
Enhanced Hybridization Selectivity Using Structured GammaPNA Probes

 ,
,  and
and
Abstract
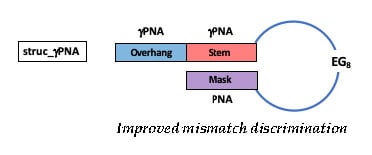
1. Introduction
2. Results
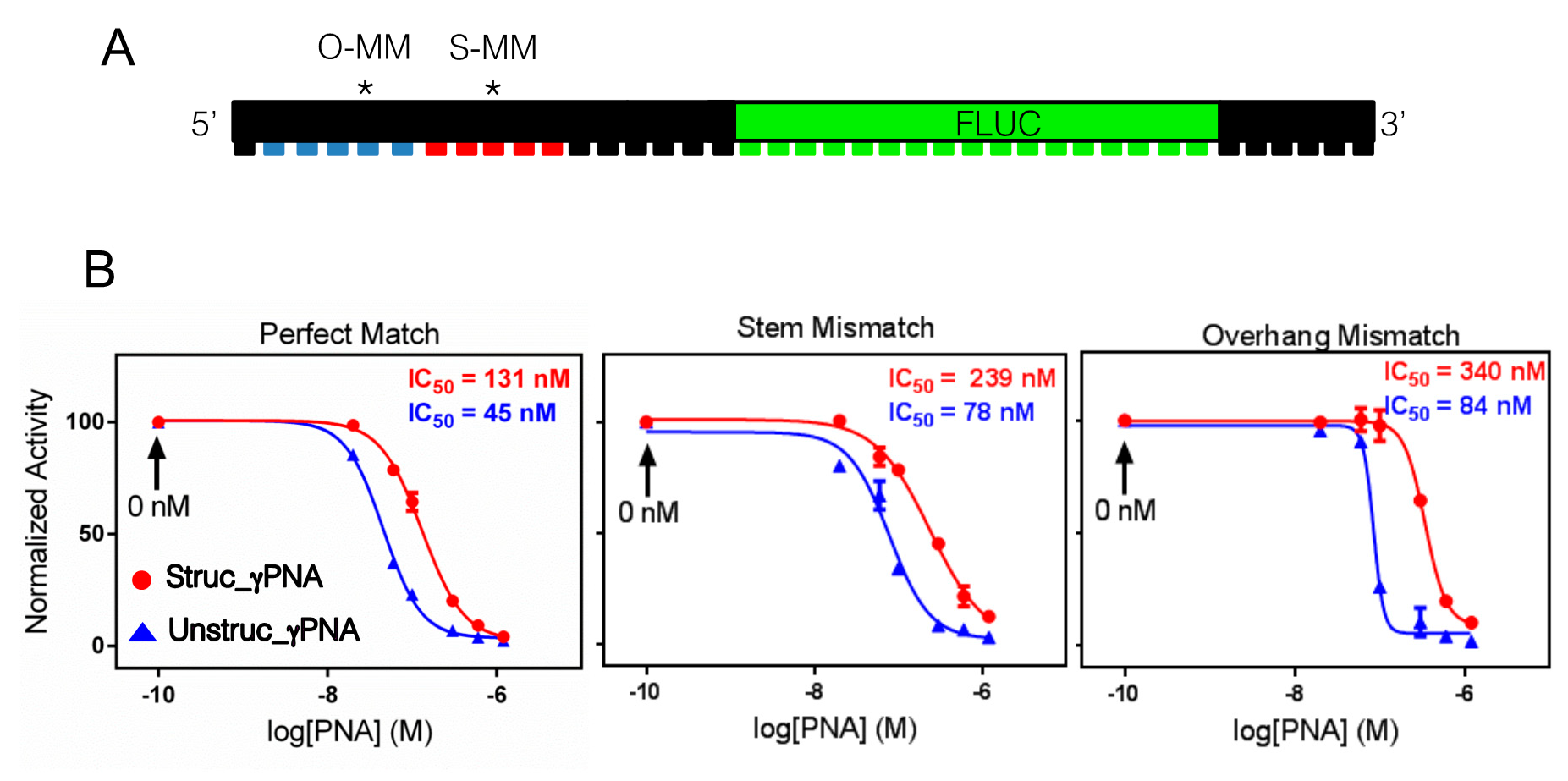
2.1. Hairpin Design and Characterization
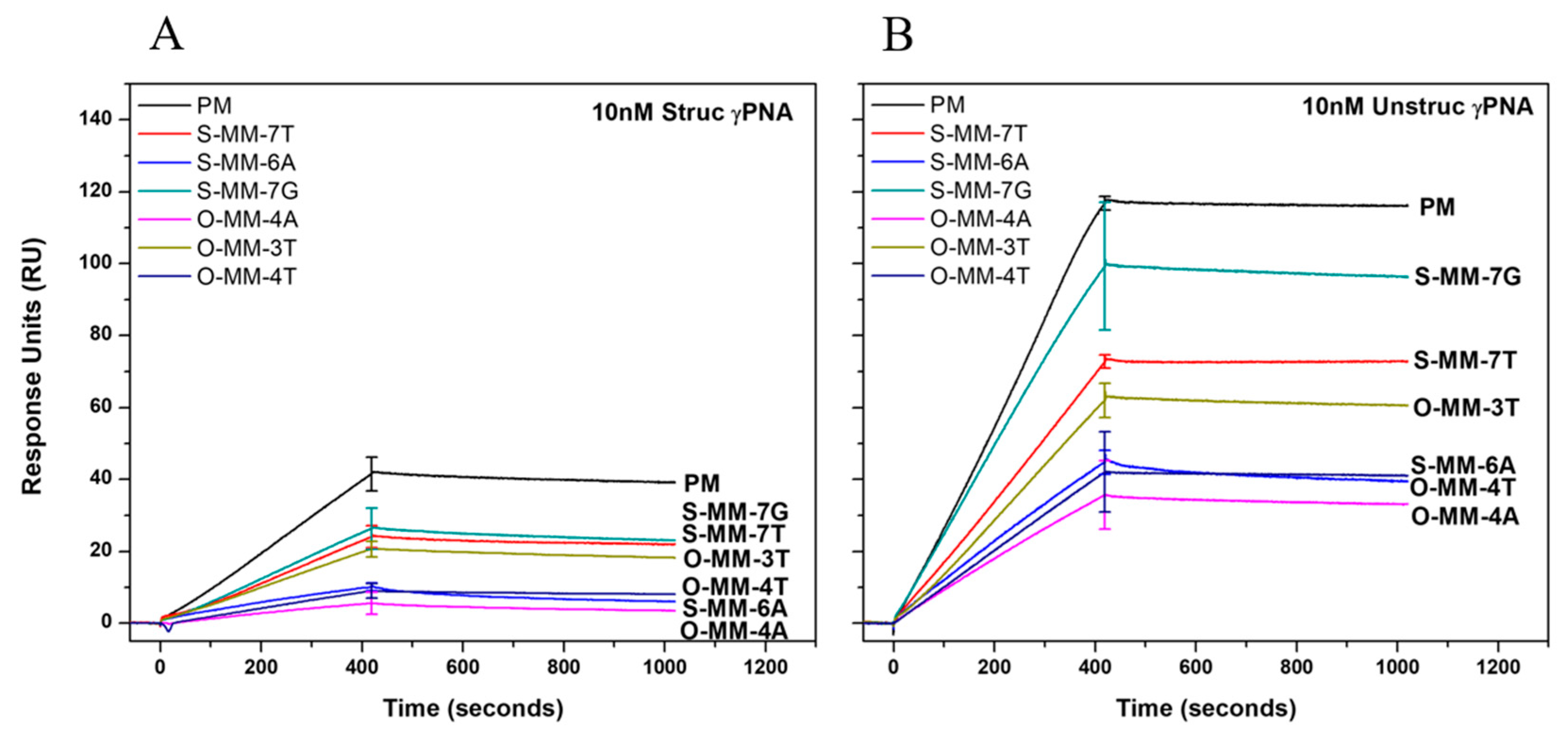
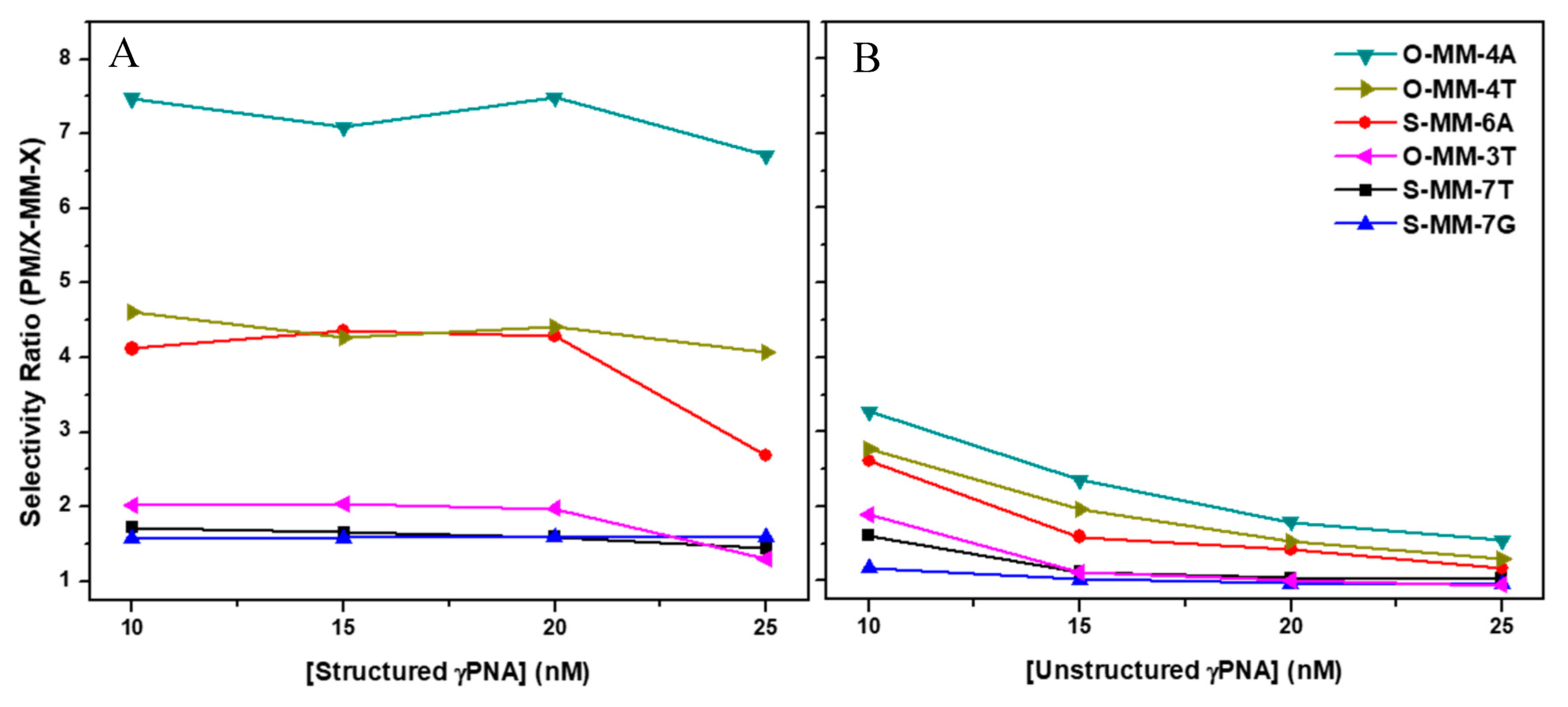
2.2. Hybridization Selectivity Analyzed by SPR
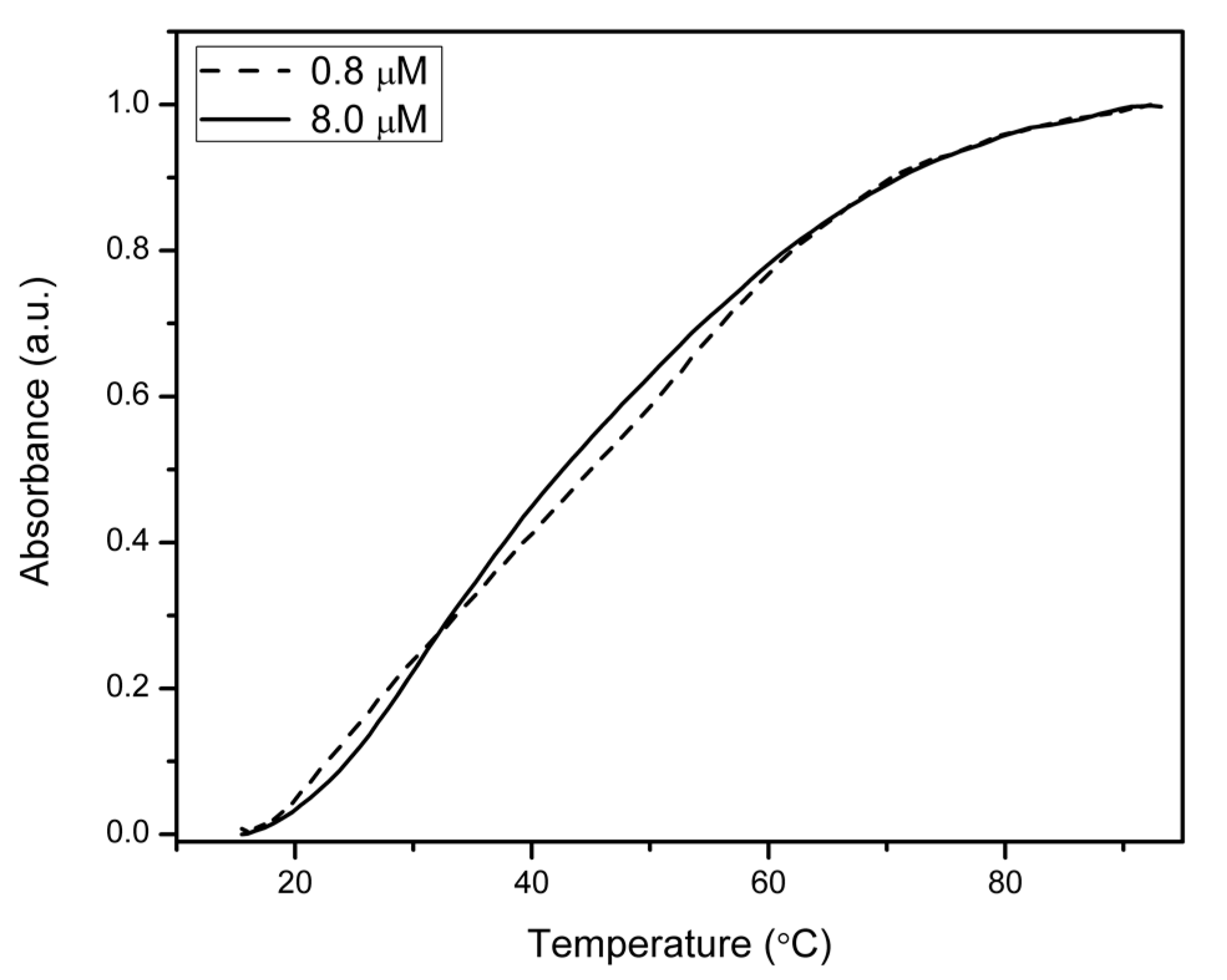
2.3. UV Melting Analysis of Select γPNA-DNA Duplexes
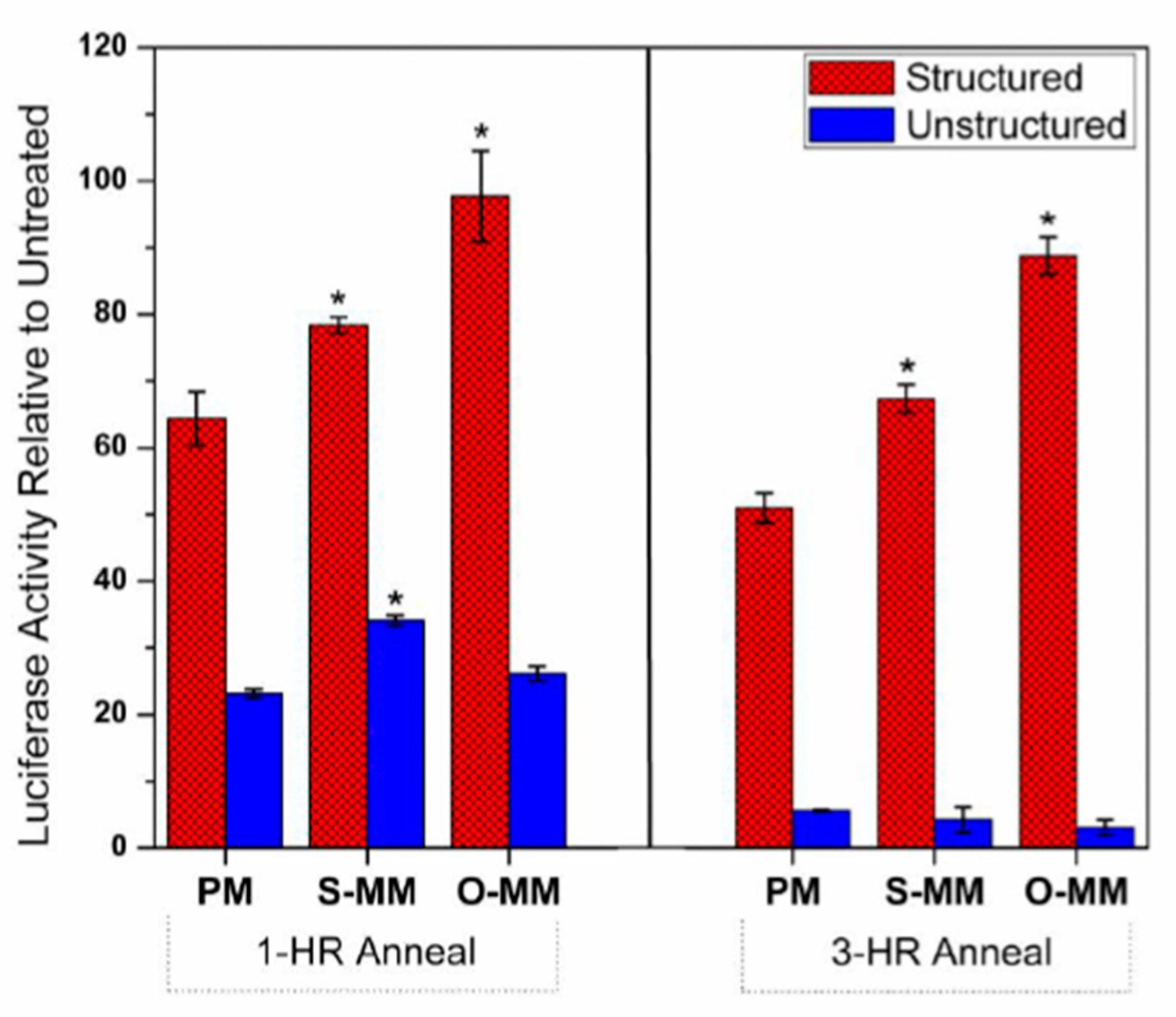
2.4. Antisense Effects in a Luciferase Reporter Assay
3. Discussion
4. Materials and Methods
4.1. γPNA/DNA Oligomers
4.2. Surface Plasmon Resonance (SPR)
4.3. UV Melting
4.4. Luciferase Assays
4.5. Transcription Reaction and Purification
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Demidov, V.V.; Frank-Kamenetskii, M.D. Two Sides of the Coin: Affinity and Specificity of Nucleic Acid Interactions. Trends Biochem. Sci 2004, 29, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Bonnet, G.; Tyagi, S.; Libchaber, A.; Kramer, F.R. Thermodynamic Basis of the Enhanced Specificity of Structured DNA Probes. PNAS 1999, 96, 6171–6176. [Google Scholar] [CrossRef] [PubMed]
