3. Materials and Methods
3.1. General
Ultraviolet (UV) spectra were obtained on a JASCO V-560 UV/VIS spectrophotometer (JASCO, Tokyo, Japan). Optical rotations were measured with a JASCO DIP-370 digital polarimeter. The ECD spectra were measured with a JASCO J-725N spectrophotometer.
1H and
13C NMR spectra were recorded on a Varian Unity Plus 500 spectrometer (Agilent Technologies, Santa Clara, CA, USA) operating at 500 and 125 MHz for the
1H and
13C nuclei, respectively. NMR spectra were also recorded on a JEOL JNM-AL 400 spectrometer (JEOL Ltd., Tokyo, Japan) operating at 400 and 100 MHz for the
1H and
13C nuclei, respectively. ESIMS were obtained using a JEOL JMS-T100TD spectrometer. FABMS were recorded on a JMS700N spectrometer (JEOL Ltd.) using
m-nitrobenzyl alcohol or glycerol as the matrix. Column chromatography was performed using Sephadex LH-20 (25–100 mm, GE Healthcare UK Ltd., Little Chalfont, UK), MCI-gel CHP20P (75–150 mm, Mitsubishi Chemical Co., Tokyo, Japan), Diaion HP20SS (Mitsubishi Chemical Co.), Toyopearl Butyl-650C (Tosoh Bioscience Japan, Tokyo, Japan), Cosmosil 75C18OPN (Nacalai Tesque Inc., Kyoto, Japan), and Chromatorex ODS (Fuji Silysia Chemical Ltd., Kasugai, Japan) columns. TLC was performed on precoated Kieselgel 60 F
254 plates (0.2-mm thickness, Merck, Darmstadt, Germany), using toluene–ethyl formate–formic acid (1:7:1,
v/
v) and CHCl
3–MeOH–H
2O (7:3:0.5,
v/
v) mixtures as the eluents. The spots were detected using ultraviolet illumination and by spraying with 2% ethanolic FeCl
3 solution (for phenolic compounds), 5% anisaldehyde in ethanolic 5% H
2SO
4 solution (for proanthocyanidins), or 5% H
2SO
4 solution followed by heating. Analytical HPLC was performed on a Cosmosil 5C18-ARII (Nacalai Tesque Inc., Kyoto, Japan) column (250 × 4.6 mm, i.d.) with a gradient elution of 4–30% (39 min) and 30–75% (15 min) CH
3CN in 50 mM H
3PO
4 at 35 °C (flow rate, 0.8 mL/min; detection, JASCO photodiode array detector MD-2018 plus). Geraniin was obtained as yellow crystalline powder from previous our studies [
33].
3.2. Plant Material
Fresh leaves of Triadica sebifera and Elaeocarpus sylvestris var. ellipticus were collected in Bunkyo campus of Nagasaki University. Fresh leaves of Carpinus laxiflora were collected Mt. Gokahara, Isahaya, Nagasaki prefecture.
