Benzoxazole Derivative K313 Induces Cell Cycle Arrest, Apoptosis and Autophagy Blockage and Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
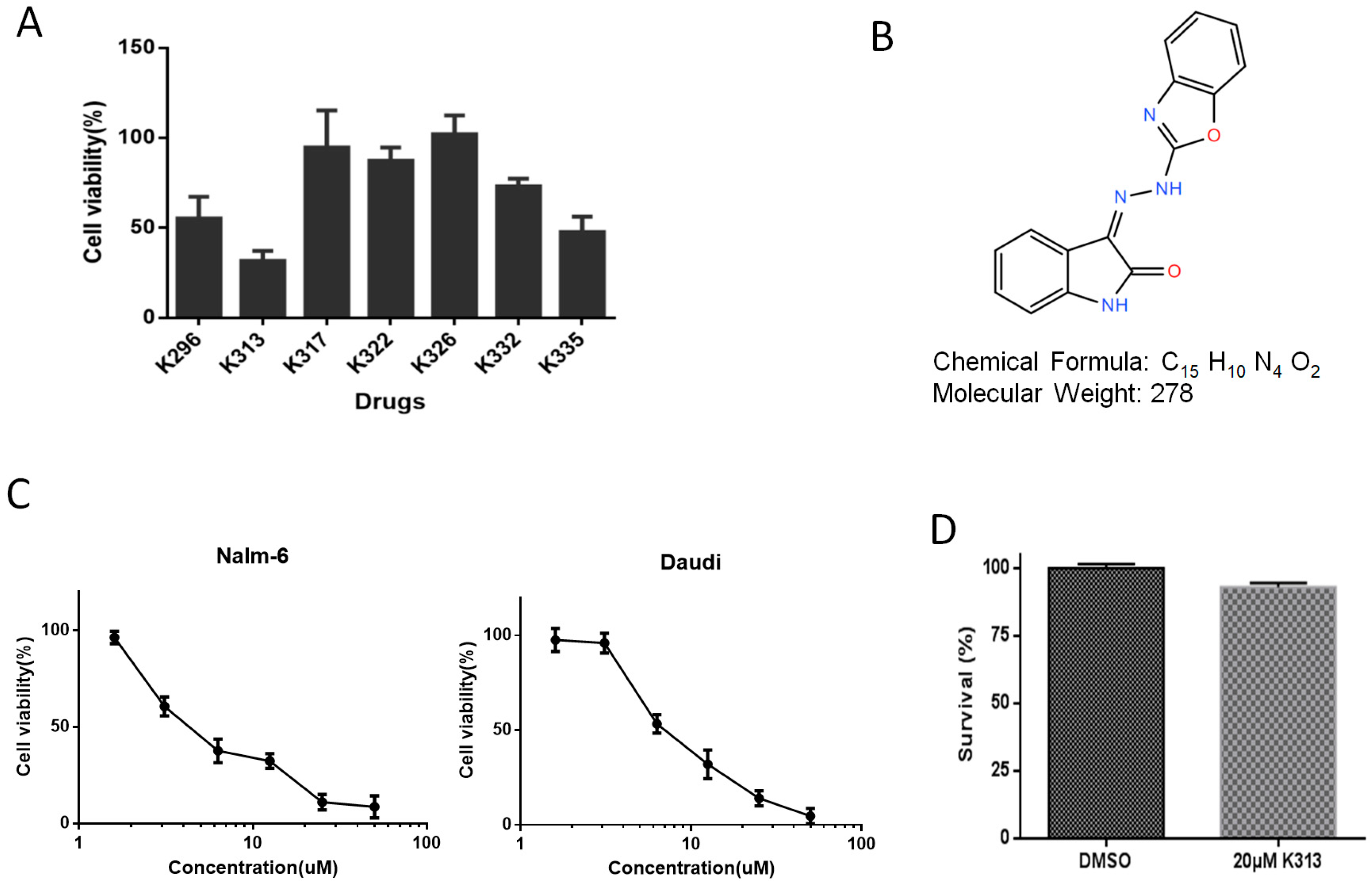
2.1. K313 Reduces the Viability of Nalm-6 and Daudi Cells without Affecting Normal Healthy PBMCs
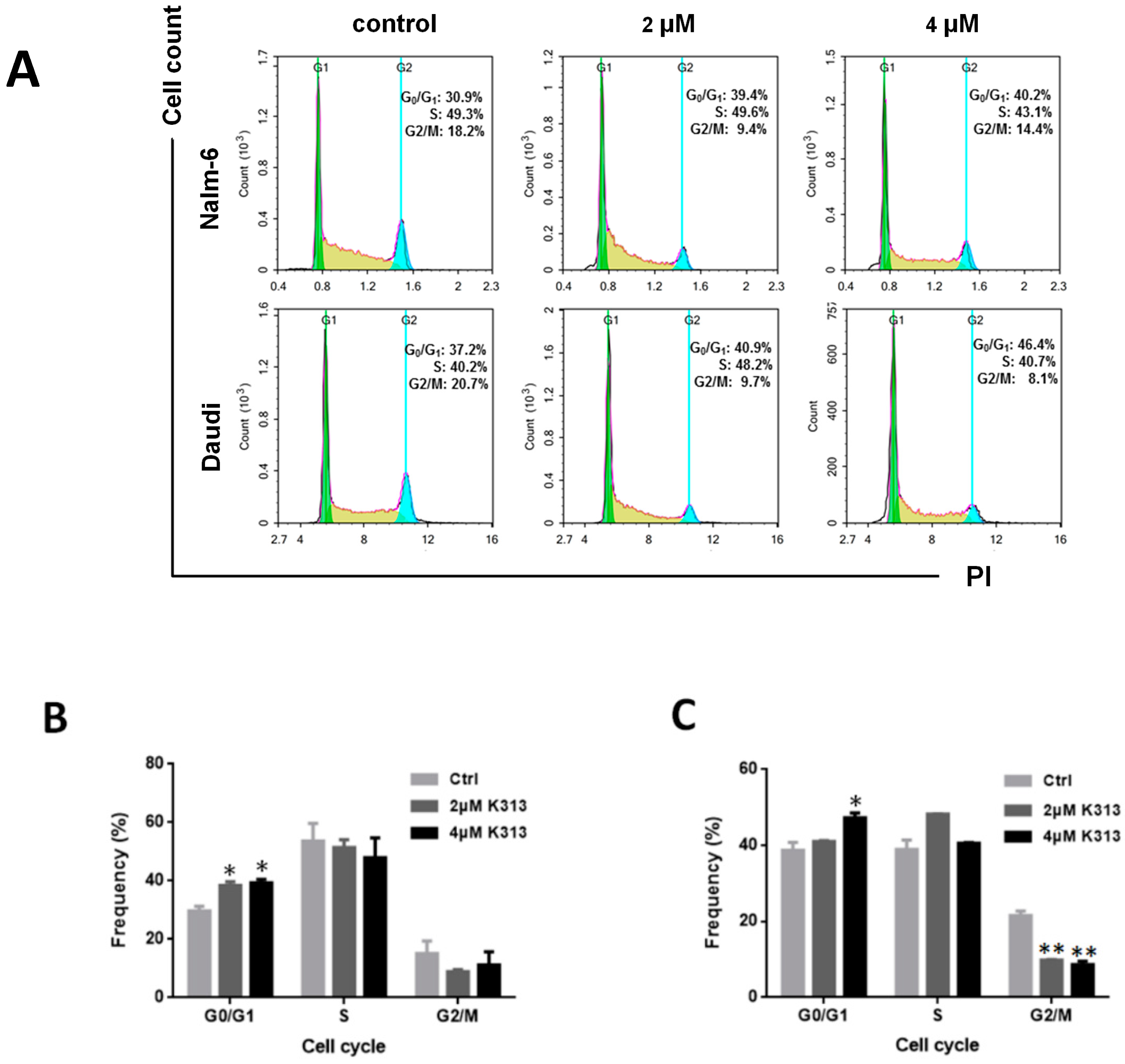
2.2. K313 Induces Moderate Cell Cycle Arrest at G0/G1 Phase in Nalm-6 and Daudi Cells
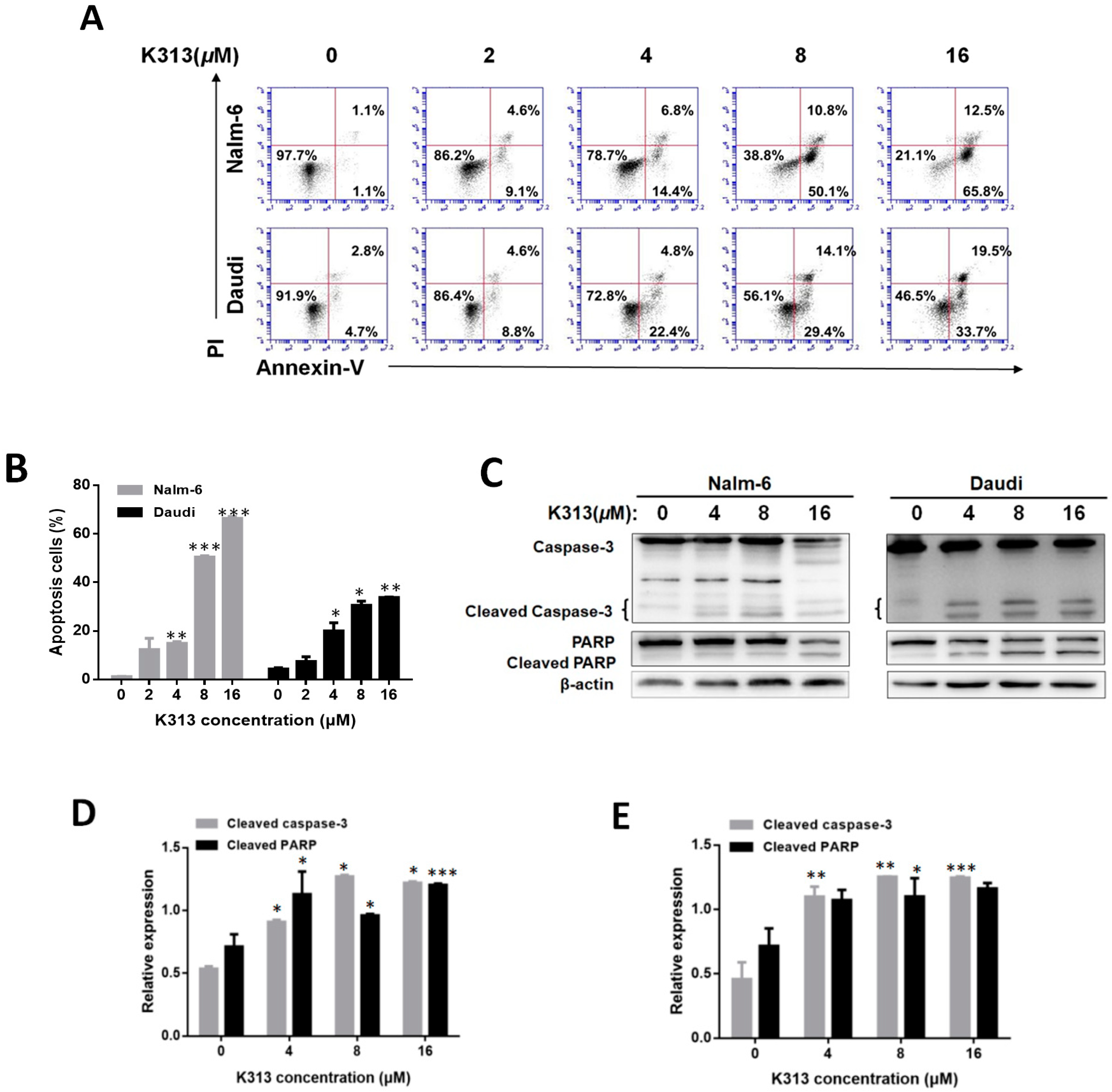
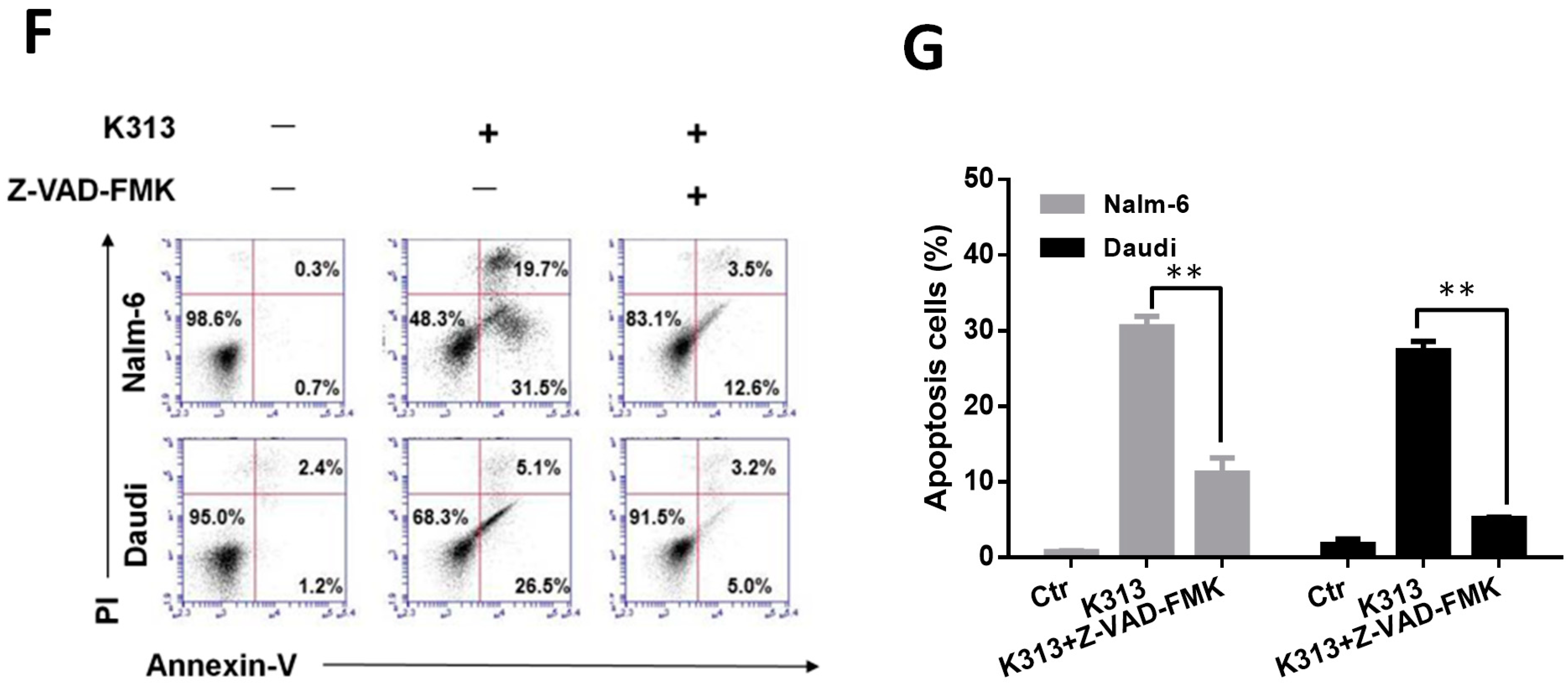
2.3. K313 Induces Apoptosis in Nalm-6 and Daudi Cells
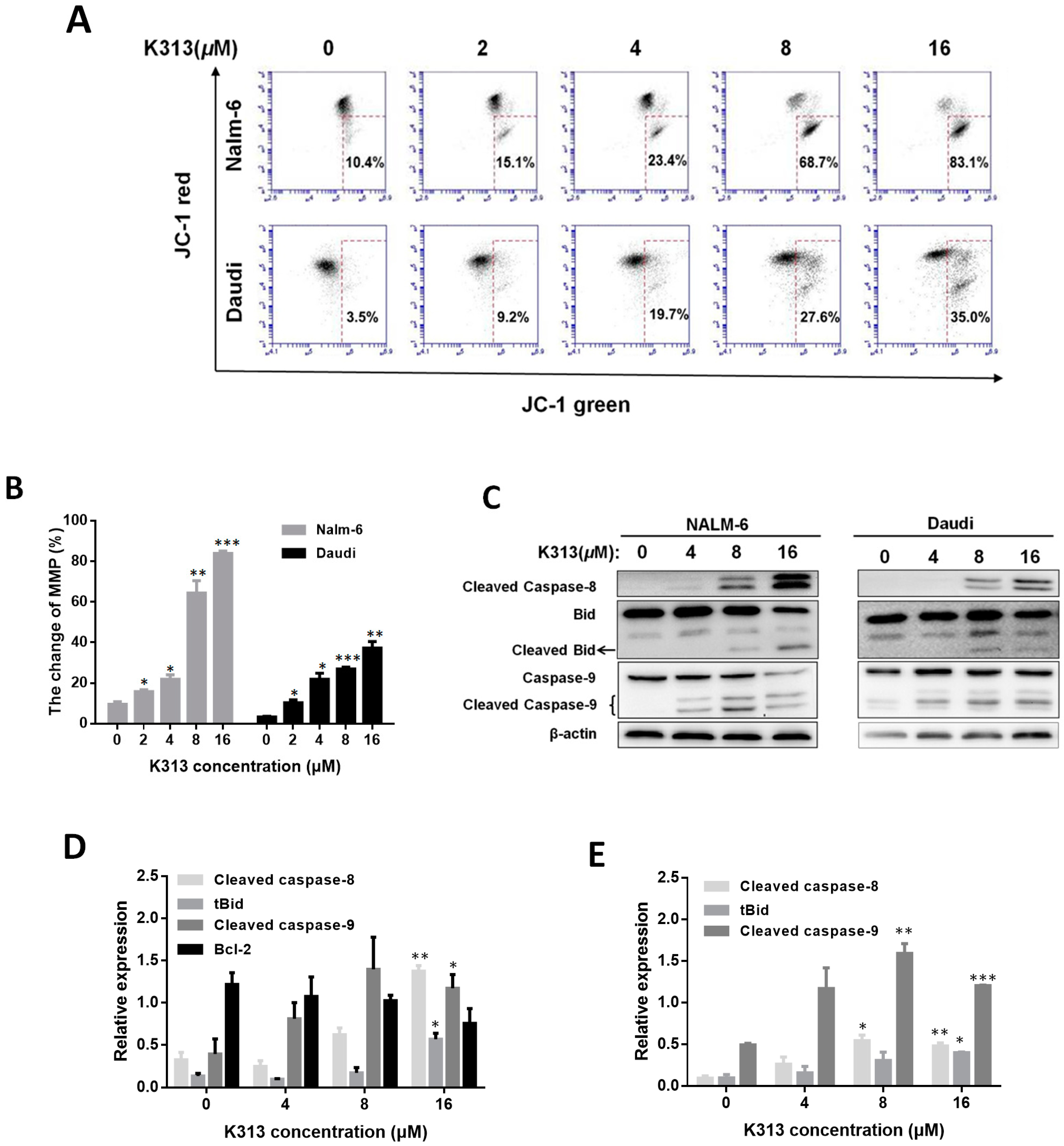
2.4. K313 Decreases Cell Mitochondrial Membrane Potential and Activates Mitochondrial Pathway of Apoptosis
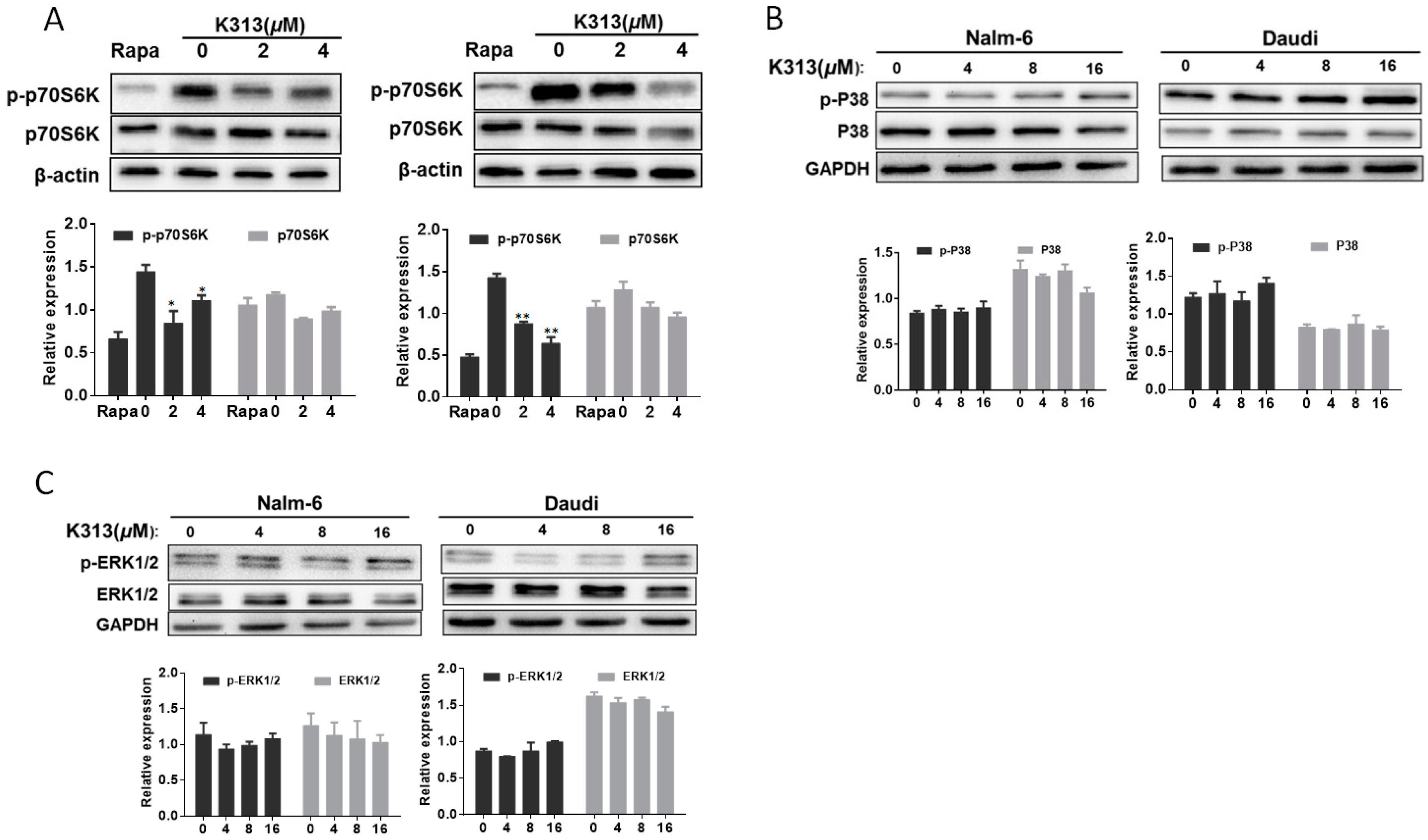
2.5. K313 Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells
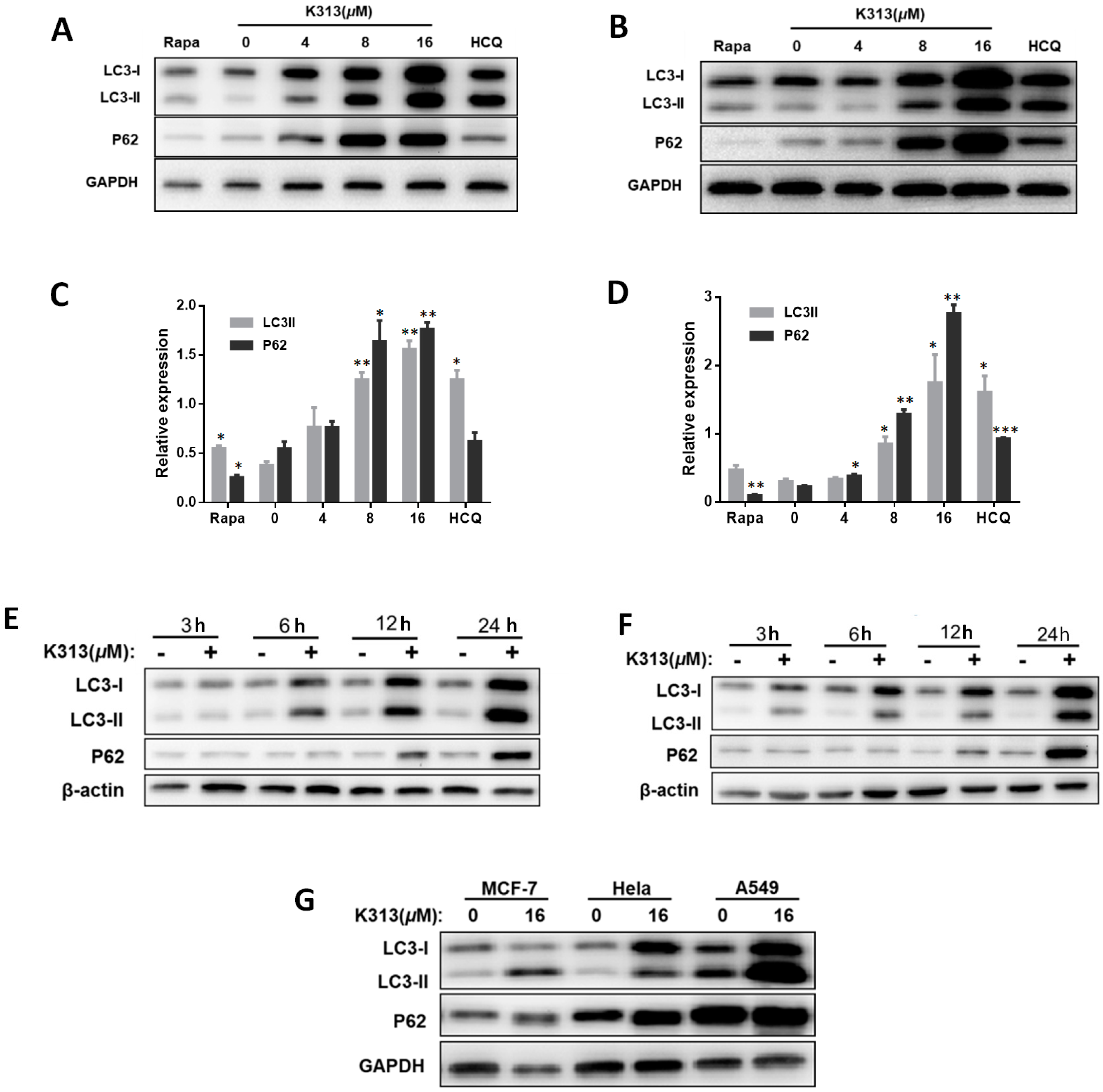
2.6. K313 Inhibits Autophagic Flux in Cancer Cells
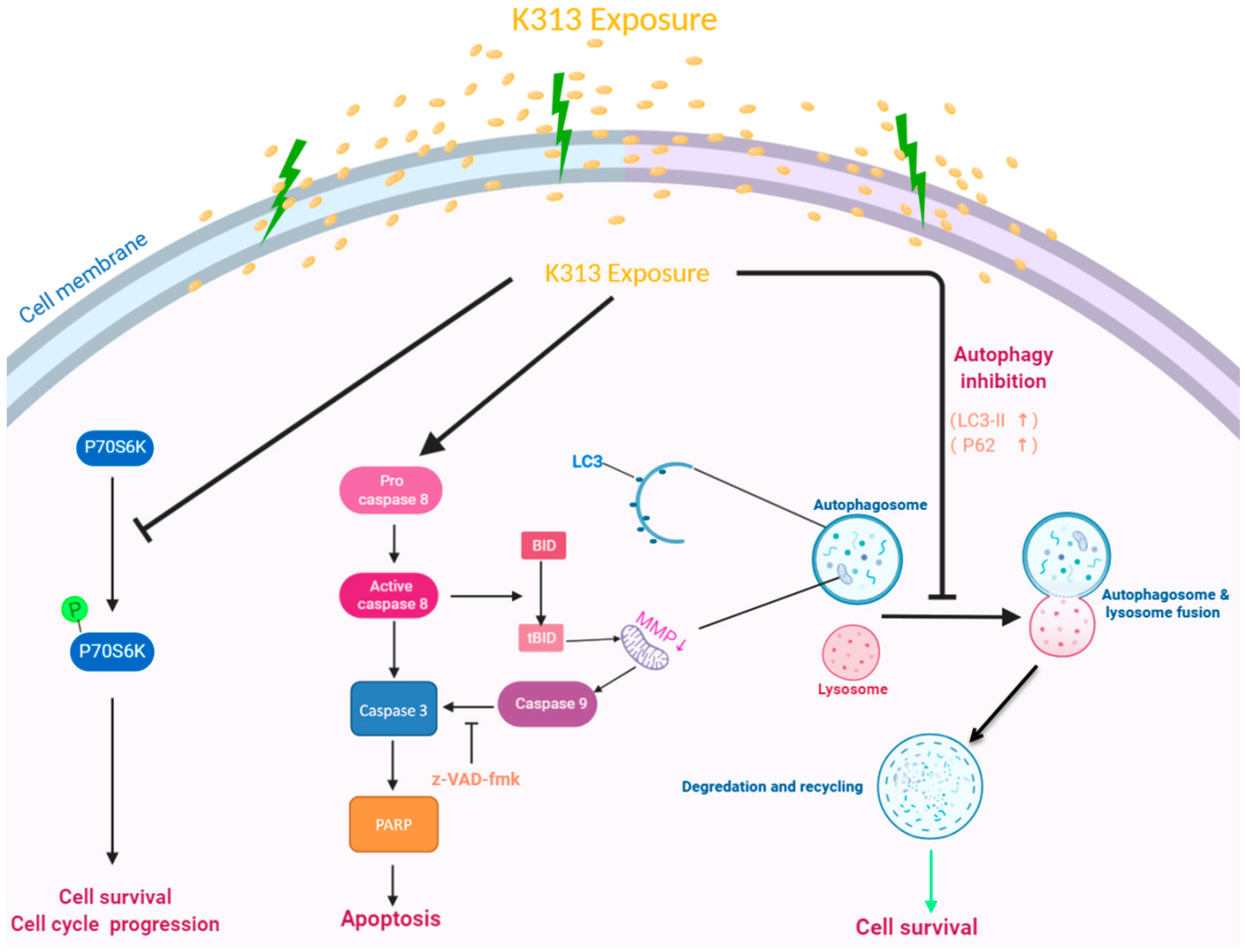
3. Discussion
4. Materials and Methods
4.1. Cells and Reagents
4.2. Cell Culture
4.3. Cell Viability Assay
4.4. Cell Cycle Analysis
4.5. Detection of Cell Apoptosis
4.6. Measurement of Mitochondrial Membrane Potential
4.7. Western Blot Analysis
4.8. Cell Immunofluorescence Staining
4.9. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Korfi, K.; Smith, M.; Swan, J.; Somervaille, T.C.P.; Dhomen, N.; Marais, R. BIM mediates synergistic killing of B-cell acute lymphoblastic leukemia cells by BCL-2 and MEK inhibitors. Cell Death Dis. 2016, 7, e2177. [Google Scholar] [CrossRef] [PubMed]
- Terwilliger, T.; Abdul-Hay, M. Acute lymphoblastic leukemia: A comprehensive review and 2017 update. Blood Cancer J. 2017, 7, e577. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.W.; Douer, D. Acute Lymphoblastic Leukemia in Adolescents and Young Adults. Acta Haematol. 2014, 132, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Pui, C. Recent Research Advances in Childhood Acute Lymphoblastic Leukemia. J. Formos. Med Assoc. 2010, 109, 777–787. [Google Scholar] [CrossRef]
