.1. NMR Elucidation
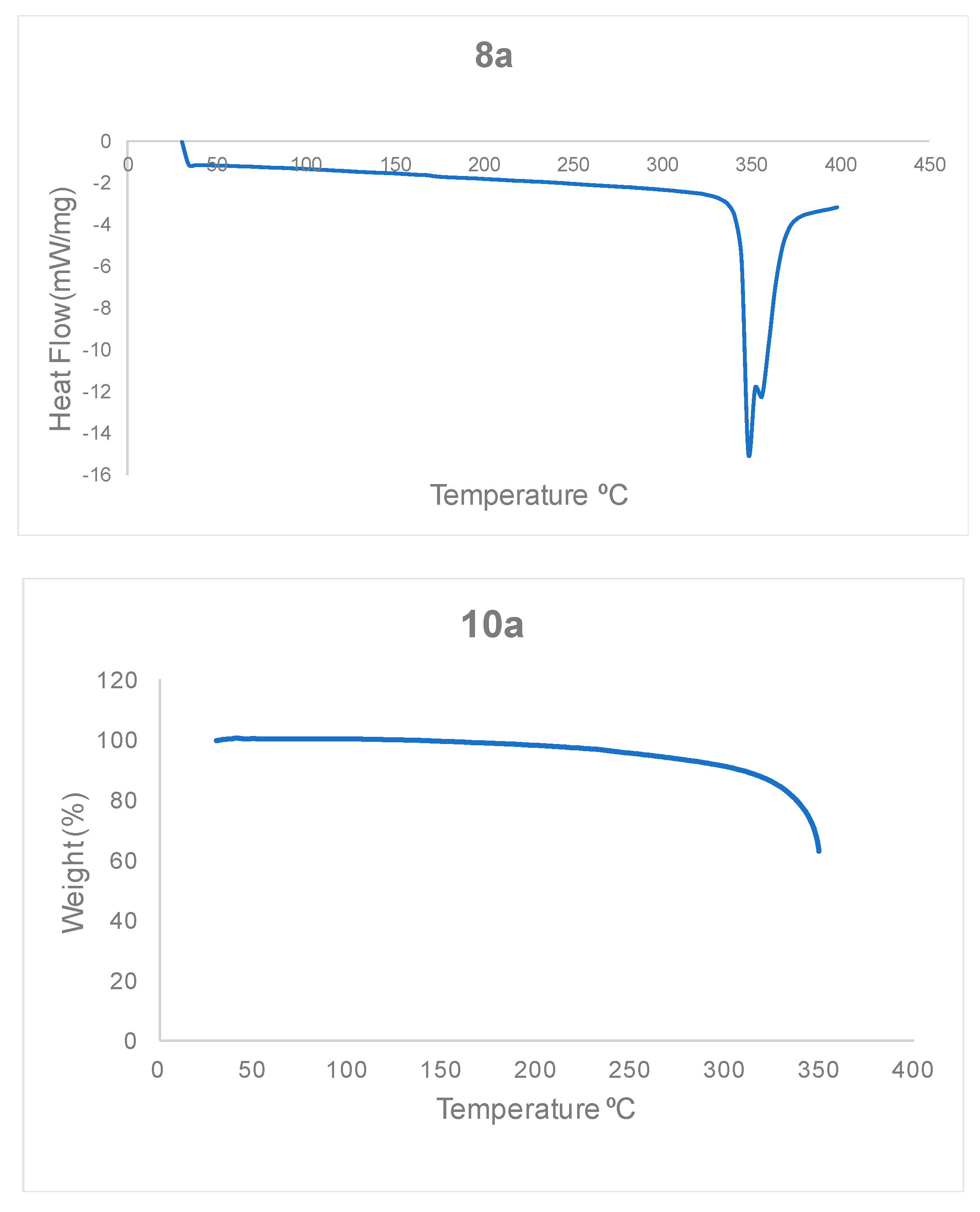
3.1.2. Thermal Stability of Bisteroidal Esters of Terephthalic Acid
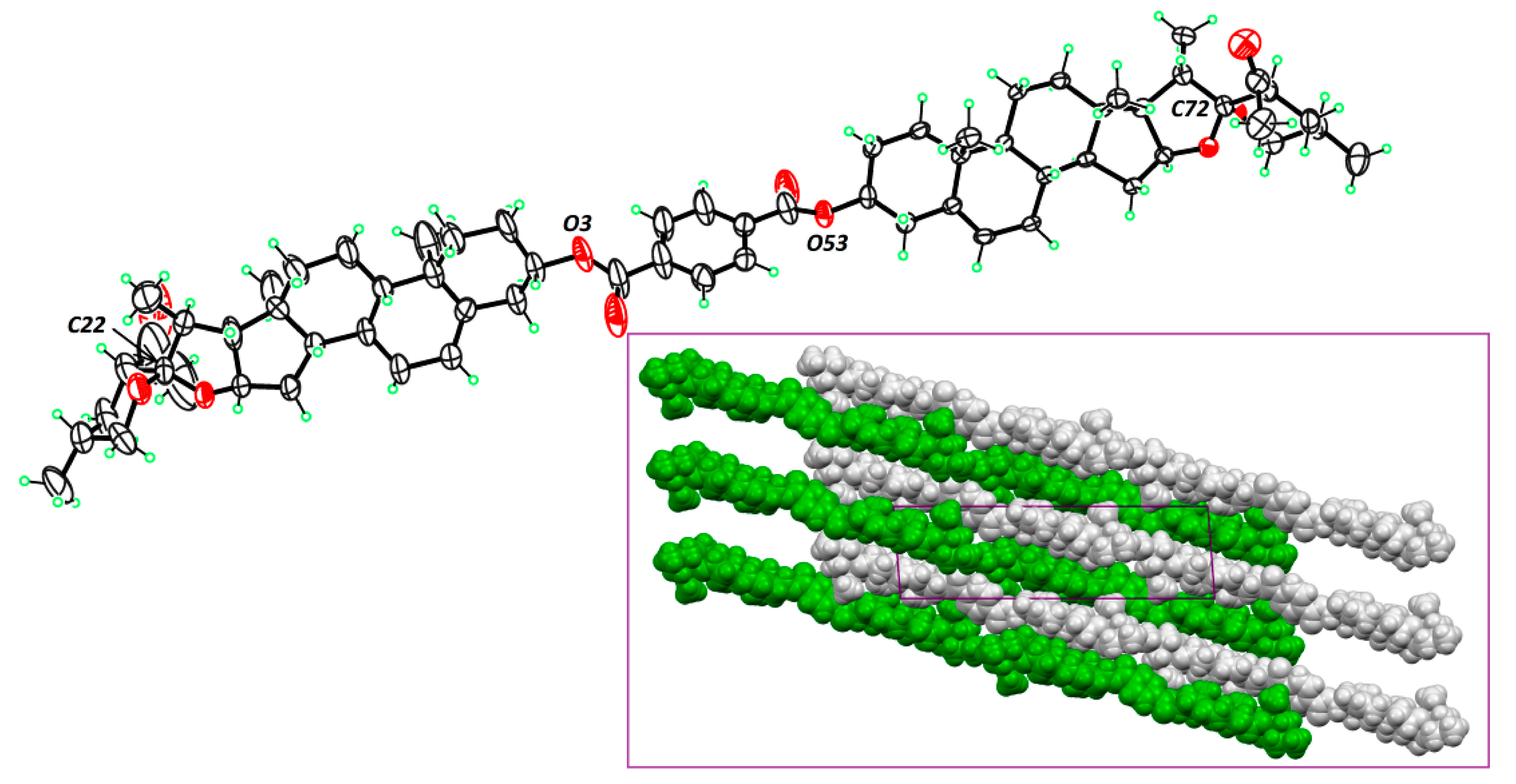
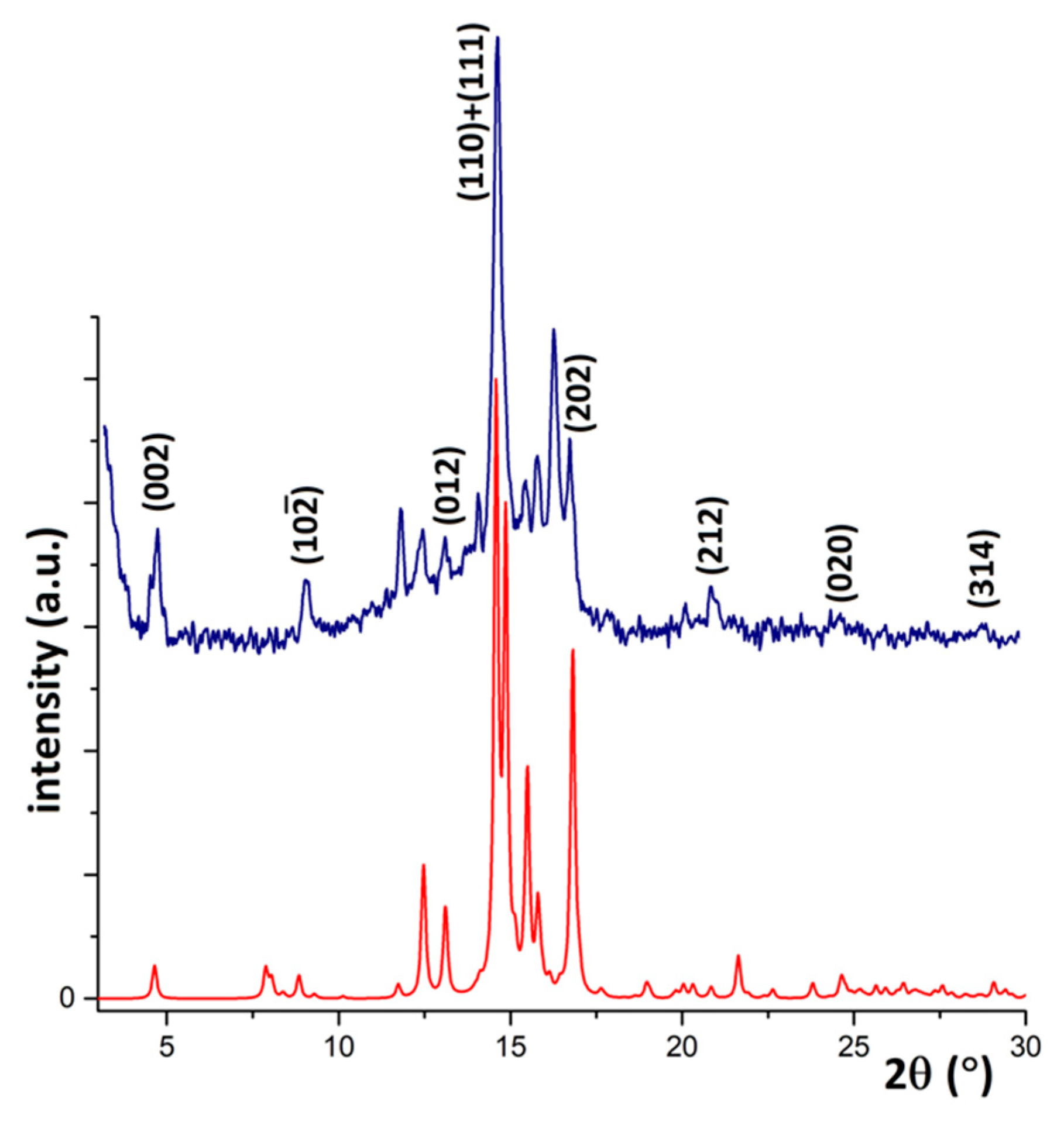
3.2. Responsive Self-Assembly of Bisteroidal Esters of Terephthalic Acid
4. Material and Methods
4.1. General Procedures and Materials
4.1.1. Scanning Electron Microscopy (SEM)
4.1.2. Differential Scanning Calorimetry (DSC)
4.1.3. Thermogravimetry/TGA
4.1.4. Powder X-ray Diffraction
4.2. Chemical Synthesis and Characterization
4.2.1. Preparation and Structural Characterization of Bisteroidal Esters of Terephthalic Acid. General Procedure
4.2.2. Bicholesteryl Terephthalate (5)
4.2.3. Bicholestan-3-yl Terephthalate (6)
4.2.4. Bidiosgeninyl 1,4-Terephthalate (8a)
4.2.5. Bihecogeninyl Terephthalate (8b)
4.2.6. Bisarsasapogeninyl Terephthalate (8c)
4.2.7. Bi-(23R)-23-acetyldiosgeninyl Terephthalate (10a)
4.2.8. Bi-(23R)-acetylhecogeninyl Terephthalate (10b)
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nahar, L.; Sarker, S.D. Steroid Dimers: Chemistry and Applications in Drug Design and Delivery; John Wiley & Sons: Hoboken, NJ, USA, 2012. [Google Scholar]
- Sarker, S.D.; Nahar, L. Chemistry for Pharmacy Students: General, Organic and Natural Product Chemistry; John Wiley & Sons: Hoboken, NJ, USA, 2007. [Google Scholar]
- Nahar, L.; Sarker, S.D.; Turner, A.B. A review on synthetic and natural steroid dimers: 1997–2006. Curr. Med. Chem. 2007, 14, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Windaus, A.; Borgeaud, P. Ober die photochemische Dehydrierung des Ergosterins. Liebig’s Annu. 1928, 460, 235–237. [Google Scholar]
- Mayorquín-Torres, M.C.; Colin-Molina, A.; Perez-Estrada, S.; Galano, A.; Rodríguez-Molina, B.; Iglesias-Arteaga, M.A. Synthesis, characterization, and solid state dynamic studies of a hydrogen bond-hindered steroidal molecular rotor with a flexible axis. J. Org. Chem. 2018, 83, 3768–3779. [Google Scholar] [CrossRef] [PubMed]