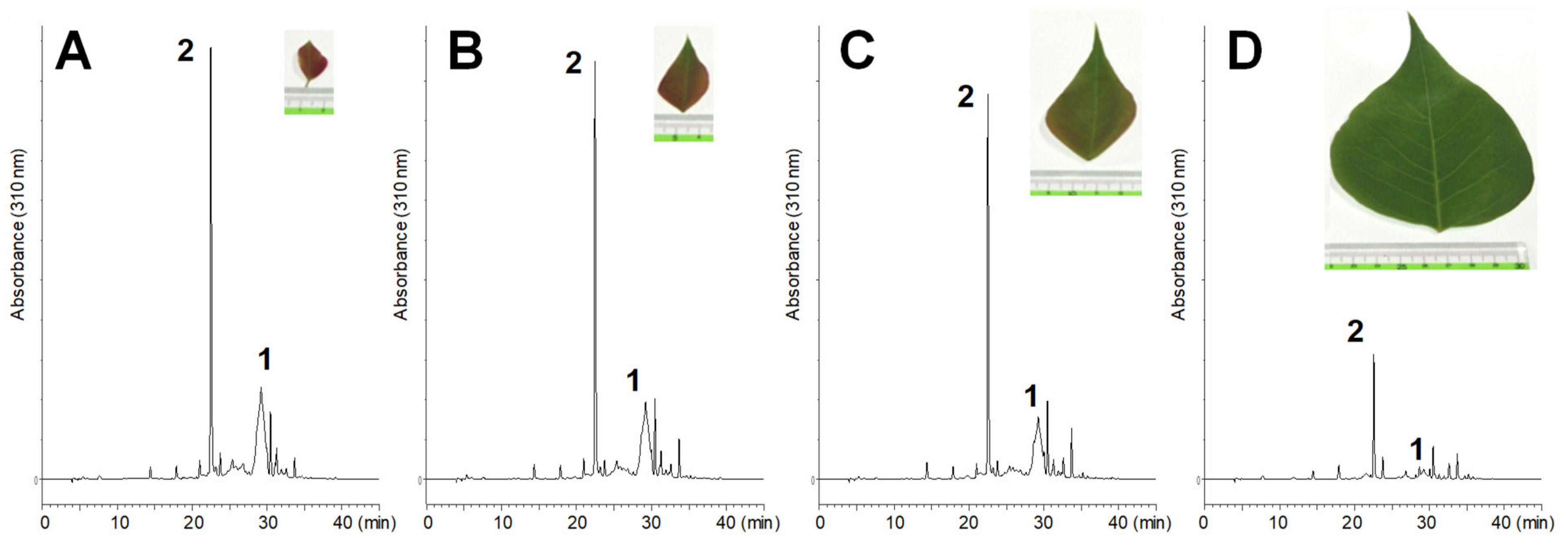
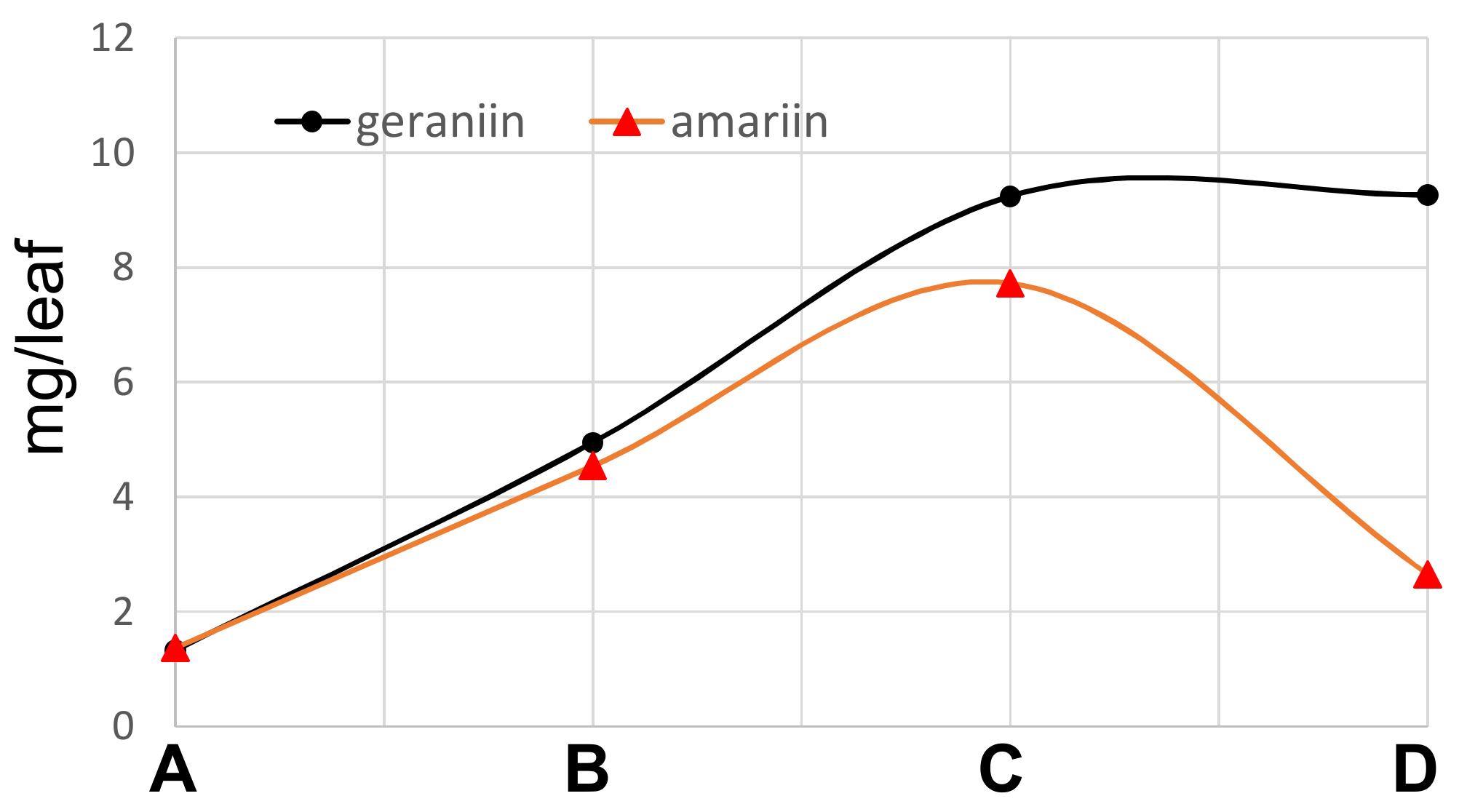
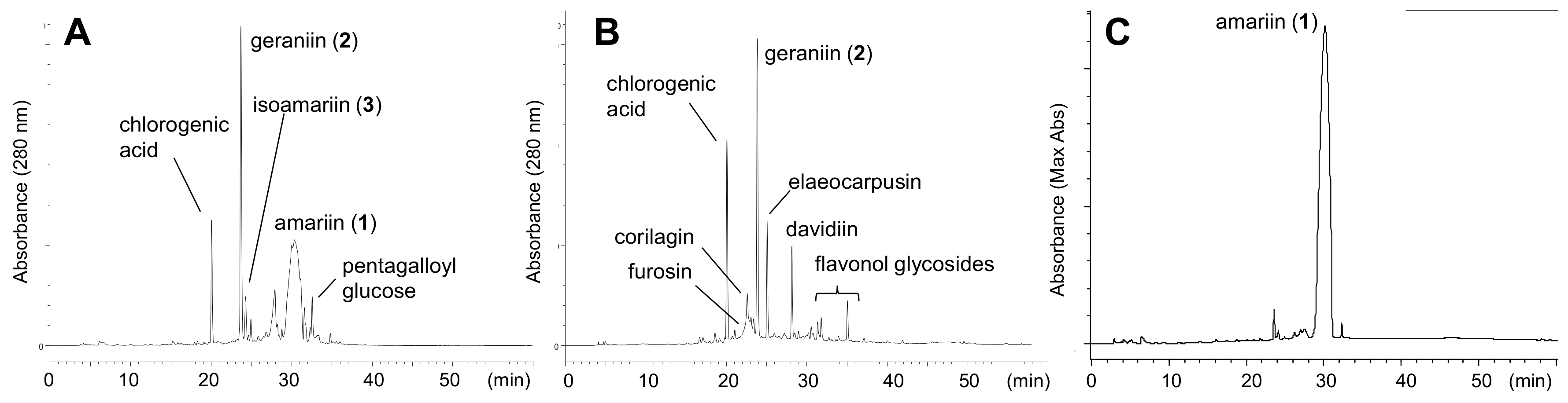
3.3. Quantification of Ellagitannins in the Leaves of T. Sebifera
Fresh leaves of
T. sebifera were lyophilized and powdered by grinding in a mortar. Aliquots (50 mg) of the powder (each 4 samples) were extracted with 60% CH
3CN containing 0.1% trifluoroacetic acid (1.5 mL) at room temperature for 7 h. The extracts were analyzed by HPLC. Calibration curves were built using geraniin (320 nm) isolated in our previous study [
24] and amariin (275 nm) isolated in this study. Average dried weights of the leaves in the
Figure 1 were as follows: A, 0.007 g/leaf (fresh: 0.022 g/leaf); B, 0.029 g/leaf (fresh: 0.089 g/leaf); C, 0.065g/leaf (fresh: 0.20 g/leaf), and D, 0.195 g/leaf (fresh: 0.63 g/leaf).
3.4. Isolation of Amariin (1) and Isoamariin (3)
Fresh young leaves of Carpinus laxiflora (750 g) collected in April 30, 2014 were homogenized with 80% acetone containing 0.005% trifluoroacetic acid (TFA) (3 L) at room temperature 2 times. The extract was filtered, and the filtrate was concentrated using rotary evaporator. Resulting precipitates formed in the aqueous solution were removed by filtration. The filtrate (about 800 mL) was applied to Diaion HP20SS (8 cm i.d. × 37 cm) column with 0.005% TFA containing increasing proportions of MeOH [0% (500 mL), 20% (1 L), 30% (1 mL), 40% (1 L), 50% (1 L), 60% (1 L), 80% (1 L), and 100% (2 L)] to give 5 fractions. Fraction (Fr.) 3 (16.8 g) mainly containing 1 was subjected to Sephadex LH-20 column chromatography (5 cm i.d. × 22 cm) with 0%–100% MeOH in 0.005% TFA (20% stepwise, each 300 mL) to yield a crude sample of 1 (10.9 g). A portion (7.5 g) of the crude sample was purified by Diaion HP20SS column chromatography (4 cm i.d. × 27 cm, 0%–60% MeOH in 0.005% TFA, 5% stepwise, each 200 mL). Tubes containing 1 were combined and concentrated by rotary evaporator (40 ˚C) to give yellow precipitates of 1, which were collected by filtration (3.7 g). The filtrate (0.76 g) was separated by Chromatorex ODS (3 cm i.d. × 25 cm, 0%–50% MeOH in 0.005% TFA, 5% stepwise, each 100 mL) to yield 3 (121 mg).
