Probing Reversible Guest Binding with Hyperpolarized 129Xe-NMR: Characteristics and Applications for Cucurbit[n]urils


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
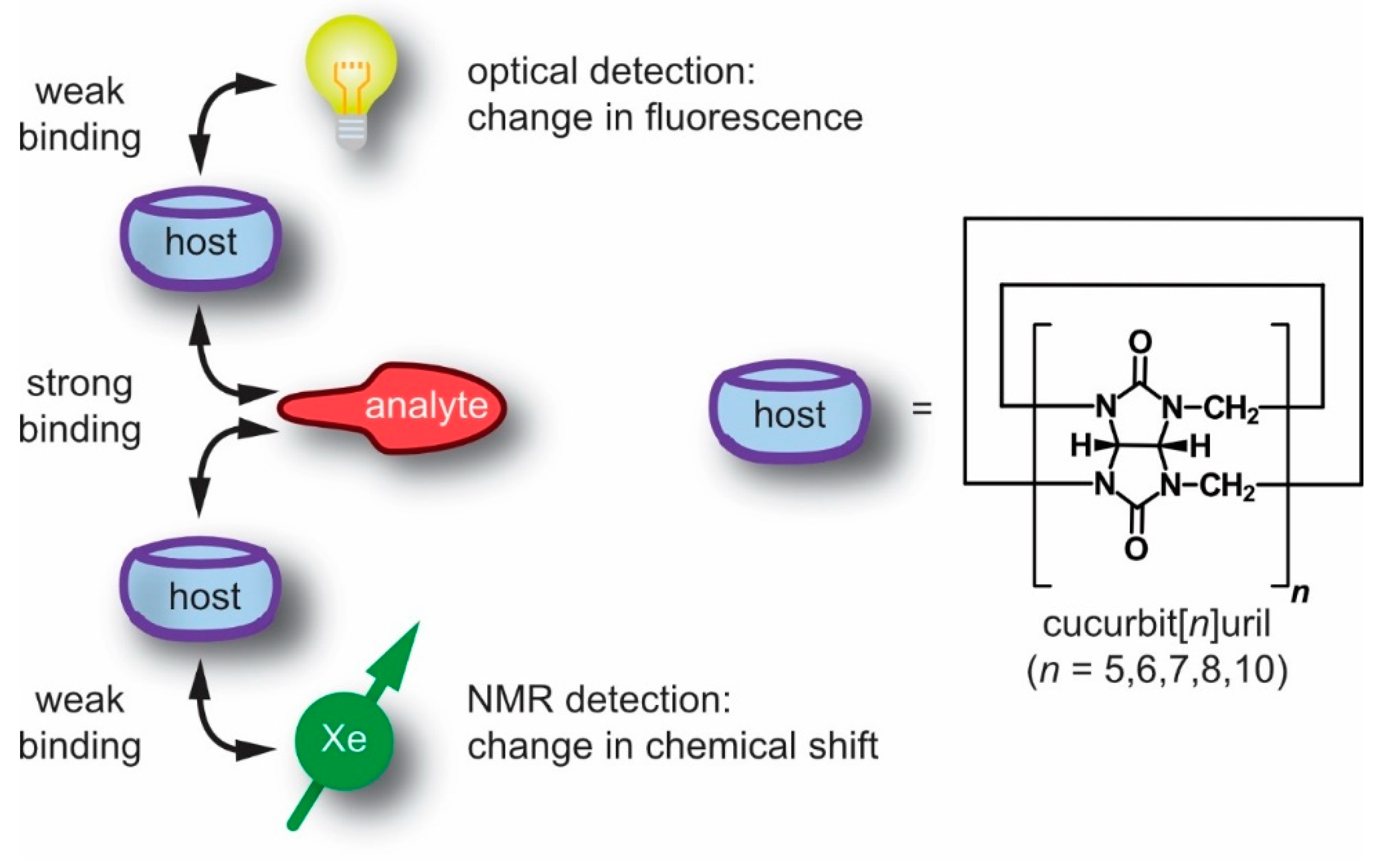
1. Introduction
- Xe participates in weak, non-covalent interactions, thus it does not significantly disturb the interaction of the supramolecular constituents;
- the NMR chemical shift range of 129Xe is rather large and provides a sensitive measure for changes in the immediate molecular environment even without engaging in more stable (or covalent) interactions;
- the noble gas can be “hyperpolarized” (hp), i.e., its spin magnetization can be artificially enhanced for significantly improved NMR sensitivity;
- in combinations with its sufficient solubility in many solvents, including water, this enables straightforward NMR detection of this monoatomic reporter for biochemical targets and many other solution samples of interest.