- Moser, B.R. Review of cytotoxic cephalostatins and ritterazines: Isolation and synthesis. J. Nat. Prod. 2008, 71, 487–491. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The dimeric steroid–pyrazine marine alkaloids: Challenges for isolation, synthesis, and biological studies. Angew. Chem. Int. Ed. Engl. 1996, 35, 611–615. [Google Scholar] [CrossRef]
- Nonappa; Maitra, U. Unlocking the potential of bile acids in synthesis, supramolecular/materials chemistry and nanoscience. Org. Biomol. Chem. 2008, 6, 657–669. [Google Scholar] [CrossRef]
- Valdez-García, R.M.; Alarcón-Manjarrez, C.; Galano, A.; Rodríguez-Molina, B.; Flores-Álamo, M.; Iglesias-Arteaga, M. Synthesis of dimeric steroid trioxabispiroacetals scaffolds by gold(i)-catalyzed hydroalkoxylation–hydration of diynediols. Eur. J. Org. Chem. 2019, 2019, 4916–4927. [Google Scholar] [CrossRef]
- Ercole, F.; Whittaker, M.R.; Quinn, J.F.; Davis, T.P. Cholesterol modified self-assemblies and their application to nanomedicine. Biomacromolecules 2015, 16, 1886–1914. [Google Scholar] [CrossRef]
- Huang, J.L.; Wang, S.; Wu, G.L.; Yan, L.; Dong, L.; Lai, X.P.; Yin, S.C.; Song, B. Mono-molecule-layer nano-ribbons formed by self-assembly of bolaamphiphiles. Soft Matter 2014, 10, 1018–1023. [Google Scholar] [CrossRef]
- Dhinakaran, M.K.; Soundarajan, K.; Das, T.M. Synthesis of novel benzimidazole-carbazole-N-glycosylamines and their self-assembly into nanofibers. N. J. Chem. 2014, 38, 4371–4379. [Google Scholar] [CrossRef]
- Zhao, H.Y.; Chen, H.B.; Gao, Y.; Li, H.M. Self-assembly of sodium 4-(4, 5-diphenyl-1H-imidazol-2-yl) benzoate into ultralong microbelts. Cryst. Eng. Comm. 2014, 16, 7507–7514. [Google Scholar] [CrossRef]
- Tambara, K.; Olsen, J.C.; Hansen, D.E.; Dan Panto, G. The thermodynamics of the self-assembly of covalently linked oligomeric naphthalenediimides into helical organic nanotubes. Org. Biomol. Chem. 2014, 12, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.X.; Wu, F.C.; Tian, Y.; Wu, M.; Zhou, Q.; Jiang, S.D.; Niu, Z.W. Size dependent cellular uptake of rod-like bionanoparticles with different aspect ratios. Sci. Rep. 2016, 6, 24567. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.X.; Lu, J.R.; Wu, J.D.; Huand, J.; Ju, Y. Water tuned nano/micro-structures in a redox-responsive supramolecular gel. RSC Adv. 2014, 4, 63539–63543. [Google Scholar] [CrossRef]