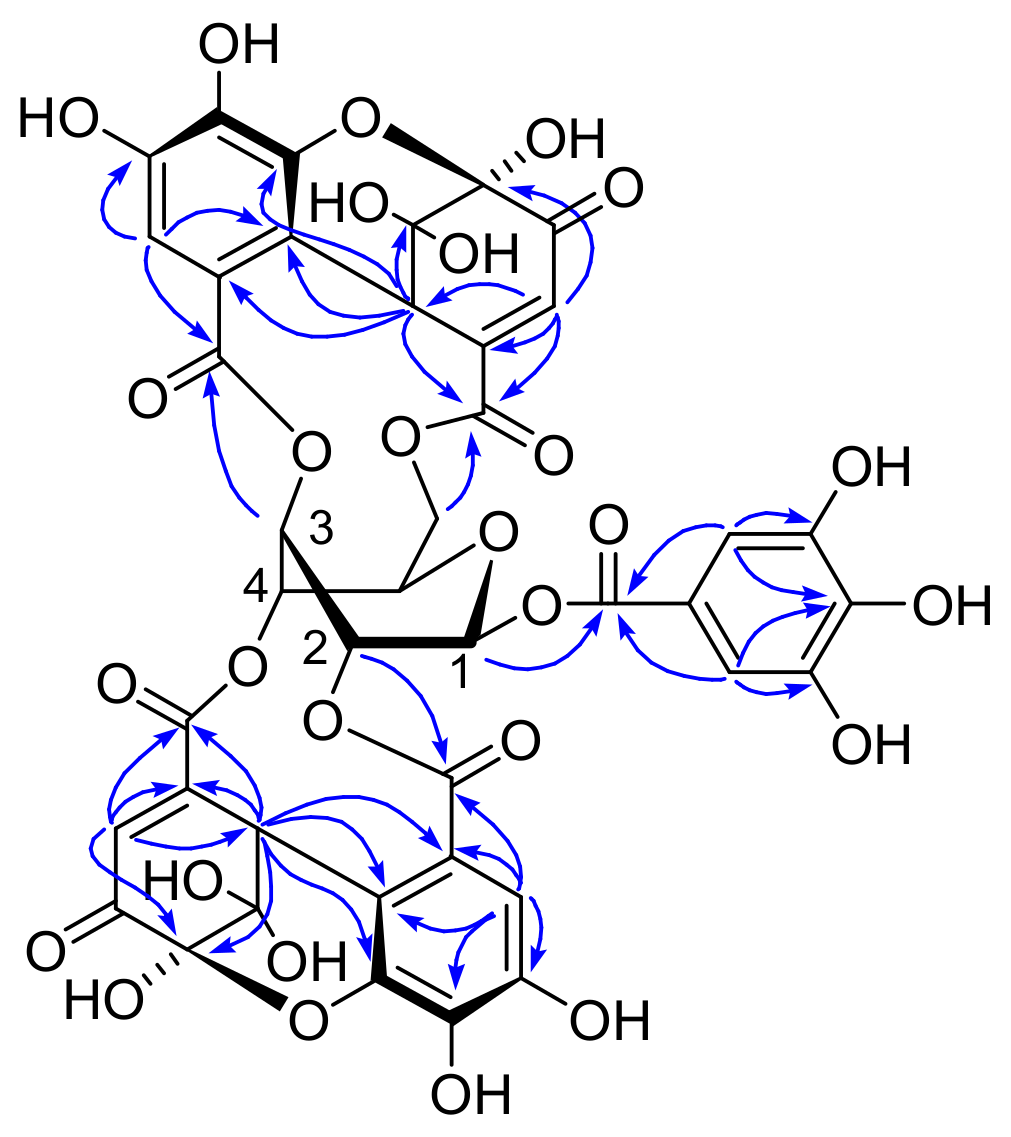
Amariin (1): Yellow powder, 1H NMR (500 MHz, acetone-d6) δ: galloyl: 7.23, 7.22 (each s, galloyl-2,6); DHHDP (5- and 6-membered-ring hemiacetal structure): 7.26, 7.238, 7.236, 7.235, 7.233 (each s, H-3’), 6.71, 6.67, 6.60, 6.58 [each s, H(6)-3], 6.31, 6.26 [each d, J = 1.5 Hz, H(5)-3], 5.31. 5.29, 5.27, 5.26 [each s, H(6)-1], 4.90, 4.88, 4.86, 4.85 [each d, J = 1,5 Hz, H(5)-1]; glucose (two major isomers among 4 isomers), isomer A: 6.40 (d, J = 5.4 Hz, glc-H-1), 5.79 (dd, J = 1.5, 3.2 Hz, glc-3), 5.50 (br d, J = 3.2 Hz, glc-4), 5.47 (dt, J = 1.5, 5.4 Hz, glc-2), 5.23 (dd, J = 2.7, 13.3 Hz, glc-6), 4.77 (br s, glc-5), 4.39 (br d, J = 13.3 Hz, glc -6); isomer B: 6.35 (d, J = 5.1 Hz, glc-1), 5.65 (dt, J = 1.3, 3.7 Hz, glc-4), 5.62 (dt, J = 1.5, 5.1 Hz, glc-2), 5.53 (dd, J = 1.5, 3.7 Hz, glc-3), 5.33 (dd, J = 2.6, 13.3 Hz, glc-6), 4.83 (br s, glc-5), 4.39 (br d, J = 13.3 Hz, glc -6); 13C NMR (125 MHz, acetone-d6) δ: only signals of major isomers COO: 168.4, 168.1, 166.2, 165.1, 165.0, 164.8, 164.7, 164.6, 164.4; galloyl: 146.21, 146.18 (C-3,5), 139.9, 139.2 (C-4), 110.40, 110.39 (C-2,6); DHHDP: 194.2 [C(5)-4], 191.6, 191.2 [C(6)-4], 153.6, 152.5, 152.1 [C(6)-2], 148.6, 147.8. 147.3 [C(5)-6’], 143.5, 143.0, 142.9 [C(6)-6’], 129.2, 128.6 [C(6)-3], 125.7 [C(5)-3], 96.6, 96.2 [C(6)-5], 92.31, 92.27, 92.0 [C(5)-5, C(6)-6], 52.2, 51.1 [C(5)-1], 46.4, 45.0, 44.8 [C(6)-1]; other aromatic and sp2 carbons: 145.93, 145.86, 145.83, 139.2, 138.5, 138.4, 137.6, 120.3, 120.0, 119.9, 119.8, 119.7, 118.7, 117.0, 116.0, 115.0, 114.9, 113.9, 113.6, 113.1; glucose (two major isomers among 4 isomers): 95.7, 94.4 (glc-1), 78.1, 77.7 (glc-5), 73.7, 72.7 (glc-2), 70.4, 69.7 (glc-4), 67.6,66.1 (glc-3), 66.0, 65.8 (glc-6).
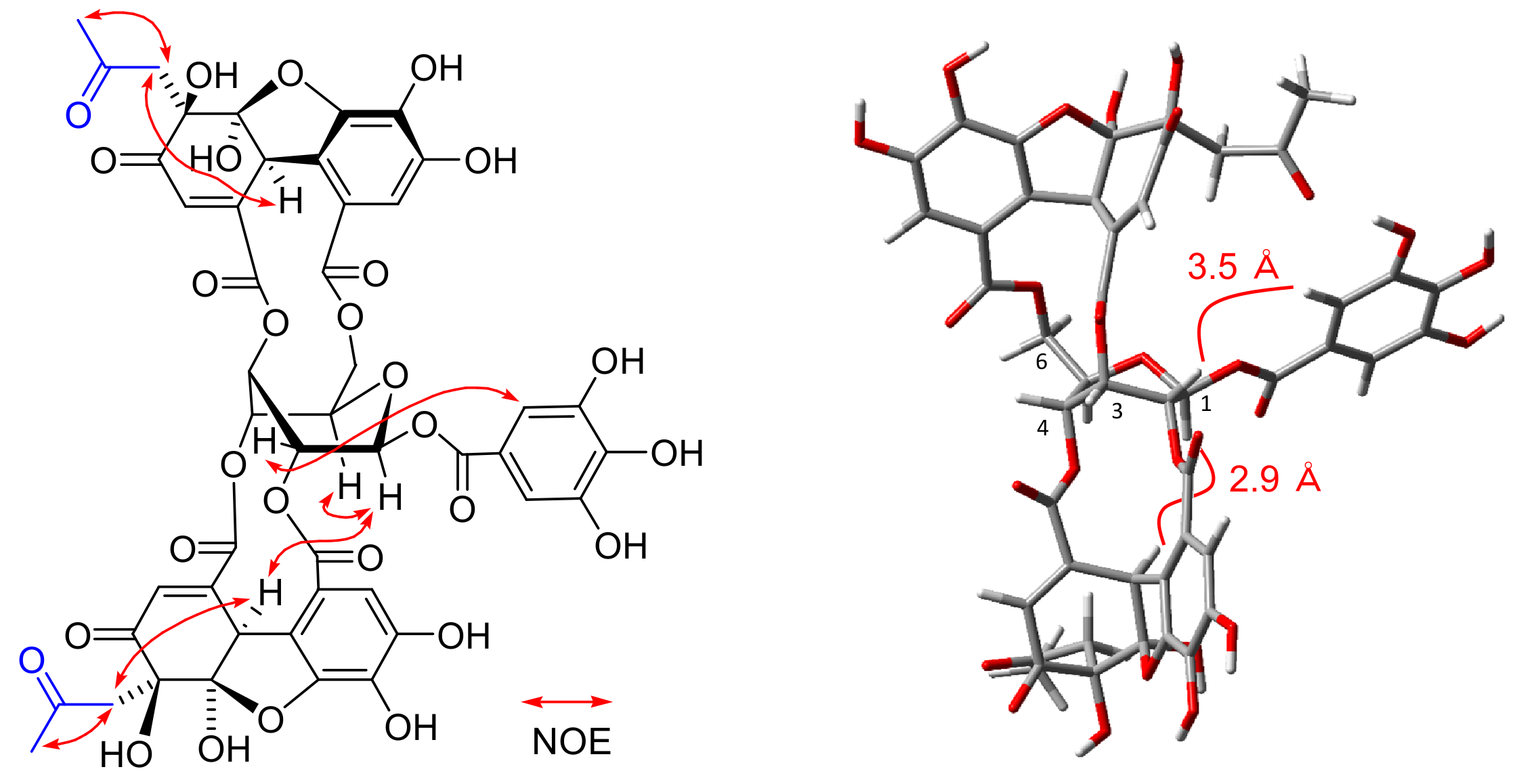
Isoamariin (3): Yellow powder, [α −136.8 (c 0.1, MeOH); FAB-MS m/z: 991 [M + Na]+; HR-FAB-MS m/z: 991.0656 [M + Na]+ (calcd. for C41H28O28Na, 991.0665); IR νmax cm−1: 3413, 1705, 1613, 1528, 1447, 1325, 1213; UV (MeOH) λmax (log ε): 279 (4.43), 221 (4.89); ECD (MeOH) λmax (Δε): 369 (−3.9), 326 (−1.4), 293 (−8.6), 270 (−2.1), 262 (−2.4), 257 (−2.1), 236 (−14.7), 228 (0), 209 (+36.4). 1H NMR (500 MHz, acetone-d6) δ: galloyl: 7.21, 7.20 (each s, galloyl-2,6); 2,4-DHHDP 5-membered-ring hemiacetal structure: 7.29 (s, H-3’), 6.26 (d, J = 1.4 Hz, H-3), 4.95 (d, J = 1.4 Hz, H-1); 2,4-DHHDP 6-membered-ring hemiacetal structure: 7.27 (s, H-3’), 6.60 (s, H-3), 5.35 (s, H-1); glucose with 5-membered-ring hemiacetal 2,4-DHHDP: 6.42 (d, J = 5.2 Hz, glc-1), 5.67 (dt, J = 1.3, 5.2 Hz, glc-2), 5.51 (dd, J = 1.3, 3.7 Hz, glc-3), 6.17 (dt, J = 1.3, 3.7 Hz, glc-4), 4.72 (br s, glc-5), 4.60 (dd, J = 3.1, 13.3 Hz, glc-6), 4.90 (dd, J = 1.7, 13.3 Hz, glc -6); glucose with 6-membered-ring hemiacetal 2,4-DHHDP: 6.36 (d, J = 4.7 Hz, glc-1), 5.78 (dt, J = 1.4, 4.7 Hz, glc-2), 5.36 (dd, J = 1.4, 3.7 Hz, glc-3), 6.28 (dt, J = 1.3, 3.5 Hz, glc-4), 4.76 (br s, glc-5), 4.62 (dd, J = 3.1, 12.3 Hz, glc-6), 4.93 (dd, J = 1.8, 12.3 Hz, glc-6); 13C