- Tsourkas, A.; Behlke, M.A.; Rose, S.D.; Bao, G. Hybridization Kinetics and Thermodynamics of Molecular Beacons. Nucleic Acids Res. 2003, 31, 1319–1330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Plakos, K.J.I.; Lou, X.; White, R.J.; Qian, J.; Plaxco, K.W.; Soh, H.T. Fluorescence Detection of Single-Nucleotide Polymorphisms with a Single, Self-Complementary, Triple-Stem DNA Probe. Angew. Chem. Int. Ed. 2009, 48, 4354–4358. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.Y.; Chen, S.X.; Yin, P. Optimizing the Specificity of Nucleic Acid Hybridization. Nat. Chem. 2012, 4, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.R.; Wang, J.S.; Fang, J.Z.; Evans, E.R.; Pinto, A.; Pekker, I.; Boykin, R.; Ngouenet, C.; Webster, P.J.; Beechem, J.; et al. Continuously Tunable Nucleic Acid Hybridization Probes. Nat. Methods 2015, 12, 1191–1196. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Nordén, B.; Nielsen, P.E. PNA Hybridizes to Complementary Oligonucleotides Obeying the Watson-Crick Hydrogen-Bonding Rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-Selective Recognition of DNA by Strand Displacement with a Thymine-Substituted Polyamide. Science 1991, 254, 1498–1500. [Google Scholar] [CrossRef]