- Gao, Y.X.; Hao, J.; Wu, J.D.; Zhang, X.; Huand, J.; Ju, Y. Solvent-directed assembly of a pyridinium-tailored methyl oleanolate amphiphile: Stepwise growth of microrods and nanofibers. Langmuir 2016, 32, 1685–1692. [Google Scholar] [CrossRef]
- Koshkina, O.; Lang, T.; Thiermann, R.; Docter, D.; Stauber, R.H.; Secker, C.; Schlaad, H.; Weidner, S.; Mohr, B.; Maskos, M.; et al. Temperature-triggered protein adsorption on polymer-coated nanoparticles in serum. Langmuir 2015, 31, 8873–8881. [Google Scholar] [CrossRef]
- Draper, E.R.; Mears, L.L.E.; Castilla, A.M.; King, S.M.; McDonald, T.O.; Akhtar, R.; Adams, D.J. Using the hydrolysis of anhydrides to control gel properties and homogeneity in pH-triggered gelation. RSC Adv. 2015, 5, 95369–95378. [Google Scholar] [CrossRef]
- Zhang, S.; Yang, S.; Zang, J.; Yang, R.; Zhao, G.; Xu, C. Self-assembly of NH2-(α, l-lysine)5-COOH and SDS into nanodiscs or nanoribbons regulated by pH. Chem. Commun. 2014, 50, 9943–9946. [Google Scholar] [CrossRef]
- Sutar, P.; Suresh, V.M.; Maji, T.K. Tunable emission in lanthanide coordination polymer gels based on a rationally designed blue emissive gelator. Chem. Commun. 2015, 51, 9876–9879. [Google Scholar] [CrossRef]
- Xie, F.; Qin, L.; Liu, M.H. A dual thermal and photo-switchable shrinking–swelling supramolecular peptide dendron gel. Chem. Commun. 2016, 52, 930–933. [Google Scholar] [CrossRef]
- Wang, Y.Y.; Zhou, D.Y.; Li, H.N.; Li, R.R.; Zhong, Y.Y.; Sun, X. Hydrogen-bonded supercoil self-assembly from achiral molecular components with light-driven supramolecular chirality. J. Mater. Chem. C 2014, 2, 6402–6409. [Google Scholar] [CrossRef]