NMR (125 MHz, acetone-d6) δ: galloyl: 119.7, 119.9 (galloyl-1), 110.3 (galloyl-2,6), 146.2 (galloyl-3,5), 139.8, 139.9 (galloyl-4), 164.8, 164.9 (galloyl-7); 2,4-DHHDP 5-membered-ring hemiacetal structure: 52.1 (C-1), 148.9 (C-2), 125.8 (C-3), 194.3 (C-4), 92.4 (C-5), 109.3 (C-6), 117.1 (C-1’), 120.3 (C-2’), 113.1 (C-3’), 147.8 (C-4’), 137.6 (C-5’), 147.3 (C-6’); 2,4-DHHDP 6-membered-ring hemiacetal structure: 46.4 (C-1), 154.0 (C-2), 129.3 (C-3), 191.7 (C-4), 96.2 (C-5), 92.4 (C-6); 116.1 (C-1’), 119.0 (C-2’), 113.6 (C-3’), 145.9 (C-4’), 139.2 (C-5’), 144.0 (C-6’); glucose with 5-membered-ring hemiacetal 2,4-DHHDP: 94.3 (glc-1), 74.2 (glc-2), 67.5 (glc-3), 69.6 (glc-4), 77.1 (glc-5), 68.0 (glc-6); glucose with 6-membered-ring hemiacetal 2,4-DHHDP: 95.4 (glc-1), 73.6 (glc-2), 69.0 (glc-3), 68.7 (glc-4), 76.5 (glc-5), 67.9 (glc-6).
3.5. Preparation of Acetonyl Derivative
Amariin (1) (130 mg) was dissolved in 80% acetone (20 mL) containing HCO2NH4 (130 mg) and stirred at r.t. for 13h. After removal of acetone by evaporation, the aqueous solution was subjected to Diaion HP20SS column chromatography (2 cm i.d. × 15 cm, 0%–60% MeOH in H2O, 5% stepwise, each 50 mL) to yield 1a (57.2 mg).
Bisacetonyl derivative (1a): off-white amorphous powder, [α −55.2 (c 0.1, MeOH); IRνmax cm−1: 3406, 1715, 1620, 1531, 1441; UV (MeOH) λmax (log ε): 282 (4.56), 221 (4.91). 1H NMR (500 MHz, acetone-d6) δ: 1H-NMR (acetone-d6, 500 MHz) δ: 2.19 (3H, s, 2,4-acyl-CH3), 2.23 (3H, s, 3,6-acyl-CH3), 2.96 (1H, d, J = 15.3 Hz, 3,6-acyl-CH2), 3.06 (1H, d, J = 15.6 Hz, 2,4-acyl-CH2), 3.46 (1H, d, J = 15.6 Hz, 2,4-acyl-CH2), 3.48 (1H, d, J = 15.3 Hz, 3,6-acyl-CH2), 4.21 (1H, dd, J = 1.2, 13.1 Hz, glc-6a), 4.83 (1H, d, J = 1.5 Hz, 3,6-acyl-1), 4.88 (1H, br. s, glc-5), 4.98 (1H, d, J = 1.5 Hz, 2,4-acyl-1’), 5.39 (1H, dt, J = 1.4, 3.8 Hz, glc-4), 5.47 (1H, dt, J = 1.5, 4.9 Hz, glc-2), 5.57 (1H, dd, J = 3.2, 13.1 Hz, glc-6b), 5.70 (1H, dd, J = 1.4, 3.8 Hz, glc-3), 6.35 (1H, d, J = 1.5 Hz, 2,4-acyl-3’), 6.44 (1H, d, J = 4.9 Hz, glc-1), 6.46 (1H, d, J = 1.5 Hz, 3,6-acyl-3), 7.20 (1H, s, 3,6-acyl-3’), 7.22 (1H, s, 2,4-acyl-3), 7.27 (2H, s, galloyl-H); 13C NMR (acetone-d6, 125 MHz) δ: 32.0 (2,4-acyl-CH3), 32.2 (3,6-acyl-CH3), 49.2 (3,6-acyl-CH2), 49.9 (2,4-acyl-CH2), 50.8 (3,6-acyl-1), 51.9 (2,4-acyl-1’), 64.5 (glc-6), 66.0 (glc-3), 69.7 (glc-4), 73.8 (glc-2), 78.1 (glc-5), 80.2 (3,6-acyl-5), 80.6 (2,4-acyl-5’), 94.3 (glc-1), 110.0 (2,4-acyl-6’), 110.4 (galloyl-2, 6), 110.6 (3,6-acyl-6), 113.0 (2,4-acyl-3, 3,6-acyl-3’), 116.9 (2,4-acyl-1), 117.9 (3,6-acyl-1’), 119.9 (3,6-acyl-2’), 120.1 (galloyl-1), 120.5 (2,4-acyl-2), 127.1 (3,6-acyl-3), 127.5 (2,4-acyl-3’), 136.5 (3,6-acyl-5’), 137.3 (2,4-acyl-5), 139.6 (galloyl-4), 144.6 (3,6-acyl-2), 144.9 (2,4-acyl-2’), 146.1 (galloyl-3, 5), 146.7 (3,6-acyl-6’), 147.0 (2,4-acyl-6), 147.4 (2,4-acyl-4, 3,6-acyl-4’), 164.3 (2,4-acyl-7), 164.9 (galloyl-7, 3,6-acyl-7), 166.1 (2,4-acyl-7’), 166.9 (3,6-acyl-7’), 196.9 (3,6-acyl-4), 197.2 (2,4-acyl-4’), 205.7 (2,4-acyl-CO), 205.9 (3,6-acyl-CO).