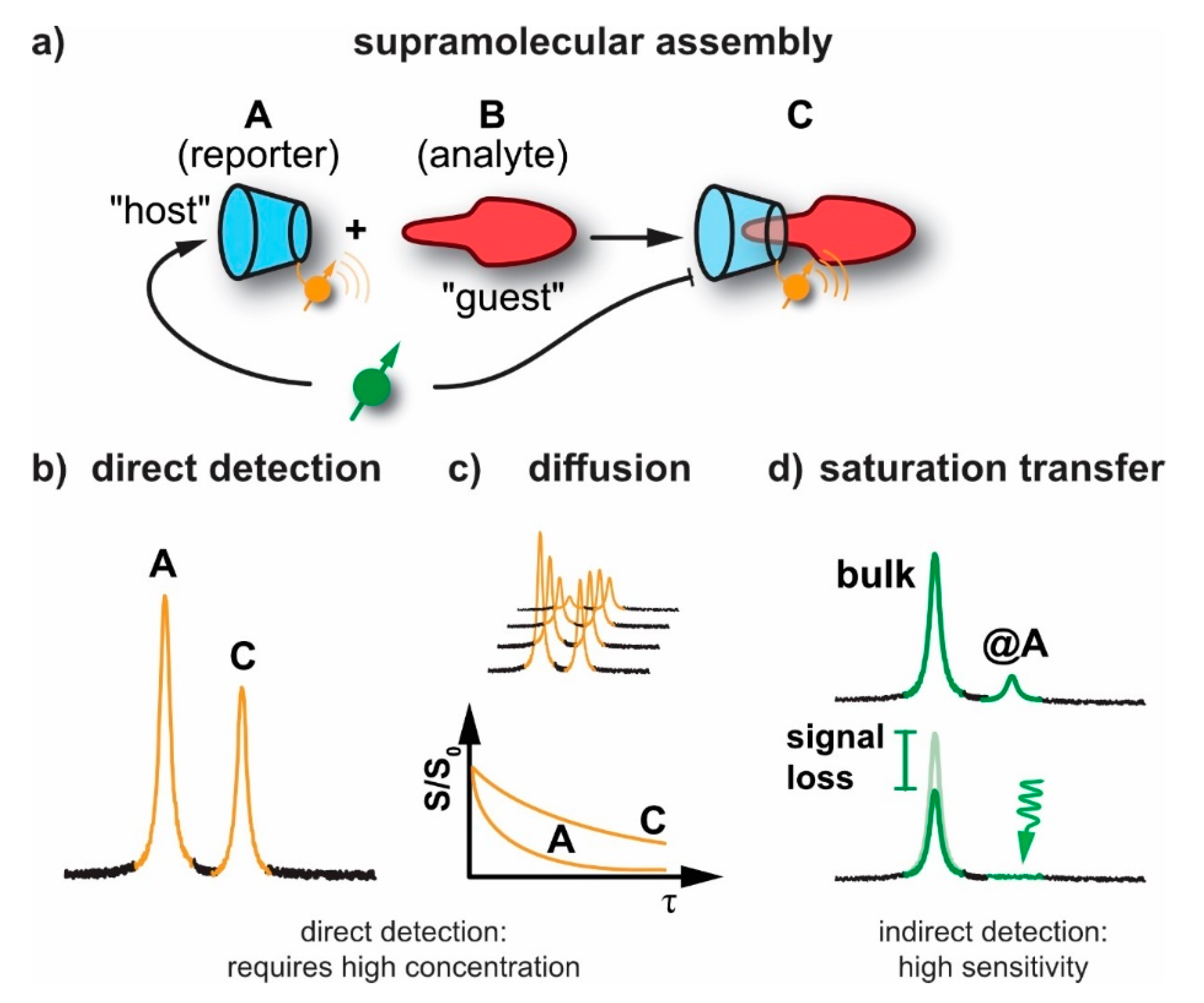
2. NMR Techniques for Studying Supramolecular Assemblies
2.1. General Sensitivity Considerations
2.2. Diffusion NMR
2.3. Detection of Exchange-Connected Pools
3. Cucurbit[n]urils and Their Detection with Xe-NMR Spectroscopy