- Dai, J.D.; Wei, X.; Cao, Z.J.; Zhou, Z.P.; Yu, P.; Pan, J.M.; TZou, T.B.; Li, C.X.; Yan, Y.S. Highly-controllable imprinted polymer nanoshell at the surface of magnetic halloysite nanotubes for selective recognition and rapid adsorption of tetracycline. RSC Adv. 2014, 4, 7967–7978. [Google Scholar] [CrossRef]
- Li, Y.; Dias, J.R. Dimeric and oligomeric steroids. Chem. Rev. 1997, 97, 283–304. [Google Scholar] [CrossRef]
- Wang, G.T.; Zhao, X.; Li, Z.T. Hydrogen bonded arylamide-linked cholesteryl dimesogenic liquid crystals: A study of the length and side chain effects. Tetrahedron 2011, 67, 48–57. [Google Scholar] [CrossRef]
- Perez-Labrada, K.; Mendez, Y.; Brouard, I.; Rivera, D.G. Multicomponent ligation of steroids: Creating diversity at the linkage moiety of bis-spirostanic conjugates by Ugi reactions. ACS Comb. Sci. 2013, 15, 320–330. [Google Scholar] [CrossRef][Green Version]
- Angelova, A.; Fajolles, C.; Céline Hocquelet, C.; Djedaïni-Pilard, F.; Lesieur, S.; Bonnet, V.; Perly, B.; Lebas, G.; Mauclaire, L. Physico-chemical investigation of asymmetrical peptidolipidyl cyclodextrins. J. Colloid Interface Sci. 2008, 322, 304–314. [Google Scholar] [CrossRef]
- Liu, D.; Angelova, A.; Liu, J.; Garamus, V.M.; Angelov, B.; Zhang, X.; Li, Y.; Feger, G.; Li, N.; Zou, A. Self-assembly of mitochondria-specific peptide amphiphiles amplifying lung cancer cell death through targeting the VDAC1–hexokinase-II complex. J. Mater. Chem. B 2019, 7, 4706–4716. [Google Scholar] [CrossRef]
- Angelova, A.; Drechsler, M.; Garamus, V.M.; Angelov, B. Pep-lipid cubosomes and vesicles compartmentalized by micelles from self-assembly of multiple neuroprotective building blocks including a large peptide hormone PACAP-DHA. Chem. Nano. Mat. 2019, 5, 1381–1389. [Google Scholar] [CrossRef]
- Yang, H.K. Structure and solvent-triggered influences in the self-assembly of polyoxometalate–steroid conjugates. RSC Adv. 2016, 6, 66431–66437. [Google Scholar] [CrossRef]
- Nakanishi, T.; Shen, Y.; Wang, J. Superstructures and superhydrophobic property in hierarchical organized architectures of fullerenes bearing long alkyl tails. J. Mater. Chem. 2010, 20, 1253–1260. [Google Scholar] [CrossRef]