- Santasusana, J.-M.R.; Oriol, A.; Sanz, M.A.; Tormo, M.; Abellan, P.F.; Del Potro, E.; Abella, E.; Bueno, J.; Parody, R.; Bastida, P.; et al. Comparison of the Results of the Treatment of Adolescents and Young Adults With Standard-Risk Acute Lymphoblastic Leukemia With the Programa Español de Tratamiento en Hematología Pediatric-Based Protocol ALL-96. J. Clin. Oncol. 2008, 26, 1843–1849. [Google Scholar]
- Nguyen, K.; Devidas, M.; Cheng, S.-C.; La, M.; Raetz, E.A.; Carroll, W.L.; Winick, N.J.; Hunger, S.P.; Gaynon, P.S.; Loh, M.L.; et al. Factors influencing survival after relapse from acute lymphoblastic leukemia: A Children’s Oncology Group study. Leukemia 2008, 22, 2142–2150. [Google Scholar] [CrossRef]
- Wong, B.J.; Berry, J.L. Acute Lymphoblastic Leukemia Relapse Presenting as Optic Nerve Infiltration. JAMA Ophthalmol. 2017, 135, e164656. [Google Scholar] [CrossRef]
- Cheson, B.D.; Canellos, G.P. The Cancer and Leukemia Group B Lymphoma Committee. Clin. Cancer Res. 2006, 12, 3572–3575. [Google Scholar] [CrossRef]
- Murawski, N.; Pfreundschuh, M. New drugs for aggressive B-cell and T-cell lymphomas. Lancet Oncol. 2010, 11, 1074–1085. [Google Scholar] [CrossRef]
- Xia, A.-L.; Wang, X.-C.; Lu, Y.-J.; Lu, X.-J.; Sun, B. Chimeric-antigen receptor T (CAR-T) cell therapy for solid tumors: Challenges and opportunities. Oncotarget 2017, 8, 90521–90531. [Google Scholar] [CrossRef]
- Lad, D.P.; Jain, A.; Varma, S. Complications and management of coagulation disorders in leukemia patients. Blood Lymphat. Cancer Targets Ther. 2017, 7, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Magrath, I.T. African Burkitt’s lymphoma. History, biology, clinical features, and treatment. Am. J. Pediatric Hematol. Oncol. 1991, 13, 222–246. [Google Scholar] [CrossRef]