3.1. Synthesis and Properties of Cucurbit[n]urils
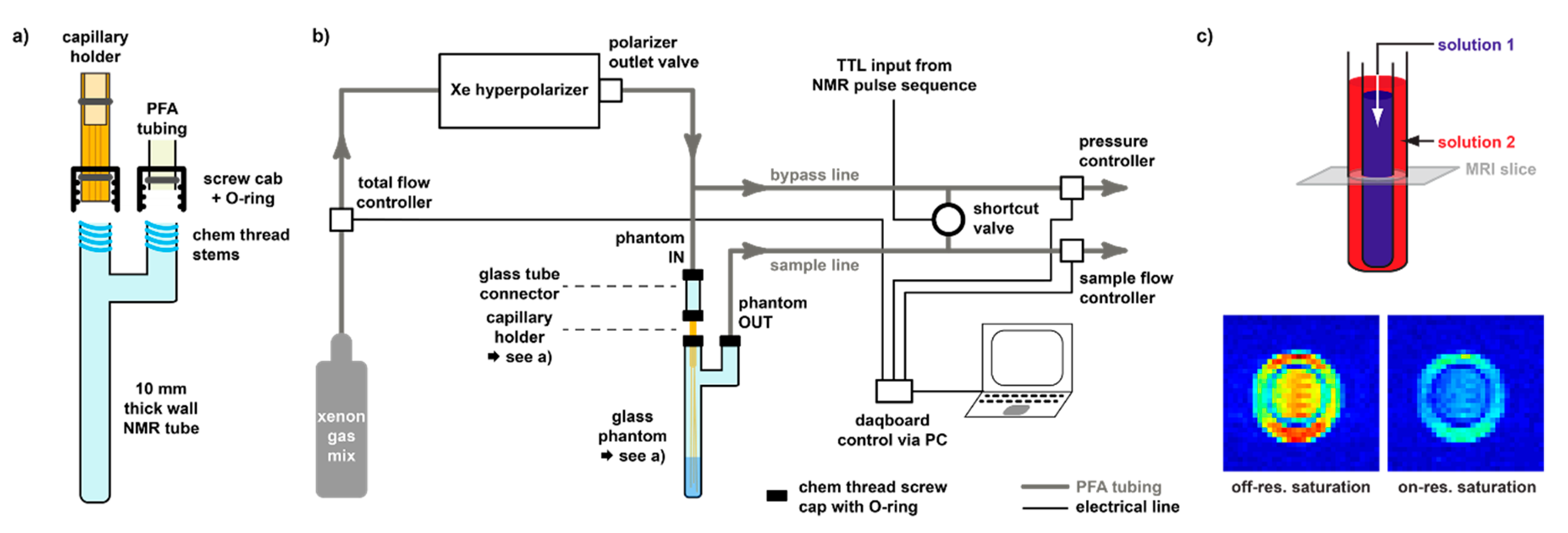
3.2. Hyperpolarized Xe for Sensitivity-Enhanced NMR
3.3. Delivery and Acquisition Considerations for hp Xe
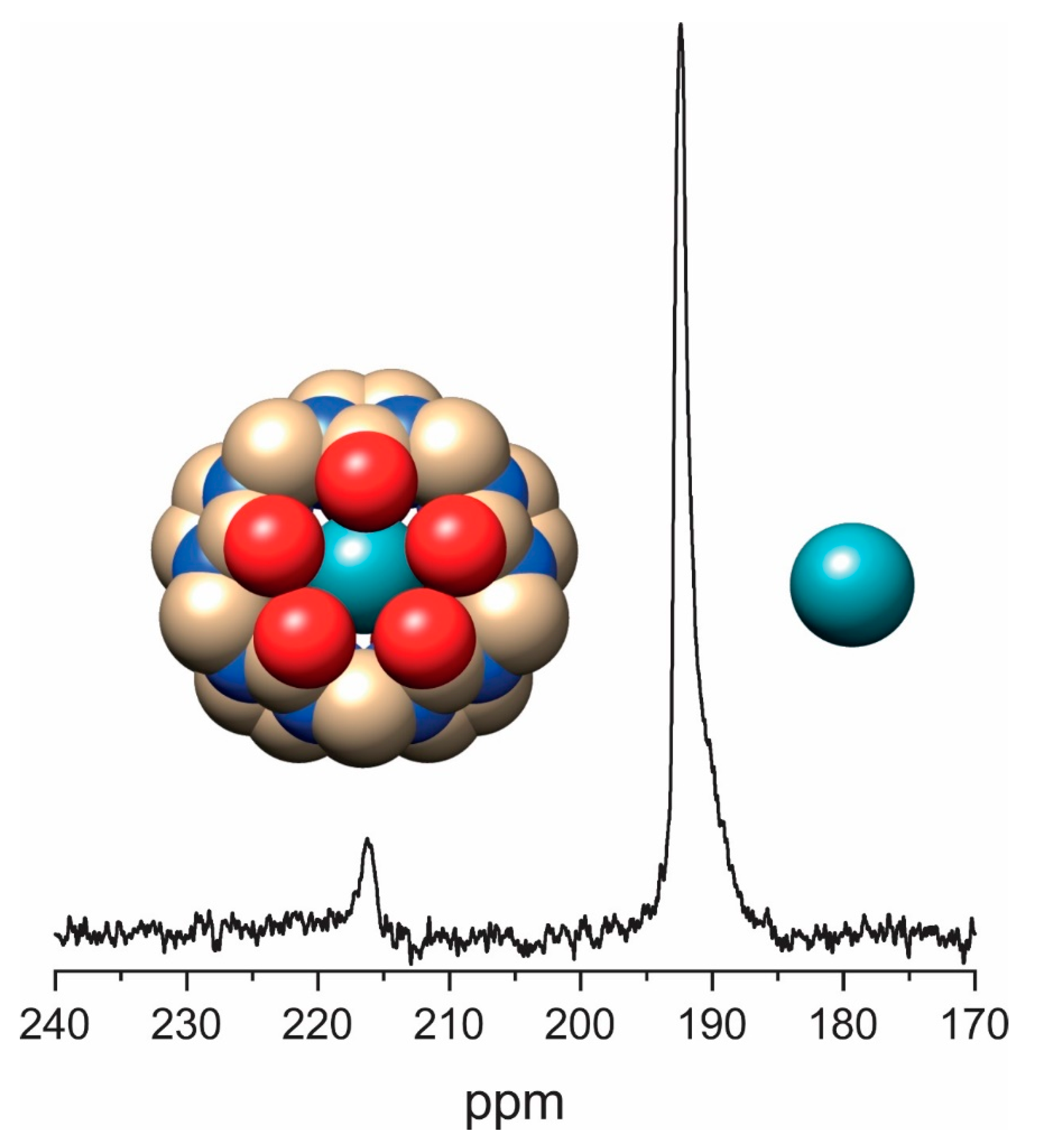