3.6. Preparation of Phenazine Derivative
Amariin (1) (50 mg) and o-phenylenediamine (10 mg) was dissolved in 10% AcOH in EtOH (2 mL) and heated at 50 ˚C for 2 h. The mixture was applied to Sephadex LH-20 (2 cm i.d. × 15 cm, 0-20% H2O in EtOH, 10% stepwise, each 100 mL) to yield 1b (30.2 mg). Phenazine derivative 3a was prepared from 3 in the same way.
Phenazinde derivative (1b): UV (MeOH) λmax (log ε) 445 (3.56), 378 (4.13), 281 (4.98), 205 (4.87); ECD (MeOH) Δε (nm) 377 (−1.48), 325 (+3.86), 284 (−67.5), 250 (+72.1), 221 (−42.0). 1H NMR (acetone-d6, 500 MHz) δ: 4.11 (1H, dd, J = 3.3, 12.0 Hz, glc-6a), 4.98 (1H, dd, J = 8.1, 12.0, glc-6b), 5.04 (1H, dd, J = 3.3, 8.1, glc-5), 5.40 (1H, d, J = 4.1, glc-4), 5.80 (1H, d, J = 4.1, glc-3), 5.86 (1H, d, J = 5.5, glc-2), 6.23 (1H, d, J = 5.5, glc-1), 7.01 (2H, s, galloyl-H), 7.03 (1H, s, 3,6-acyl-3’), 7.52 (1H, s, 2,4-acyl-3), 8.24 (1H, s, 3,6-acyl-3), 8.31 (1H, s, 2,4-acyl-3’); 13C-NMR (acetone-d6, 125 MHz) δ: 64.7 (glc-6), 67.7 (glc-3), 68.3 (glc-4), 76.2 (glc-2), 77.0 (glc-5), 91.7 (glc-1), 109.7 (2,4-acyl-3), 110.1 (galloyl-2, 6), 113.2 (3,6-acyl-3’), 115.2 (3,6-acyl-1’), 116.4 (2,4-acyl-1), 116.7 (3,6-acyl-1), 116.8 (2,4-acyl-1’), 119.8 (galloyl-1), 120.1 (2,4-acyl-3’), 120.6 (2,4-acyl-2), 120.7 (3,6-acyl-3), 123.1 (3,6-acyl-2’), 130.0, 130.4, 130.5 (2,4-acyl-4”, 5”, 3,6-acyl-4”, 5”), 132.2, 132.3 (2,4-acyl-3”, 6”, 3,6-acyl-3”, 6”), 136.0 (2,4-acyl-2’), 136.3 (3,6-acyl-2), 137.4 (3.6-acyl-5’), 138.0, 139.0 (2,4-acyl-5’, 3,6-acyl-5), 139.3 (2,4-acyl-5), 139.6 (galloyl-4), 142.8, 143.0, 143.1, 145.1, 145.3, 145.4, 145.6, 145.9 (2,4-acyl-6, 4’, 1”, 2”, 3,6-acyl-4, 4’, 6’, 1”, 2”), 145.3 (2,4-acyl-4), 146.0 (galloyl-3, 5), 152.1 (3,6-acyl-6), 152.3 (2,4-acyl-6’), 164.7 (galloyl-7), 165.9 (3,6-acyl-7), 166.6 (2,4-acyl-7’), 167.8 (3,6-acyl-7’), 167.9 (2,4-acyl-7).
Phenazine derivative (3a): Brown amorphous powder, [α −323.9 (c 0.06, MeOH); FAB-MS m/z: 1077 [M + H]+, 1099 [M + Na]+; HR-FAB-MS m/z: 1099.1403 [M + Na]+, (calcd. for C53H32O22N4Na, 1099.1406); IR vmax cm−1: 3336, 1732, 1613, 1509, 1463, 1355, 1335, 1308, 1189; UV (MeOH) λmax (log ε): 446 sh (3.51), 375 (4.15), 279 (5.00), 223 sh (4.86), 207 (4.89); ECD (MeOH) λmax (Δε): 381 (+3.8), 359 (+1.3), 304 (+16.9), 292 (0), 274 (−79.0), 257 (0), 247 (+29.4), 230 (0), 220 (−24.8), 212 (0); 1H NMR (acetone-d6, 500 MHz) δ: 4.12 (1H, t, J = 5.1, 11.6 Hz, glc-6a), 4.84 (1H, dd, J = 8.5, 11.6, glc-6b), 5.11 (1H, dd, J = 5.1, 8.5, glc-5), 5.48 (1H, d, J = 4.1, glc-3), 5.63 (1H, dd, J = 0.7, 4.1, glc-4), 5.71 (1H, d, J = 5.7, glc-2), 6.22 (1H, d, J = 5.7, glc-1), 6.99 (2H, s, galloyl-H), 7.16, 7.46, 7.86, 8.27 (each 1H, each s, aromatic) 7.93–8.01, 8.15–8.36 (each m, phenazine-H); 13C NMR (acetone-d6, 125 MHz) δ: 66.7, 68.2, 68.7, 76.2, 76.8 (glc-2~6), 91.7 (glc-1), 110.2 (galloyl-2,6), 113.5 (acyl-3), 115.7, 116.0, 116.6, 117.0, 118.5 (galloyl-1, acyl-1, 1’), 120.1, 120.2, 120.5, 122.7 (acyl-2, 3’), 130.2, 130.6, 130.7 (acyl-4”, 5”), 132.2, 132.4, 132.5 (acyl-3”, 6”), 136.1, 136.2 (acyl-2’), 138.2, 139.2, 139.5, 139.8, 139.9, 142.8, 142.9, 143.3, 143.5, 145.2, 145.3, 145.5, 145.7, 145.9 (galloyl-4, acyl-4, 5, 6, 4’, 5’, 1”, 2”), 146.2 (galloyl-3, 5), 152.3, 152.4 (acyl-6’), 164.9, 166.2, 166.6, 167.5, 167.9 (esters).