- Molyneux, E.; Rochford, R.; Griffin, B.; Newton, R.; Jackson, G.; Menon, G.; Harrison, C.; Israels, T.; Bailey, S. Burkitt’s lymphoma. Lancet 2012, 379, 1234–1244. [Google Scholar] [CrossRef]
- Perkins, A.S.; Friedberg, J.W. Burkitt lymphoma in adults. ASH Educ. Program Book 2008, 1, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Rida, S.; Ashour, F.; El-Hawash, S.; ElSemary, M.; Badr, M.; Shalaby, M. Synthesis of some novel benzoxazole derivatives as anticancer, anti-HIV-1 and antimicrobial agents. Eur. J. Med. Chem. 2005, 40, 949–959. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Liu, J.; Macho, J.M.; Jiang, X.; Xie, D.; Jiang, F.; Liu, W.; Fu, L. Design, synthesis and antimicrobial evaluation of novel benzoxazole derivatives. Eur. J. Med. Chem. 2017, 126, 7–14. [Google Scholar] [CrossRef]
- AbdelGawad, M.; Belal, A.; Omar, H.A.; Hegazy, L.; Rateb, M.E. Synthesis, Anti-Breast Cancer Activity, and Molecular Modeling of Some Benzothiazole and Benzoxazole Derivatives. Arch. Pharm. 2013, 346, 534–541. [Google Scholar] [CrossRef]
- Kim, D.; Won, H.Y.; Hwang, E.S.; Kim, Y.-K.; Choo, H.-Y.P. Synthesis of benzoxazole derivatives as interleukin-6 antagonists. Bioorganic Med. Chem. 2017, 25, 3127–3134. [Google Scholar] [CrossRef]
- Xiang, P.; Zhou, T.; Wang, L.; Sun, C.-Y.; Hu, J.; Zhao, Y.-L.; Yang, L. Novel Benzothiazole, Benzimidazole and Benzoxazole Derivatives as Potential Antitumor Agents: Synthesis and Preliminary in Vitro Biological Evaluation. Molecules 2012, 17, 873–883. [Google Scholar] [CrossRef]
- Iyer, V.B.; Gurupadayya, B.M.; Sairam, K.V.; Inturi, B.; Chandan, R.S.; Tengli, A.K. Anti-Inflammatory Activity of 1,3,4-Oxadiazoles Derived from Benzoxazole. J. Pharm. Sci. Pharmacol. 2015, 2, 233–241. [Google Scholar] [CrossRef]