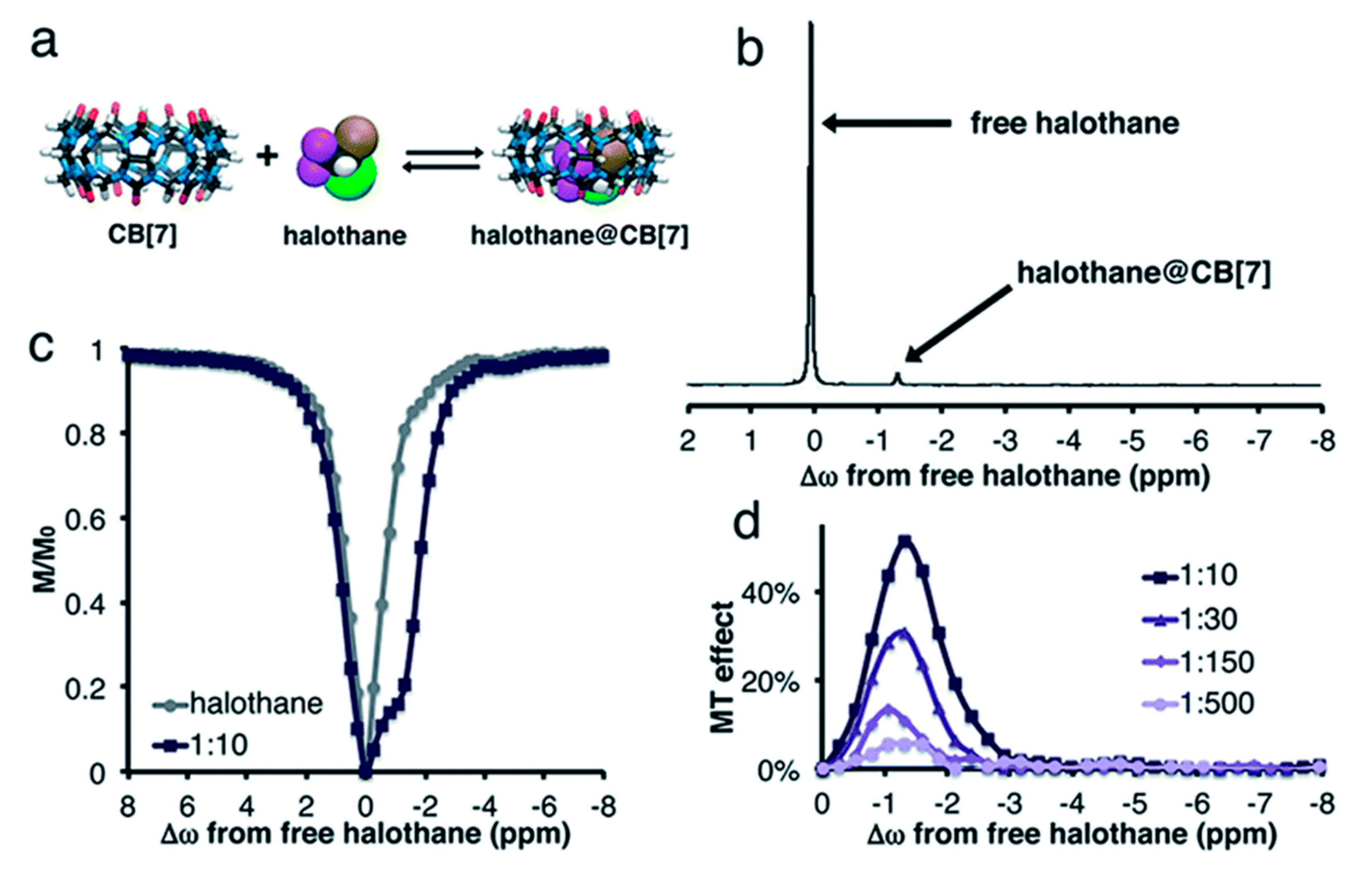
3.4. Direct Detection of Xe•CB[n] Inclusion Complexes
4. Saturation Transfer-Based Detection (CEST + GEST)
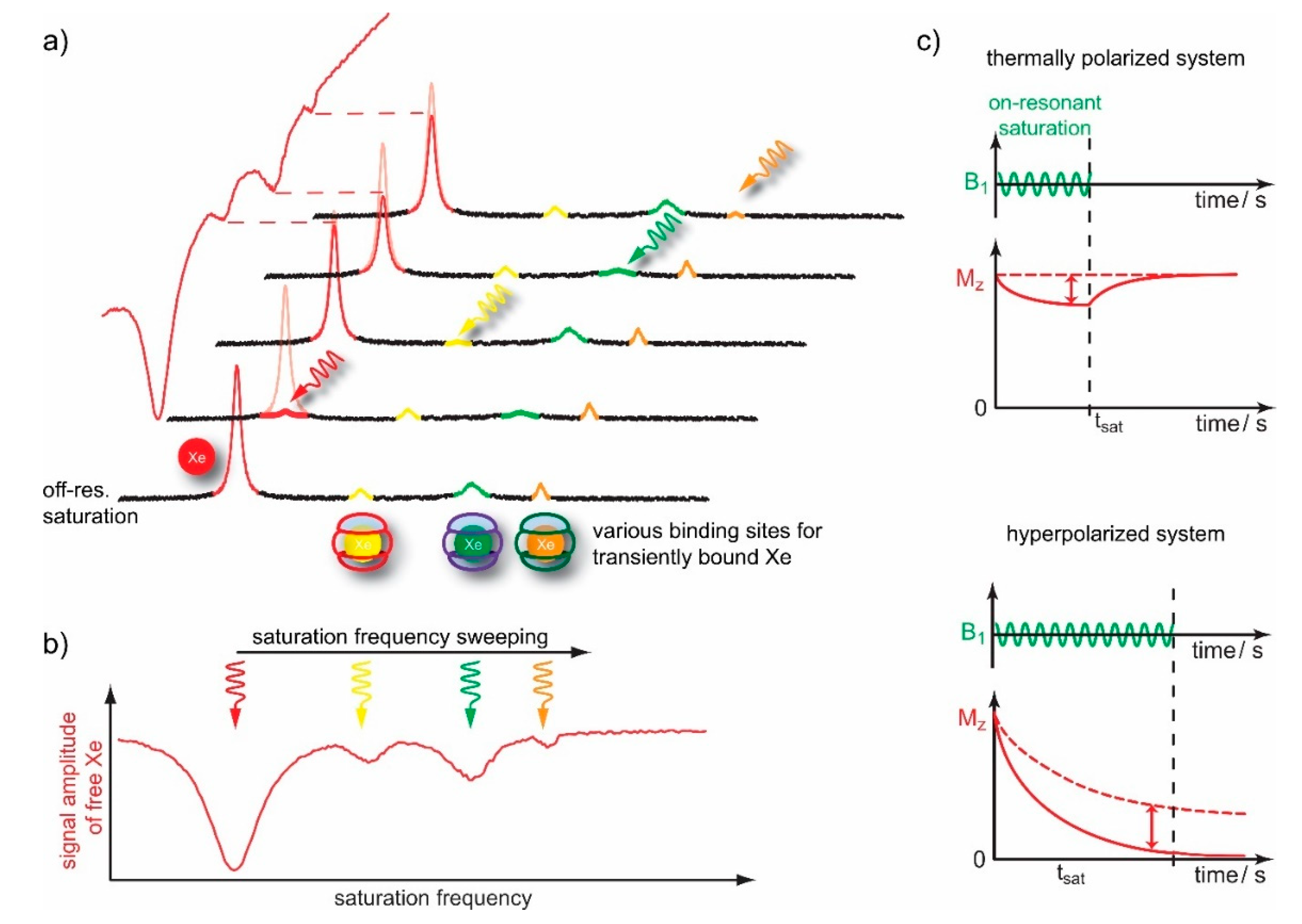
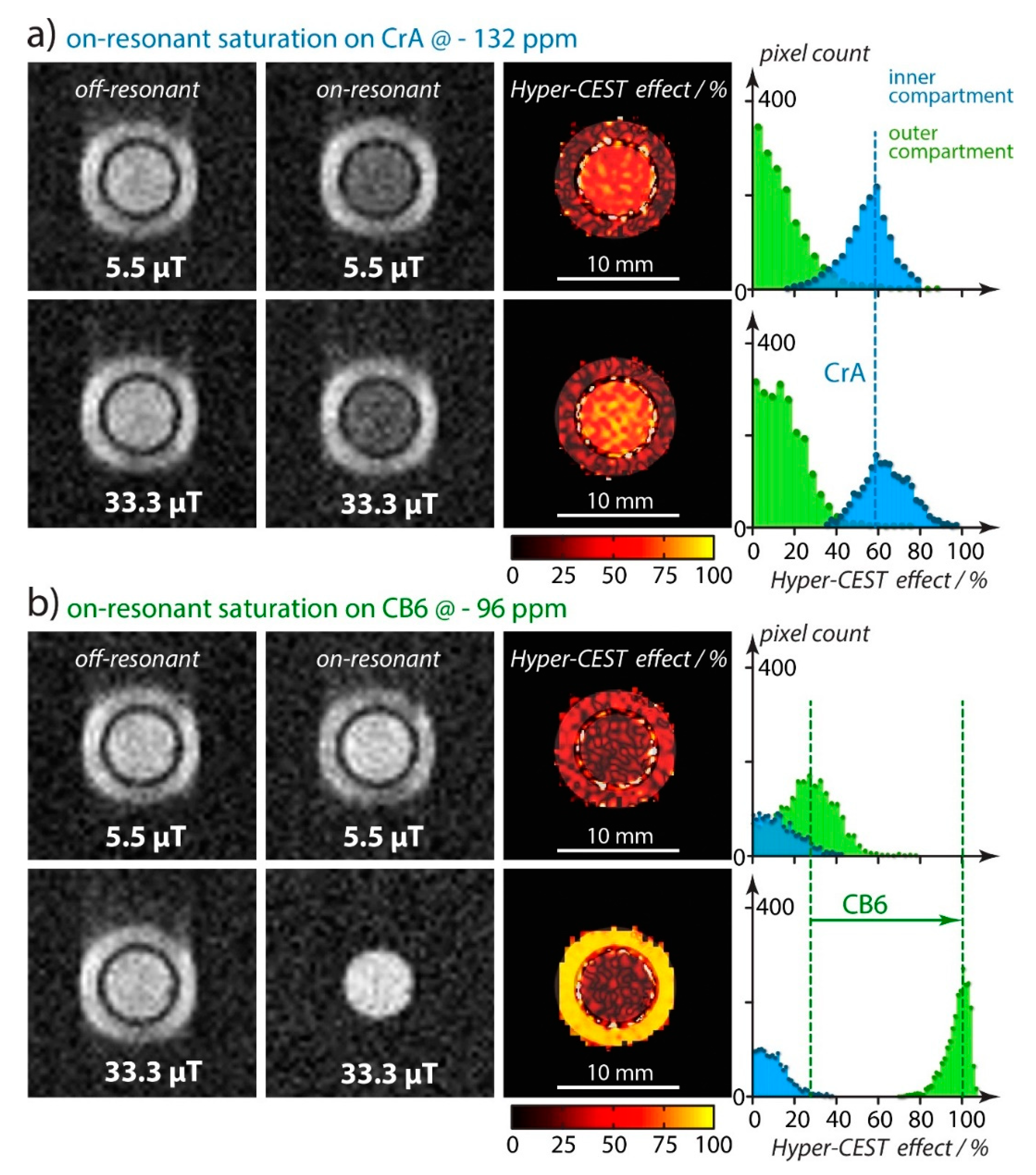
4.1. HyperCEST Characterics of CB[n]s
4.2. Optimizing CEST Detection
- B1 = kBA/γ yields 50 % of maximum depolarization rate possible, namely on-res = (fB kBA)/2;
- B1 ≤ kBA/γ yields on-res ≅ (fB kBA)(γ B1)2 which is parabolic in B1 for this low power regime.