- Ariga, K.; Watanabe, S.; Mori, T.; Takeya, J. Soft 2D nanoarchitectonics. NPG Asia Mater. 2018, 10, 90–106. [Google Scholar] [CrossRef]
- Wang, J.; Qi, W.; Lei, N.; Chen, X. Lamellar hydrogel fabricated by host-guest interaction between α- cyclodextrin and amphiphilic phytosterol ethoxylates. Colloids Surf. A Physicochem. Eng. Asp. 2019, 570, 462–470. [Google Scholar] [CrossRef]
- Hwang, J.J.; Iyer, S.N.; Li, L.S.; Claussen, R.; Harrington, D.A.; Stupp, S.I. Self-assembling biomaterials: Liquid crystal phases of cholesteryl oligo (L-lactic acid) and their interactions with cells. Proc. Natl. Acad. Sci. USA 2002, 99, 9662–9667. [Google Scholar] [CrossRef] [PubMed]
- Dahl, J.S.; Dahl, C.E.; Bloch, K. Effect of cholesterol on macromolecular synthesis and fatty acid uptake by Mycoplasma capricolum. J. Biol. Chem. 1981, 256, 87–91. [Google Scholar] [PubMed]
- Dahl, J.S.; Dahl, C.E. Proteolipid formation in Mycoplasma capricolum. Influence of cholesterol on unsaturated fatty acid acylation of membrane proteins. Proc. Natl. Acad. Sci. USA 1983, 80, 692–696. [Google Scholar] [CrossRef] [PubMed]
- Yeagle, P.L. Cholesterol and the cell membrane. Biochim. Biophys. Acta 1985, 822, 267–287. [Google Scholar] [CrossRef]
- Yeagle, P.L. Modulation of membrane function by cholesterol. Biochimie 1991, 73, 1303–1310. [Google Scholar] [CrossRef]
- Guo, Z.H.; Liu, X.F.; Hu, J.S.; Yang, L.Q.; Chen, Z.P. Synthesis and Self-Assembled Behavior of pH-Responsive Chiral Liquid Crystal Amphiphilic Copolymers Based on Diosgenyl-Functionalized Aliphatic Polycarbonate. Nanomaterials 2017, 7, 169. [Google Scholar] [CrossRef]
- Meza-Reyes, S.; Sandoval-Ramírez, J.; Montiel-Smith, S.; Hernández-Linares, G.; Viñas-Bravo, O.; Martínez-Pascual, R.; Fernández-Herrera, M.A.; Vega-Báez, J.L.; Merino-Montiel, P.; Santillán, R.; et al. β-Alkoxy-α, β-unsaturated ketone systems in steroidal frameworks, and their conversion to 23, 24-bisnorlactones. Arkivoc 2005, 6, 307–320. [Google Scholar]