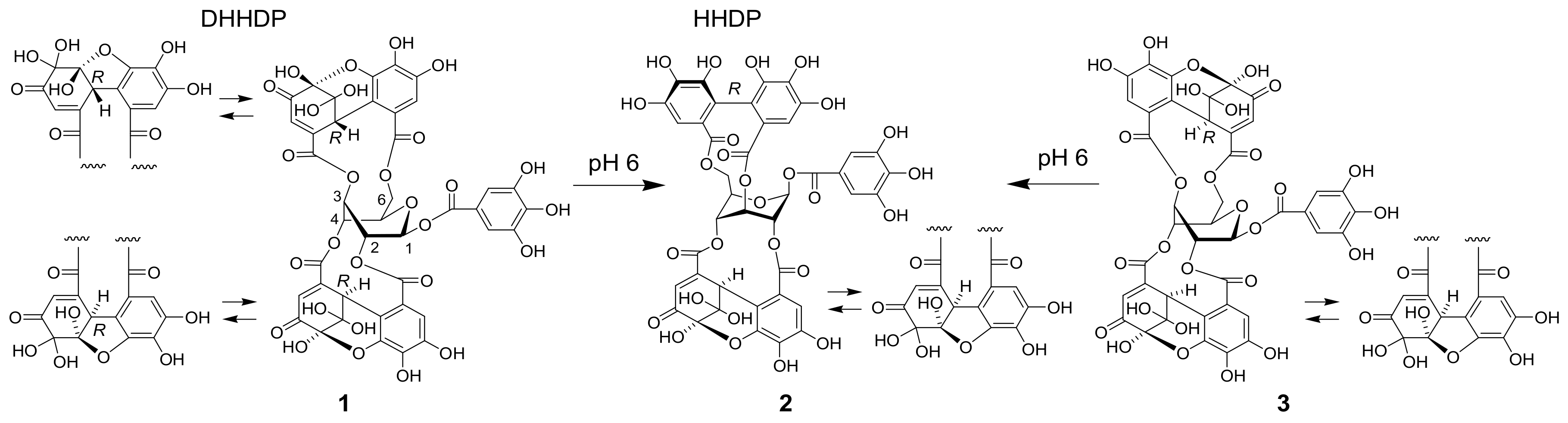
3.7. Treatment of 1 at pH 6
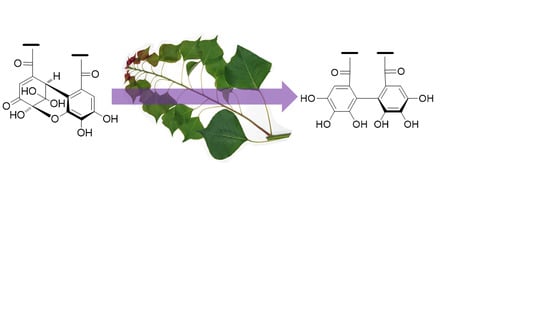
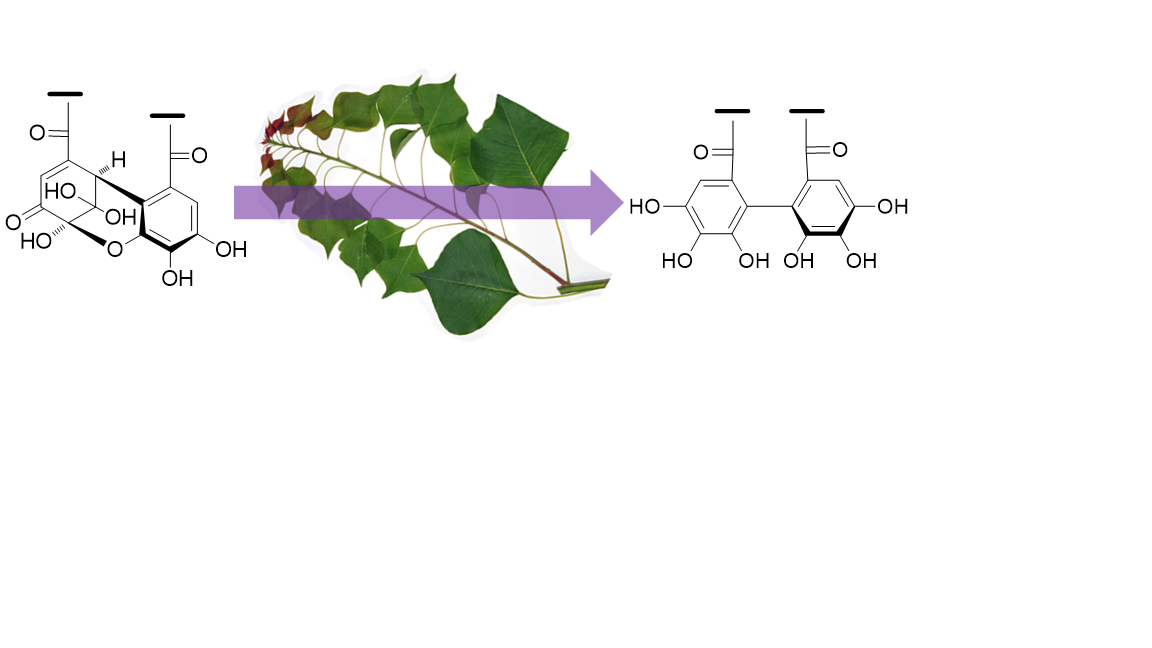
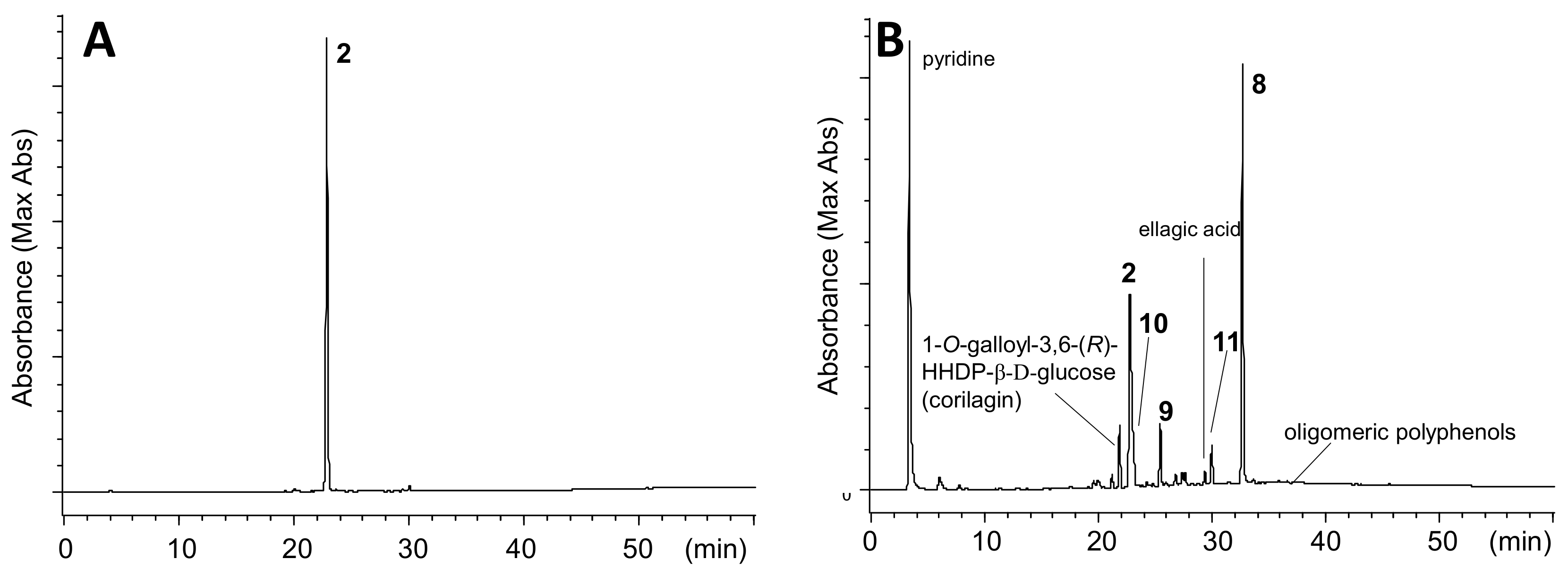
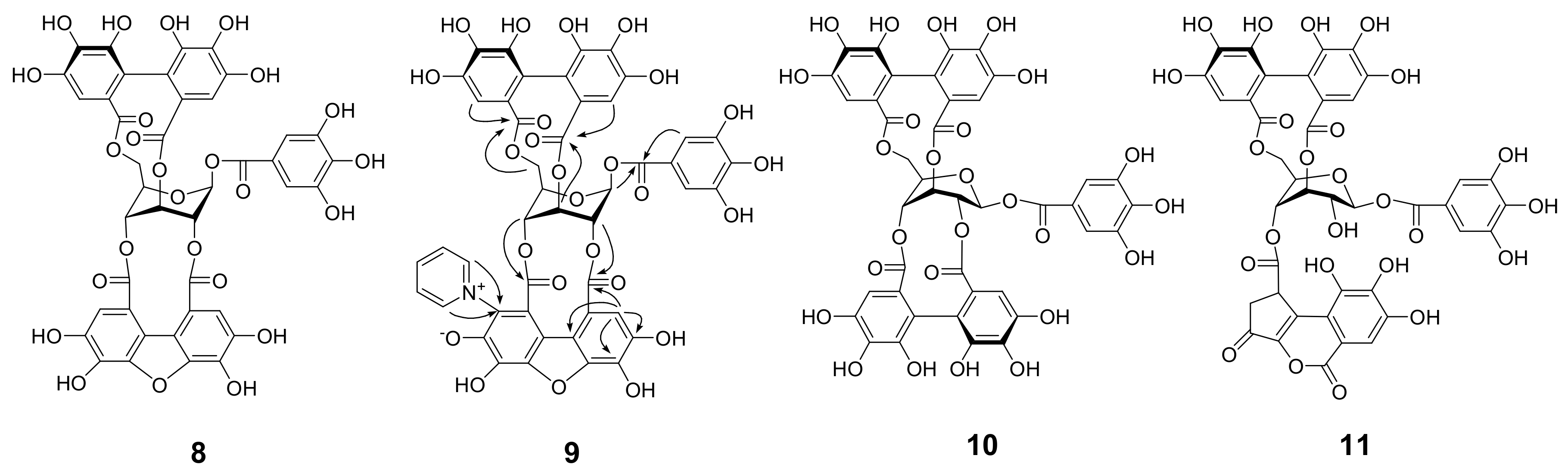
Amariin (1) (1.0 g) was dissolved in citrate-phosphate buffer (0.05 M, pH 6.0) and stirred at r.t. for 20 h. The mixture was acidified by addition of 5% TFA to pH 2 and applied to Sephadex LH-20 column chromatography (3 cm i.d. × 30 cm, 0%–100% MeOH in H2O, 20% stepwise, each 200 mL, followed by MeOH-acetone-H2O, 90:6:4, 80:12:8, 70:18:12, each 200 mL) to yield 11 fractions. Fr. 3 (23.5 mg) was identified as gallic acid. Fr. 4 (60.6 mg) was subjected to Chromatorex ODS column chromatography (2 cm i.d. × 17 cm, 0–70% MeOH in H2O, 5% stepwise, each 100 mL) to give 1-O-galloyl-3,6-(S)-HHDP-β-d-glucose (17.6 mg). Fr. 5 (43.7 mg) was purified by MCI gel CHP 20P column chromatography (2 cm i.d. × 17 cm, 0-50% MeOH in H2O, 5% stepwise, each 100 mL) to afford 1-O-galloyl-3,6-(R)-HHDP-β-d-glucose (corilagin, 24.0 mg). Similar separation of Fr. 7 (63.9 mg) yielded 1-O-galloyl-2,3-(R)-DHHDP-β-d-glucose (furosin, 34.4 mg). Fractions 9 and 10 were identified to be geraniin (2)(463.9 mg) and ellagic acid (48.0 mg), respectively.
1-O-Galloyl-3,6-(S)-HHDP-β-d-glucose: Brown amorphous powder; [α +20.7° (c 0.10, MeOH); UV (MeOH) λmax (log ε): 293 (4.36), 273 (4.49), 227 (4.80), 219 (4.88); IR νmax cm−1: 3378, 1709, 16213; ESI-MS (negative) m/z: 633 [M − H]−; HR-FAB-MS m/z: 657.0705 (calcd for C27H22O18Na, 657.0703); ECD (MeOH) Δε (nm): +3.38 (229), −2.16 (267), +1.45 (293); 1H NMR (acetone-d6, 500 MHz) δ: 7.21 (2H, s, galloyl-H-2,6), 7.23 (1H, s, HHDP-H-3), 7.01 (1H, s, HHDP-H-3’), 6.29 (1H, d, J = 9.0 Hz, glc-1), 5.10 (1H, dt, J = 1.1, 5.7 Hz, glc-3), 4.88 (1H, d, J = 12.3 Hz, glc-6), 4.25 (1H, br d, J = 2.4 Hz, glc-5), 3.96 (1H, dt, J = 5.7, 9.0 Hz, glc-2), 3.84 (1H, dd, J = 3.4, 12.3 Hz, glc-6), 3.59 (1H, br s, glc-4); 13C NMR (acetone-d6, 125 MHz) δ: 