- relative strong saturation with B1 ≥ kBA/γ shows a linear dependence Γ ≅ 2γB1;
- weaker saturation with B1 ≤ kBA/γ yields Γ ≅ 2kBA, which is the minimum possible width that is governed by the Xe exchange rate.
4.3. GEST NMR with 19F-bearing Guests
4.4. Quantitative Saturation Transfer Analysis (qHyperCEST)
5. Cucurbit[n]uril-Based Supramolecular Systems
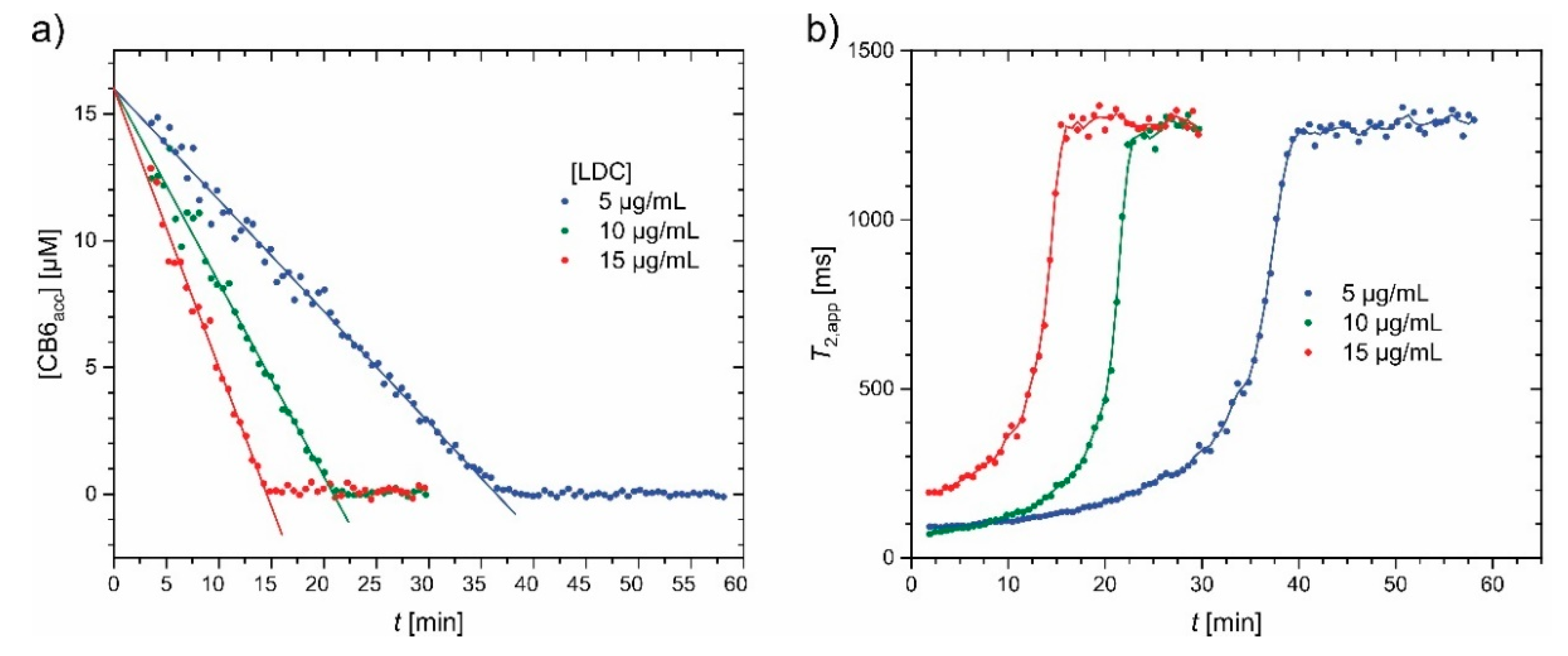
5.1. Displacement Assays
5.2. Molecular Relays with Two-Faced Guests
5.3. Molecular Rotaxanes
5.4. Supramolecular Assemblies Between two Hosts
5.5. Functionalized Supramolecular Assemblies
6. Conclusions and Future Directions
Funding
Conflicts of Interest
References
- Lehn, J.-M. Perspectives in Supramolecular Chemistry—From Molecular Recognition towards Molecular Information Processing and Self-Organization. Angew. Chem. Int. Ed. Engl. 1990, 29, 1304–1319. [Google Scholar] [CrossRef]
- Palmer, L.; Stupp, S.I. Molecular Self-Assembly into One-Dimensional Nanostructures. Accounts Chem. Res. 2008, 41, 1674–1684. [Google Scholar] [CrossRef] [PubMed]
- Ruben, M.; Ziener, U.; Lehn, J.-M.; Ksenofontov, V.; Gütlich, P.; Vaughan, G.B.M. Hierarchical Self-Assembly of Supramolecular Spintronic Modules into 1D- and 2D-Architectures with Emergence of Magnetic Properties. Chemistry 2005, 11, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Vriezema, D.M.; Comellas Aragonès, M.; Elemans, J.A.A.W.; Cornelissen, J.J.L.M.; Rowan, A.E.; Nolte, R.J.M. Self-assembled nanoreactors. Chem. Rev. 2005, 105, 1445–1489. [Google Scholar] [CrossRef]
- Kataoka, K.; Harada, A.; Nagasaki, Y. Block copolymer micelles for drug delivery: Design, characterization and biological significance. Adv. Drug Deliv. Rev. 2001, 47, 113–131. [Google Scholar] [CrossRef]
- Lorenzo, M.O.; Baddeley, C.; Muryn, C.; Raval, R. Extended surface chirality from supramolecular assemblies of adsorbed chiral molecules. Nature 2000, 404, 376–379. [Google Scholar] [CrossRef]
- Benloucif, M.R.; Brewster, M.E. Cyclodextrin-based pharmaceutics: Past, present and future. Nat. Rev. Drug Discov. 2004, 3, 1023–1035. [Google Scholar]
- Kang, J.; Kumar, V.; Yang, D.; Chowdhury, P.R.; Hohl, R.J. Cyclodextrin complexation: Influence on the solubility, stability, and cytotoxicity of camptothecin, an antineoplastic agent. Eur. J. Pharm. Sci. 2002, 15, 163–170. [Google Scholar] [CrossRef]
- Pollard, T.D.; Cooper, J.A. Actin, a Central Player in Cell Shape and Movement. Science 2009, 326, 1208–1212. [Google Scholar] [CrossRef]
- Lemon, B.; Tjian, R. Orchestrated response: A symphony of transcription factors for gene control. Genome Res. 2000, 14, 2551–2569. [Google Scholar] [CrossRef]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Mechanical aspects of apoptosome assembly. Curr. Opin. Cell Boil. 2006, 18, 677–684. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Xu, B. Enzyme-Instructed Self-Assembly: A Multistep Process for Potential Cancer Therapy. Bioconjugate Chem. 2015, 26, 987–999. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Feng, Z.; Wu, D.; Fritzsching, K.; Rigney, M.; Zhou, J.; Jiang, Y.; Schmidt-Rohr, K.; Xu, B. Enzyme-Regulated Supramolecular Assemblies of Cholesterol Conjugates against Drug-Resistant Ovarian Cancer Cells. J. Am. Chem. Soc. 2016, 138, 10758–10761. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Fukuoka, Y.; Morimoto, Y.; Honjo, T.; Koda, D.; Goto, M.; Maruyama, T. Cancer Cell Death Induced by the Intracellular Self-Assembly of an Enzyme-Responsive Supramolecular Gelator. J. Am. Chem. Soc. 2015, 137, 770–775. [Google Scholar] [CrossRef]