- Kantari, C.; Walczak, H. Caspase-8 and Bid: Caught in the act between death receptors and mitochondria. Biochim. Biophys. Acta Bioenerg. 2011, 1813, 558–563. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of Cells and Tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Cuervo, A.M. Lipophagy: Connecting Autophagy and Lipid Metabolism. Int. J. Cell Boil. 2012, 2012, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Xilouri, M.; Stefanis, L. Chaperone mediated autophagy in aging: Starve to prosper. Ageing Res. Rev. 2016, 32, 13–21. [Google Scholar] [CrossRef]
- Janku, F.; McConkey, D.J.; Hong, D.S.; Kurzrock, R. Autophagy as a target for anticancer therapy. Nat. Rev. Clin. Oncol. 2011, 8, 528–539. [Google Scholar] [CrossRef] [PubMed]
- Degenhardt, K.; Mathew, R.; Beaudoin, B.; Bray, K.; Anderson, D.; Chen, G.; Mukherjee, C.; Shi, Y.; Gélinas, C.; Fan, Y.; et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell 2006, 10, 51–64. [Google Scholar] [CrossRef]
- Kimura, T.; Takabatake, Y.; Takahashi, A.; Isaka, Y. Chloroquine in Cancer Therapy: A Double-Edged Sword of Autophagy. Cancer Res. 2013, 73, 3–7. [Google Scholar] [CrossRef]
- White, E.; DiPaola, R.S. The double-edged sword of autophagy modulation in cancer. Clin. Cancer Res. 2009, 15, 5308–5316. [Google Scholar] [CrossRef]
- Aita, V.M.; Liang, X.H.; Murty, V.; Pincus, D.L.; Yu, W.; Cayanis, E.; Kalachikov, S.; Gilliam, T.; Levine, B. Cloning and Genomic Organization of Beclin 1, a Candidate Tumor Suppressor Gene on Chromosome 17q21. Genomics 1999, 59, 59–65. [Google Scholar] [CrossRef]
- Choi, A.M.K.; Ryter, S.W.; Levine, B.; Doria, A.; Gatto, M.; Punzi, L. Autophagy in human health and disease. N. Engl. J. Med. 2013, 368, 1845–1846. [Google Scholar] [CrossRef]
- Yue, Z.; Jin, S.; Yang, C.; Levine, A.J.; Heintz, N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc. Natl. Acad. Sci. USA 2003, 100, 15077–15082. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Yu, J.; Bhagat, G.; Furuya, N.; Hibshoosh, H.; Troxel, A.; Rosen, J.; Eskelinen, E.-L.; Mizushima, N.; Ohsumi, Y.; et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J. Clin. Investig. 2003, 112, 1809–1820. [Google Scholar] [CrossRef]