168.0 (HHDP-7), 167.7 (HHDP-7’), 166.1 (galloyl-7), 145.8 (galloyl-3,5), 144.6, 144.5 (HHDP-4,4’,6,6’), 139.1 (galloyl-4), 138.1 (HHDP-5), 136.4 (HHDP-5’), 126.4 (HHDP-2, 2’), 121.2 (galloyl-1), 117.5 (HHDP-1), 116.7 (HHDP-1’), 112.4 (HHDP-3’), 110.3 (galloyl-2,6), 108.1 (HHDP-3’), 92.3 (glc-1), 81.2 (glc-3), 80.1 (glc-5), 72.7 (glc-4), 70.2 (glc-2), 63.3 (glc-6).
3.8. Heating of 2 with Pyridine in CH3CN
A solution of
2 (2.0 g) in 4% pyridine in CH
3CN (50 mL) was heated at 80 ˚C for 90 min. After cooling, the solution was mixed with 2% aqueous TFA (25 mL) and concentrated. Resulting aqueous solution was applied to Sephadex LH-20 column chromatography (3 cm i.d. × 25 cm, 60%–100% MeOH in H
2O, 10% stepwise, each 100 mL, followed by MeOH-acetone-H
2O, 80:10:10, 200 mL) to give 7 fractions. Fr. 3 (123.8 mg) was mainly composed of 1-
O-galloyl-3,6-(
R)-HHDP-β-
d-glucose (corilagin) and product
9. Fr. 4 (78.7 mg) contained corilagin,
9, and ellagic acid. Fr. 5 (473.5 mg) was separated by Chromatorex ODS (4 cm i.d. × 25 cm, 0%–55% MeOH in H
2O, 5% stepwise, each 100 mL) to give
2 (290.6 mg) and a mixture of
9 and
11, which were separated by Diaion HP20SS (3 cm i.d. × 17 cm, 10–60% MeOH in H
2O, 5% stepwise, each 50 mL) followed by purification using Chromatorex ODS chromatography (3 cm i.d. × 28 cm) to yield
9 (23.2 mg) and
11 (39.3 mg). Fr. 6 was separated H
2O soluble and insoluble parts, and the soluble fraction was purified by Avicel cellulose column chromatography (2 cm i.d. × 15 cm) with 2% AcOH to give 1-
O-galoyl-2,3;4,6-bis-(
R)-HHDP-β-
d-glucose (
10) (30.9 mg). The H
2O insoluble precipitates were combined with Fr. 7 and subjected to size-exclusion column chromatography using Sephadex LH-20 (2 cm i.d. × 55 cm) with 7M urea:acetone (2:3,
v/
v, containing conc. HCl at 5 mL/L)[
34] to give fractions containing oligomeric polyphenols and
8. The removal of urea by Diaion HP20SS column chromatography afforded
8 (595.8 mg) and oligomeric polyphenols (188.3 mg).