- Pires, R.; Abul-Haija, Y.M.; Da Costa, D.S.; Novoa-Carballal, R.; Reis, R.; Ulijn, R.V.; Pashkuleva, I. Controlling Cancer Cell Fate Using Localized Biocatalytic Self-Assembly of an Aromatic Carbohydrate Amphiphile. J. Am. Chem. Soc. 2015, 137, 576–579. [Google Scholar] [CrossRef]
- Du, X.; Zhou, J.; Wang, H.; Shi, J.; Kuang, Y.; Zeng, W.; Yang, Z.; Xu, B. In situ generated D-peptidic nanofibrils as multifaceted apoptotic inducers to target cancer cells. Cell Death Dis. 2017, 8, e2614. [Google Scholar] [CrossRef]
- Wang, H.; Feng, Z.; Wang, Y.; Zhou, R.; Yang, Z.; Xu, B. Integrating Enzymatic Self-Assembly and Mitochondria Targeting for Selectively Killing Cancer Cells without Acquired Drug Resistance. J. Am. Chem. Soc. 2016, 138, 16046–16055. [Google Scholar] [CrossRef]
- Gao, Y.; Shi, J.; Yuan, D.; Xu, B. Imaging enzyme-triggered self-assembly of small molecules inside live cells. Nat. Commun. 2012, 3, 1033. [Google Scholar] [CrossRef]
- Yang, Z.; Liang, G.; Xu, B. Enzymatic Hydrogelation of Small Molecules. Accounts Chem. Res. 2008, 41, 315–326. [Google Scholar] [CrossRef]
- Boekhoven, J.; Hendriksen, W.E.; Koper, G.; Eelkema, R.; Van Esch, J.H. Transient assembly of active materials fueled by a chemical reaction. Science 2015, 349, 1075–1079. [Google Scholar] [CrossRef] [PubMed]
- Wiskur, S.; Ait-Haddou, H.; Lavigne, J.; Anslyn, E.V. Teaching Old Indicators New Tricks. Accounts Chem. Res. 2001, 34, 963–972. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, B.T.; Anslyn, E.V. Indicator–displacement assays. Co-ord. Chem. Rev. 2006, 250, 3118–3127. [Google Scholar] [CrossRef]
- Zhang, T.; Anslyn, E.V. Using an Indicator Displacement Assay to Monitor Glucose Oxidase Activity in Blood Serum. Org. Lett. 2007, 9, 1627–1629. [Google Scholar] [CrossRef]
- Leung, D.; Folmer-Andersen, J.F.; Lynch, V.M.; Anslyn, E.V. Using Enantioselective Indicator Displacement Assays To Determine the Enantiomeric Excess of α-Amino Acids. J. Am. Chem. Soc. 2008, 130, 12318–12327. [Google Scholar] [CrossRef]
- Guo, D.-S.; Liu, Y. Supramolecular Chemistry of p-Sulfonatocalix[n]arenes and Its Biological Applications. Accounts Chem. Res. 2014, 47, 1925–1934. [Google Scholar] [CrossRef]
- D’Souza, R.; Hennig, A.; Nau, W.M. Supramolecular Tandem Enzyme Assays. Chemistry 2012, 18, 3444–3459. [Google Scholar] [CrossRef]
- Hennig, A.; Bakirci, H.; Nau, W.M. Label-free continuous enzyme assays with macrocycle-fluorescent dye complexes. Nat. Methods 2007, 4, 629–632. [Google Scholar] [CrossRef]
- Bailey, D.M.; Hennig, A.; Uzunova, V.D.; Nau, W.M. Supramolecular Tandem Enzyme Assays for Multiparameter Sensor Arrays and Enantiomeric Excess Determination of Amino Acids. Chemistry 2008, 14, 6069–6077. [Google Scholar] [CrossRef]
- Nau, W.M.; Ghale, G.; Hennig, A.; Bakirci, H.; Bailey, D.M. Substrate-Selective Supramolecular Tandem Assays: Monitoring Enzyme Inhibition of Arginase and Diamine Oxidase by Fluorescent Dye Displacement from Calixarene and Cucurbituril Macrocycles. J. Am. Chem. Soc. 2009, 131, 11558–11570. [Google Scholar] [CrossRef]