- Nahar, L.; Turner, A.B. Synthesis of ester-linked lithocholic acid dimers. Steroids 2003, 68, 1157–1161. [Google Scholar] [CrossRef]
- Soto, V.H.; Alvarez, M.; Meijide, F.; Trillo, J.V.; Antelo, A.; Jover, A.; Galantini, L.; Vázquez-Tato, J. Ice-like encapsulated water by two cholic acid moieties. Steroids 2012, 77, 1228–1232. [Google Scholar] [CrossRef]
- Campanelli, A.R.; Candeloro de Sanctis, S.; Giglio, E.; Pavel, N.V.; Quagliata, C. From crystal to micelle: A new approach to the micellar structure. J. Incl. Phenom. Macr. Chem. 1989, 7, 391–400. [Google Scholar] [CrossRef]
- Soto, V.H.; Jover, A.; Galantini, L.; Meijide, F.; Vázquez Tato, J. Crystal structure of the supramolecular linear polymer formed by the self-assembly of mono-6-deoxy- 6-adamantylamide-b-cyclodextrin. Acta Crystalogr. B 2004, 60, 204–210. [Google Scholar] [CrossRef] [PubMed]
- Bertolasi, V.; Bortolini, O.; Fantin, G.; Fogagnolo, M.; Perrone, D. Preparation and characterization of some keto-bile acid azines. Steroids 2007, 72, 756–764. [Google Scholar] [CrossRef]
- Bai, X.; Barnes, C.; Pascal, R.A.; Chen, X.; Dias, J.R. Bile acid-based cage compounds with lipophilic outer shells and inner cavities. Org. Lett. 2011, 13, 3064–3067. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Choudhury, A.R.; Guru, R.T.N.; Maitra, U. Selective and unusual fluoride ion complexation by a steroidal receptor using OH-F and CH-F interactions: A new motif for anion coordination? Org. Lett. 2005, 7, 1441–1444. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.R.; Pascal, R.A., Jr.; Morrill, J.; Holder, A.J.; Gao, H.; Barnes, C. Remarkable structures of cyclotri(deoxycholate) and cyclotetra(24-norcholate) acetate esters. J. Am. Chem. Soc. 2002, 124, 4647–4652. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. Structure validation in chemical crystallography. Acta Cryst. 2009, 65, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Spek, A.L. PLATON SQUEEZE: A tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, 71, 9–18. [Google Scholar]