- White, E. The role for autophagy in cancer. J. Clin. Investig. 2015, 125, 42–46. [Google Scholar] [CrossRef]
- Kimmelman, A.C.; White, E. Autophagy and Tumor Metabolism. Cell Metab. 2017, 25, 1037–1043. [Google Scholar] [CrossRef] [PubMed]
- White, E. Exploiting the bad eating habits of Ras-driven cancers. Genes Dev. 2013, 27, 2065–2071. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.-H.; Piao, S.; Wang, D.; McAfee, Q.W.; Nathanson, K.L.; Lum, J.J.; Li, L.Z.; Amaravadi, R.K. Measurements of tumor cell autophagy predict invasiveness, resistance to chemotherapy, and survival in melanoma. Clin. Cancer Res. 2011, 17, 3478–3489. [Google Scholar] [CrossRef] [PubMed]
- Kundu, M.; Thompson, C.B. Autophagy: Basic Principles and Relevance to Disease. Annu. Rev. Pathol. Mech. Dis. 2008, 3, 427–455. [Google Scholar] [CrossRef]
- Kondo, Y.; Kanzawa, T.; Sawaya, R.; Kondo, S. The role of autophagy in cancer development and response to therapy. Nat. Rev. Cancer 2005, 5, 726–734. [Google Scholar] [CrossRef]
- Pagotto, A.; Pilotto, G.; Mazzoldi, E.L.; Nicoletto, M.O.; Frezzini, S.; Pastò, A.; Amadori, A. Autophagy inhibition reduces chemoresistance and tumorigenic potential of human ovarian cancer stem cells. Cell Death Dis. 2017, 8, e2943. [Google Scholar] [CrossRef]
- Liu, L.; Yang, M.; Kang, R.; Wang, Z.; Zhao, Y.; Yu, Y.; Xie, M.; Yin, X.; Livesey, K.M.; Lotze, M.T.; et al. DAMP-mediated autophagy contributes to drug resistance. Autophagy 2011, 7, 112–114. [Google Scholar] [CrossRef]
- Eritja, N.; Chen, B.-J.; Rodríguez-Barrueco, R.; Santacana, M.; Gatius, S.; Vidal, A.; Martí, M.D.; Ponce, J.; Bergadà, L.; Yeramian, A.; et al. Autophagy orchestrates adaptive responses to targeted therapy in endometrial cancer. Autophagy 2017, 13, 608–624. [Google Scholar] [CrossRef] [PubMed]