- Ørum, H.; Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O.; Stanley, C. Single Base Pair Mutation Analysis by PNA Directed PCR Clamping. Nucleic Acids Res. 1993, 21, 5332–5336. [Google Scholar] [CrossRef]
- Stender, H.; Fiandaca, M.; Hyldig-Nielsen, J.J.; Coull, J. PNA for Rapid Microbiology. J. Microbiol. Methods 2002, 48, 1–17. [Google Scholar] [CrossRef]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A Simple γ-Backbone Modification Preorganizes Peptide Nucleic Acid into a Helical Structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef] [PubMed]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and Characterization of Conformationally Preorganized, (R)-Diethylene Glycol-Containing γ-Peptide Nucleic Acids with Superior Hybridization Properties and Water Solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef] [PubMed]
- Bahal, R.; McNeer, N.A.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.-C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In vivo correction of anaemia in b-thalassemic mice by gPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016, 7, 13304. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coşkun, S.; Song, E.; et al. In Utero Nanoparticle Delivery for Site-Specific Genome Editing. Nat. Commun. 2018, 9, 2481. [Google Scholar] [CrossRef]
- Pham, H.H.; Murphy, C.T.; Sureshkumar, G.; Ly, D.H.; Opresko, P.L.; Armitage, B.A. Cooperative Hybridization of γPNA Miniprobes to a Repeating Sequence Motif and Application to Telomere Analysis. Org. Biomol. Chem. 2014, 12, 7345–7354. [Google Scholar] [CrossRef]
- Orenstein, A.; Berlyoung, A.S.; Rastede, E.E.; Pham, H.H.; Fouquerel, E.; Murphy, C.T.; Leibowitz, B.J.; Yu, J.; Srivastava, T.; Armitage, B.A.; et al. γPNA FRET Pair Miniprobes for Quantitative Fluorescent In Situ Hybridization to Telomeric DNA in Cells and Tissue. Molecules 2017, 22, 2117. [Google Scholar] [CrossRef]
- Armitage, B.; Ly, D.; Koch, T.; Frydenlund, H.; Ørum, H.; Schuster, G.B. Hairpin-Forming Peptide Nucleic Acid Oligomers. Biochemistry 1998, 37, 9417–9425. [Google Scholar] [CrossRef]
- Zhang, D.Y.; Winfree, E. Control of DNA Strand Displacement Kinetics Using Toehold Exchange. J. Am. Chem. Soc. 2009, 131, 17303–17314. [Google Scholar] [CrossRef]