- Jehannin, M.; Rao, A.; Cölfen, H. New horizons of nonclassical crystallization. J. Am. Chem. Soc. 2019, 141, 10120–10136. [Google Scholar] [CrossRef]
- Dollase, W.A. Correction of intensities for preferred orientation in powder diffractometry: Application of the March model. J. Appl. Cryst. 1986, 19, 267–272. [Google Scholar] [CrossRef]
- Zolotoyabko, E. Determination of the degree of preferred orientation within the March–Dollase approach. J. Appl. Cryst. 2009, 42, 513–518. [Google Scholar] [CrossRef]
- Macrae, C.F.; Bruno, I.J.; Chisholm, J.A.; Edgington, P.R.; McCabe, P.; Pidcock, E.; Rodriguez-Monge, L.; Taylor, R.; van de Streek, J.; Wood, P.A. Mercury CSD 2.0—New features for the visualization and investigation of crystal structures. J. Appl. Cryst. 2008, 41, 466–470. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 5, 6, 8a–c and 10a,b are available from the authors. |














{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter | Time | Temp | Solvent | Base | Yield |
|---|---|---|---|---|---|
| entry | |||||
| a | 1 h | rt | Py | Py | nr ** |
| b | 2 h | rt | DMF | K2CO3 | nr ** |
| c | 24 h | reflux | THF | Et3N | 10% |
| d | 2 h | reflux | Py | Py | 70% |
| e | 2 h | reflux | Py | Py | 80% |
| 1H NMR (CDCl3) | 13C NMR (CDCl3) | ||||
|---|---|---|---|---|---|
| signals | 5 | 6 | signals | 5 | 6 |
| H-3, H-3′ | 4.90 | 4.96 | C-3, C-3′ | 75.1 | 74.9 |
| H-18, H-18′ | 0.69 | 0.87 | C-18, C-18′ | 18.7 | 12.3 |
| H-19, H-19′ | 1.07 | 0.66 | C-19, C-19′ | 19.3 | 12.0 |
| H-21, H-21′ | 0.93 | 0.91 | C-21, C-21′ | 11.8 | 18.6 |
| HAr-HAr′ | 8.08 | 8.07 | Cipso-Cipso′ | 137.1 | 134.4 |
| Signals | 8a * | 8b * | 8c * | 10a * | 10b  |
|---|---|---|---|---|---|
| H-3, H-3′ | 4.91 | 4.95 | 5.36 | 4.80 | 3.81 |
| H-6, H-6′ | 5.44 | 5.34 | |||
| H-18, H-18′ | 0.80 | 1.06 | 0.78 | 0.61 | 0.90 |
| H-19, H-19′ | 1.10 | 0.98 | 1.04 | 1.00 | 0.82 |
| H-21, H-21′ | 0.99 | 1.08 | 1.00 | 0.92 | 1.37 |
| H-26a, H-26a′ | 3.38 | 3.35 | 3.31 | 3.36 | 3.44 |