- Zou, Y.-L.; Luo, W.-B.; Xie, L.; Mao, X.-B.; Wu, C.; You, Z.-P. Targeting human 8-oxoguanine DNA glycosylase to mitochondria protects cells from high glucose-induced apoptosis. Endocrine 2018, 60, 445–457. [Google Scholar] [CrossRef] [PubMed]
- Keil, V.C.; Funke, F.; Zeug, A.; Schild, D.; Müller, M. Ratiometric high-resolution imaging of JC-1 fluorescence reveals the subcellular heterogeneity of astrocytic mitochondria. Pflügers Arch. Eur. J. Physiol. 2011, 462, 693–708. [Google Scholar] [CrossRef] [PubMed]
- Tormo, M.; Tari, A.M.; McDonnell, T.J.; Cabanillas, F.; Garcia-Conde, J.; Lopez-Berestein, G. Apoptotic Induction in Transformed Follicular Lymphoma Cells by Bcl-2 Downregulation. Leuk. Lymphoma 1998, 30, 367–380. [Google Scholar] [CrossRef]
- Kawasome, H.; Papst, P.; Webb, S.; Keller, G.; Johnson, G.L.; Gelfand, E.W.; Terada, N. Targeted disruption of p70s6k defines its role in protein synthesis and rapamycin sensitivity. Proc. Natl. Acad. Sci. USA 1998, 95, 5033–5038. [Google Scholar] [CrossRef]
- Kwon, H.-K.; Bae, G.-U.; Yoon, J.-W.; Kim, Y.K.; Lee, H.Y.; Lee, H.-W.; Han, J.-W. Constitutive activation of p70S6k in cancer cells. Arch. Pharmacal Res. 2002, 25, 685–690. [Google Scholar] [CrossRef]
- Hou, G.; Xue, L.; Lu, Z.; Fan, T.; Tian, F.; Xue, Y. An activated mTOR/p70S6K signaling pathway in esophageal squamous cell carcinoma cell lines and inhibition of the pathway by rapamycin and siRNA against mTOR. Cancer Lett. 2007, 253, 236–248. [Google Scholar] [CrossRef]
- Li, H.; Kong, X.; Cui, G.; Ren, C.; Fan, S.; Sun, L.; Zhang, Y.; Cao, R.; Li, Y.; Zhou, J. Rapamycin restores p14, p15 and p57 expression and inhibits the mTOR/p70S6K pathway in acute lymphoblastic leukemia cells. Int. J. Hematol. 2015, 102, 558–568. [Google Scholar] [CrossRef]
- Geng, J.; Klionsky, D.J. The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 2008, 9, 859–864. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, J.; Abeliovich, H.; Acevedo-Arozena, A.; Adachi, H.; Adams, C.; Adams, P.D.; Adeli, K.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef]