- Sacui, I.; Hsieh, W.-C.; Manna, A.; Sahu, B.; Ly, D.H. Gamma Peptide Nucleic Acids: As Orthogonal Nucleic Acid Recognition Codes for Organizing Molecular Self-Assembly. J. Am. Chem. Soc. 2015, 137, 8603–8610. [Google Scholar] [CrossRef]
- Tan, X.; Bruchez, M.P.; Armitage, B.A. Closing the Loop: Constraining TAT Peptide by gammaPNA Hairpin for Enhanced Cellular Delivery of Biomolecules. Bioconjug. Chem. 2018, 29, 2892–2898. [Google Scholar] [CrossRef]
- Lusvarghi, S.; Murphy, C.T.; Roy, S.; Tanious, F.A.; Sacui, I.; Wilson, W.D.; Ly, D.H.; Armitage, B.A. Loop and Backbone Modifications of PNA Improve G Quadruplex Binding Selectivity. J. Am. Chem. Soc. 2009, 131, 18415–18424. [Google Scholar] [CrossRef] [PubMed]
- SantaLucia, J.J.; Hicks, D. The Thermodynamics of DNA Structural Motifs. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 415–440. [Google Scholar] [CrossRef] [PubMed]
- Datta, B.; Armitage, B.A. Hybridization of PNA to Structured DNA Targets: Quadruplex Invasion and the Overhang Effect. J. Am. Chem. Soc. 2001, 123, 9612–9619. [Google Scholar] [CrossRef]
- Canady, T.D.; Telmer, C.A.; Oyaghire, S.N.; Armitage, B.A.; Bruchez, M.P. In Vitro Reversible Translation Control Using γPNA Probes. J. Am. Chem. Soc. 2015, 137, 10268–10275. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of γPNAs and plasmids will be made available upon request from the authors. |