| H-26e, H-26e′ | 3.48 | 3.50 | 3.95 | 3.43 | 3.54 |
| H-27, H-27′ | 0.79 | 0.78 | 1.09 | 0.80 | 0.71 |
| HAr-HAr′ | 8.08 | 8.06 | 8.11 | 8.01 | 7.56 |
in Pyridine-d5.| Signals | 8a * | 8b * | 8c * | 10a * | 10b |
|---|---|---|---|---|---|
| C-3, C-3′ | 75.2 | 74.3 | 71.9 | 75.2 | 70.0 |
| C-5, C-5′ | 139.5 | -- | -- | 139.6 | -- |
| C-6, C-6′ | 122.7 | -- | -- | 122.5 | -- |
| C-18, C-18′ | 16.2 | 16.0 | 16.0 | 16.2 | 12.7 |
| C-19, C-19′ | 19.3 | 17.1 | 24.0 | 19.4 | 16.9 |
| C-21, C-21′ | 14.4 | 13.2 | 14.3 | 14.2 | 14.3 |
| C-22, C-22′ | 109.5 | 109.2 | 109.7 | 108.1 | 108.2 |
| C-23, C-23′ | 31.3 | 31.4 | -- | 29.8 | 29.8 |
| C-26, C-26′ | 66.9 | 66.8 | 65.1 | 66.2 | 66.0 |
| C-27, C-27′ | 17.1 | 11.9 | 16.5 | 17.0 | 17.8 |
| Cipso-Cipso′ | 134.4 | 134.3 | 134.7 | 134.4 | 135.1 |
in Pyridine-d5.© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guerrero-Luna, G.; Hernández-Linares, M.G.; Bernès, S.; Carrasco-Carballo, A.; Montalvo-Guerrero, D.; Fernández-Herrera, M.A.; Sandoval-Ramírez, J. Mesoscale Assembly of Bisteroidal Esters from Terephthalic Acid. Molecules 2020, 25, 1213. https://doi.org/10.3390/molecules25051213
Guerrero-Luna G, Hernández-Linares MG, Bernès S, Carrasco-Carballo A, Montalvo-Guerrero D, Fernández-Herrera MA, Sandoval-Ramírez J. Mesoscale Assembly of Bisteroidal Esters from Terephthalic Acid. Molecules. 2020; 25(5):1213. https://doi.org/10.3390/molecules25051213
Chicago/Turabian StyleGuerrero-Luna, Gabriel, María Guadalupe Hernández-Linares, Sylvain Bernès, Alan Carrasco-Carballo, Diana Montalvo-Guerrero, María A. Fernández-Herrera, and Jesús Sandoval-Ramírez. 2020. "Mesoscale Assembly of Bisteroidal Esters from Terephthalic Acid" Molecules 25, no. 5: 1213. https://doi.org/10.3390/molecules25051213
APA StyleGuerrero-Luna, G., Hernández-Linares, M. G., Bernès, S., Carrasco-Carballo, A., Montalvo-Guerrero, D., Fernández-Herrera, M. A., & Sandoval-Ramírez, J. (2020). Mesoscale Assembly of Bisteroidal Esters from Terephthalic Acid. Molecules, 25(5), 1213. https://doi.org/10.3390/molecules25051213