- Germain, M.; Nguyen, A.P.; Le Grand, J.N.; Arbour, N.; Vanderluit, J.L.; Park, D.S.; Opferman, J.T.; Slack, R. MCL-1 is a stress sensor that regulates autophagy in a developmentally regulated manner. EMBO J. 2010, 30, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Ravikumar, B.; Floto, R.A.; Rubinsztein, D.C. Rapamycin and mTOR-independent autophagy inducers ameliorate toxicity of polyglutamine-expanded huntingtin and related proteinopathies. Cell Death Differ. 2008, 16, 46–56. [Google Scholar] [CrossRef] [PubMed]
- Kriegsmann, K.; Klee, J.S.; Hensel, M.; Buss, E.C.; Kriegsmann, M.; Cremer, M.; Ho, A.D.; Egerer, G.; Wuchter, P.; Witzens-Harig, M. Patients With Malignant Lymphoma and HIV Infection Experiencing Remission After First-Line Treatment Have an Excellent Prognosis. Clin. Lymphoma Myeloma Leuk. 2019, 19, e581–e587. [Google Scholar] [CrossRef] [PubMed]
- Onea, A.S.; Jazirehi, A.R. CD19 chimeric antigen receptor (CD19 CAR)-redirected adoptive T-cell immunotherapy for the treatment of relapsed or refractory B-cell Non-Hodgkin’s Lymphomas. Am. J. Cancer Res. 2016, 6, 403–424. [Google Scholar]
- Zhao, B.; Guo, H.-J.; Liu, Y.; Luo, X.; Yang, S.-X.; Wang, Y.-T.; Leng, X.; Mo, C.-F.; Zou, Q. K313, a novel benzoxazole derivative, exhibits anti-inflammatory properties via inhibiting GSK3β activity in LPS-induced RAW264.7 macrophages. J. Cell. Biochem. 2018, 119, 5382–5390. [Google Scholar] [CrossRef]
- Sharpe, J.C.; Arnoult, D.; Youle, R.J. Control of mitochondrial permeability by Bcl-2 family members. Biochim. Biophys. Acta Bioenerg. 2004, 1644, 107–113. [Google Scholar] [CrossRef]
- Høyer-Hansen, M.; Jäättelä, M. Autophagy: An emerging target for cancer therapy. Autophagy 2008, 4, 574–580. [Google Scholar] [CrossRef]