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sequence Name | Sequence a,b | Mismatch |
|---|---|---|
| struc_γPNA | H2N-K-TCTGGGTTCG-EG8-cgaac-H | N/A |
| unstruc_γPNA | H2N-K-TCTGGGTTCG-H | N/A |
| Perfect Match (PM) | 5′-AGACCCAAGC-3′ | N/A |
| Stem Mismatch (S-MM-7T) | 5′-AGACCCTAGC-3′ | (T-T) |
| Stem Mismatch (S-MM-6A) | 5′-AGACCAAAGC-3′ | (A-G) |
| Stem Mismatch (S-MM-7G) | 5′-AGACCCGAGC-3′ | (G-T) |
| Overhang Mismatch (O-MM-4A) | 5′-AGAACCAAGC-3′ | (A-G) |
| Overhang Mismatch (O-MM-3T) | 5′-AGTCCCAAGC-3′ | (T-T) |
| Overhang Mismatch (O-MM-4T) | 5′-AGATCCAAGC-3′ | (T-G) |
| Target | unstruc_γPNA | struc_γPNA |
|---|---|---|
| PM | 77.6 ± 0.1 | 80.0 ± 1.4 |
| O-MM-4A | 54.3 ± 1.3 | 58.7 ± 0.1 |
| O-MM-4T | 61.9 ± 0.1 | 66.0 ± 1.4 |
| DNA Sequence | Sequence a |
|---|---|
| Perfect Match (PM) | 5′-Bt-TTTTTAGACCCAAGC-3′ |
| Stem Mismatch (S-MM-7T) | 5′-Bt-TTTTTAGACCCTAGC-3′ |
| Stem Mismatch (S-MM-6A) | 5′-Bt-TTTTTAGACCAAAGC-3′ |
| Stem Mismatch (S-MM-7G) | 5′-Bt-TTTTTAGACCCGAGC-3′ |
| Overhang Mismatch (O-MM-4A) | 5′-Bt-TTTTTAGAACCAAGC-3′ |