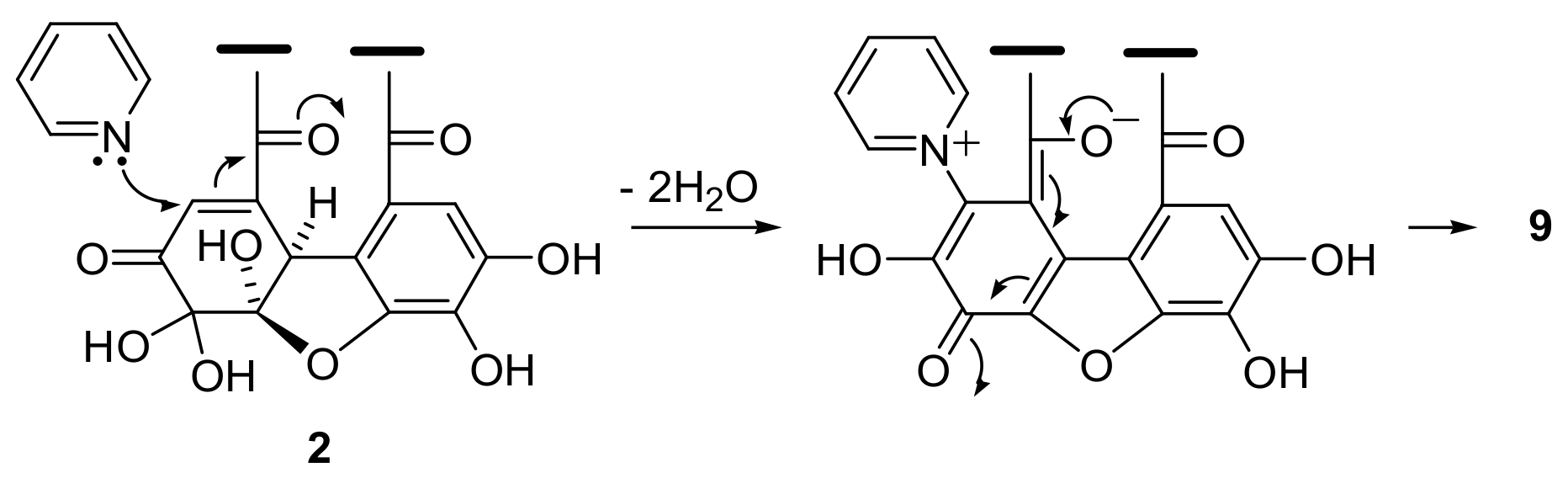
Pyridine adduct (9): A tan amorphous powder, [α −47.5 (c 0.1, MeOH); IR νmax cm−1: 3383, 1730, 1616, 1344; UV (MeOH) λmax (log ε): 349 (3.88), 280 (4.48), 217 (4.78). ESI-MS m/z: 996 [M + H]+, m/z: 995 [M − H]−; HRFABMS: m/z 996.1108 [M + H]+ (calcd for C46H30NO25, 996.1107); 1H NMR (acetone-d6, 500 MHz) δ: 9.53, 9.08 [each 1H, br d, J = 6.4 Hz, pyridine (py)-2,6], 8.62, 8.45 (each 1H, br t, J = 6.4 Hz, py-3,5), 7.37 [1H, s, benzofuran (BF)-3], 7.09 (2H, s, galloyl-2,6), 7.02 (1H, s, HHDP-3), 6.67 (1H, s, HHDP-3’), 6.39 (1H, br s, glc-3), 6.20 (1H, d, J = 3.1 Hz, glc-1), 5.39 (1H, br s, glc-2), 4.70 (1H, d, J = 3.8 Hz, glc-4), 4.41 (2H, m, glc-5, 6), 4.18 (1H, dd, J = 5.1, 10.6 Hz, glc-6); 13C NMR (acetone-d6, 125 MHz) δ: 168.3 (HHDP-7’), 166.7 (BF-7), 166.6 (HHDP-7), 165.2 (BF-7’), 165.1 (galloyl-7), 149.7, 149.2 (py-2,6), 147.9, 147.7, 147.5 (py-4, BF-4’,6’), 146.0 (galloyl-3,5), 145.9 (BF-6), 145.13, 145.06, 145.0, 144.6 (HHDP-4,4’,6,6’), 140.8 (BF-5’), 139.9 (galloyl-4), 137.3 (HHDP-5), 136.7 (HHDP-5’), 135.8 (BF-5), 134.6 (BF-2’), 129.8, 129.0 (py-3,5), 125.9 (BF-3’), 124.4, 123.8 (HHDP-2,2’), 119.6 (galloyl-1), 117.0 (BF-3), 116.7 (HHDP-1), 115.8 (BF-2), 115.7 (HHDP-1’), 115.1 (BF-1’), 114.3 (BF-1), 110.3 (galloyl-2,6), 109.5 (HHDP-3), 108.8 (HHDP-3’), 91.8 (glc-1), 74.7 (glc-5), 73.6 (glc-2), 69.3 (glc-4), 64.2 (glc-6), 62.1 (glc-3).
3.9. Methylation and Alkaline Degradation of Oligomeric Product
Oligomeric products (100 mg) in methanol (2 mL) was treated with etherial diazomethane for 2 days. The mixture was heated with 5% NaOH at 80 °C for 12 h, acidified with diluted HCl, and then partitioned with diethyl ether. The ether layer was concentrated and treated with diaomethane, and the products were separated by silica gel column chromatography (30%–100% acetone in hexane, 10% stepwise, each 50 mL) to yield methyl trimethoxybenzoate (5 mg), dimethyl (R)-hexamethoxydiphenate (2.0 mg) and dimethoxycarbonyl tetramethoxydibenzofuran (1.7 mg).
3.10. Acid Hydrolysis of DHHDP Esters
A portion (0.50 mL) of an acetone solution of
2 (10.0 mg/mL), 1-
O-galloyl-2,4-(
R)-DHHDP-β-
d-glucopyranose (6.0 mg/mL), and 1-
O-galloyl-3,6-(
R)-HHDP-β-
d-glucopyranose (6.0 mg/mL) was dispensed into 5 mL screw-capped vials, and acetone was removed in vacuo. To each vial, 0.50 mL of 2% H
2SO
4 was added and heated at 100 °C for 7 h. The reaction mixture was mixed with DMSO (4.5 mL) to dissolve ellagic acid and analyzed by HPLC (
Figure S9). Yield of the products was estimated based on the peak area and calibration curve obtained by using standard compounds: 4.76 μmol of gallic acid and 10.09 μmol of ellagic acid was produced from 5.25 μmol of
2. Similarly, 4.16 μmol of gallic acid and 3.05 μmol of ellagic acid was produced from 4.61 μmol of 1-
O-galloyl-2,4-(
R)-DHHDP-β-
d-glucopyranose, and 4.84 μmol of gallic acid and 5.59 μmol of ellagic acid was produced from 4.72 μmol of 1-
O-galloyl-3,6-(
R)-HHDP-β-
d-glucopyranose.
3.11. Acid Hydrolysis of 2
Geraniin (2) (1.00 g, 1.05 mmol, dried over P4O10) was hydrolyzed with 2% H2SO4 (50 mL) under reflux for 2 days. Insoluble precipitates were collected by filtration and washed with MeOH to give ellagic acid (504.5 mg, 1.67 mmol). The filtrate was directly subjected to Diaion HP20SS column chromatography (3 cm i.d. × 25 cm, 0%–80% MeOH in H2O, 10% stepwise, each 100 mL) to yield gallic acid (206.2 mg, 1.21 mmol), flavogallonic acid (9.8 mg, 0.02 mmol), and ellagic acid (66.0 mg, 0.22 mol). Total isolation yield of ellagic acid was 1.89 mmol.
3.12. Computational Methods
A conformational search was performed using the Monte Carlo method and the MMFF94 force field with Spartan ′14 (Wavefunction, Irvine, CA, USA). The obtained low-energy conformers within 6 kcal/mol were optimized at the B3LYP/6-31G(d,p) level in acetone (PCM). The vibrational frequencies were also calculated at the same level to confirm their stability, and no imaginary frequencies were found.
1H NMR coupling constants of the low-energy conformers with Boltzmann populations greater than 1% were calculated at the B3LYP/6-31G(d,p)u + 1s (using only the Fermi contact term) level in acetone (PCM) and scaled by using the slope parameter 0.94 [
35]. The calculated data for each conformer were averaged according to the Boltzmann distribution theory at 298 K based on their relative Gibbs free energies. All DFT calculations were performed using Gaussian 09 [
36]. GaussView was used to draw the three-dimensional molecular structures [
37].



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