- Xi, G.; Hu, X.; Wu, B.; Jiang, H.; Young, C.Y.; Pang, Y.; Yuan, H. Autophagy inhibition promotes paclitaxel-induced apoptosis in cancer cells. Cancer Lett. 2011, 307, 141–148. [Google Scholar] [CrossRef]
- Carew, J.S.; Nawrocki, S.T.; Cleveland, J.L. Modulating autophagy for therapeutic benefit. Autophagy 2007, 3, 464–467. [Google Scholar] [CrossRef]
- Wang, Z.; Zhu, S.; Zhang, G.; Liu, S. Inhibition of autophagy enhances the anticancer activity of bortezomib in B-cell acute lymphoblastic leukemia cells. Am. J. Cancer Res. 2015, 5, 639–650. [Google Scholar]
- Wang, K.; Klionsky, D.J. Mitochondria removal by autophagy. Autophagy 2011, 7, 297–300. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhong, W.; Tang, X.; Liu, Y.; Zhou, C.; Liu, P.; Li, E.; Zhong, P.; Lv, H.; Zou, Q.; Wang, M. Benzoxazole Derivative K313 Induces Cell Cycle Arrest, Apoptosis and Autophagy Blockage and Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells. Molecules 2020, 25, 971. https://doi.org/10.3390/molecules25040971
Zhong W, Tang X, Liu Y, Zhou C, Liu P, Li E, Zhong P, Lv H, Zou Q, Wang M. Benzoxazole Derivative K313 Induces Cell Cycle Arrest, Apoptosis and Autophagy Blockage and Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells. Molecules. 2020; 25(4):971. https://doi.org/10.3390/molecules25040971
Chicago/Turabian StyleZhong, Wenying, Xinwen Tang, Yang Liu, Chunyu Zhou, Pan Liu, Enhui Li, Peilin Zhong, Haoxue Lv, Qiang Zou, and Maolin Wang. 2020. "Benzoxazole Derivative K313 Induces Cell Cycle Arrest, Apoptosis and Autophagy Blockage and Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells" Molecules 25, no. 4: 971. https://doi.org/10.3390/molecules25040971
APA StyleZhong, W., Tang, X., Liu, Y., Zhou, C., Liu, P., Li, E., Zhong, P., Lv, H., Zou, Q., & Wang, M. (2020). Benzoxazole Derivative K313 Induces Cell Cycle Arrest, Apoptosis and Autophagy Blockage and Suppresses mTOR/p70S6K Pathway in Nalm-6 and Daudi Cells. Molecules, 25(4), 971. https://doi.org/10.3390/molecules25040971