| Overhang Mismatch (O-MM-3T) | 5′-Bt-TTTTTAGTCCCAAGC-3′ |
| Overhang Mismatch (O-MM-4T) | 5′-Bt-TTTTTAGATCCAAGC-3′ |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Canady, T.D.; Berlyoung, A.S.; Martinez, J.A.; Emanuelson, C.; Telmer, C.A.; Bruchez, M.P.; Armitage, B.A. Enhanced Hybridization Selectivity Using Structured GammaPNA Probes. Molecules 2020, 25, 970. https://doi.org/10.3390/molecules25040970
Canady TD, Berlyoung AS, Martinez JA, Emanuelson C, Telmer CA, Bruchez MP, Armitage BA. Enhanced Hybridization Selectivity Using Structured GammaPNA Probes. Molecules. 2020; 25(4):970. https://doi.org/10.3390/molecules25040970
Chicago/Turabian StyleCanady, Taylor D., April S. Berlyoung, Joe A. Martinez, Cole Emanuelson, Cheryl A. Telmer, Marcel P. Bruchez, and Bruce A. Armitage. 2020. "Enhanced Hybridization Selectivity Using Structured GammaPNA Probes" Molecules 25, no. 4: 970. https://doi.org/10.3390/molecules25040970
APA StyleCanady, T. D., Berlyoung, A. S., Martinez, J. A., Emanuelson, C., Telmer, C. A., Bruchez, M. P., & Armitage, B. A. (2020). Enhanced Hybridization Selectivity Using Structured GammaPNA Probes. Molecules, 25(4), 970. https://doi.org/10.3390/molecules25040970