- Ghale, G.; Ramalingam, V.; Urbach, A.R.; Nau, W.M. Determining Protease Substrate Selectivity and Inhibition by Label-Free Supramolecular Tandem Enzyme Assays. J. Am. Chem. Soc. 2011, 133, 7528–7535. [Google Scholar] [CrossRef] [PubMed]
- Florea, M.; Nau, W.M. Implementation of anion-receptor macrocycles in supramolecular tandem assays for enzymes involving nucleotides as substrates, products, and cofactors. Org. Biomol. Chem. 2010, 8, 1033–1039. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.-S.; Uzunova, V.D.; Su, X.; Liu, Y.; Nau, W.M. Operational calixarene-based fluorescent sensing systems for choline and acetylcholine and their application to enzymatic reactions. Chem. Sci. 2011, 2, 1722. [Google Scholar] [CrossRef]
- Goobes, G. Past and Future Solid-State NMR Spectroscopy Studies at the Convergence Point between Biology and Materials Research. Isr. J. Chem. 2014, 54, 113–124. [Google Scholar] [CrossRef]
- Comellas, G.; Rienstra, C.M. Protein Structure Determination by Magic-Angle Spinning Solid-State NMR, and Insights into the Formation, Structure, and Stability of Amyloid Fibrils. Annu. Rev. Biophys. 2013, 42, 515–536. [Google Scholar] [CrossRef]
- Naito, A.; Kawamura, I.; Javkhlantugs, N. Recent Solid-State NMR Studies of Membrane-Bound Peptides and Proteins. Annu. Rep. NMR Spectrosc. 2015, 86, 333–411. [Google Scholar]
- Tycko, R. Solid-state NMR studies of amyloid fibril structure. Annu. Rev. Phys. Chem. 2011, 62, 279–299. [Google Scholar] [CrossRef]
- Jaroniec, C.P. Solid-state nuclear magnetic resonance structural studies of proteins using paramagnetic probes. Solid State Nucl. Magn. Reson. 2012, 43, 1–13. [Google Scholar] [CrossRef]
- Knight, M.J.; Felli, I.C.; Pierattelli, R.; Emsley, L.; Pintacuda, G. Magic Angle Spinning NMR of Paramagnetic Proteins. Accounts Chem. Res. 2013, 46, 2108–2116. [Google Scholar] [CrossRef]
- Knight, M.J.; Pell, A.J.; Bertini, I.; Felli, I.C.; Gonnelli, L.; Pierattelli, R.; Herrmann, T.; Emsley, L.; Pintacuda, G. Structure and backbone dynamics of a microcrystalline metalloprotein by solid-state NMR. Proc. Natl. Acad. Sci. 2012, 109, 11095–11100. [Google Scholar] [CrossRef]
- Nose, T. Pulsed-Field-Gradient NMR Studies of the Diffusion of Chain Molecules in Polymer Matrices. Annu. Rep. NMR Spectrosc. 1993, 27, 217–253. [Google Scholar]
- Kärger, H.P.J. NMR and Catalysis; Pines, A., Bell, A., Eds.; Dekker: New York, NY, USA, 1994. [Google Scholar]
- Callaghan, A.C. NMR Probes and Molecular Dynamics; Tycko, R., Ed.; Kluwer: Dordrecht, The Netherland, 1993; pp. 490–523. [Google Scholar]
- Kärger, J.D.M.R. Diffusion in Zeolites and other Microporous Solids; J. Wiley & Sons INC: New York, NY, USA, 1993. [Google Scholar]
- Söderman, O.; Stilbs, P. NMR studies of complex surfactant systems. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 445–482. [Google Scholar] [CrossRef]
- Lindblom, G.; Orädd, G. NMR Studies of translational diffusion in lyotropic liquid crystals and lipid membranes. Prog. Nucl. Magn. Reson. Spectrosc. 1994, 26, 483–515. [Google Scholar] [CrossRef]
- Jones, J.A.; Wilkins, D.K.; Smith, L.J.; Dobson, C.M. Characterisation of protein unfolding by NMR diffusion measurements. J. Biomol. NMR 1997, 10, 199–203. [Google Scholar]
- Zhang, W.; Smithgall, T.E.; Gmeiner, W.H. Self-Association and Backbone Dynamics of the Hck SH2 Domain in the Free and Phosphopeptide-Complexed Forms†. Biochemistry 1998, 37, 7119–7126. [Google Scholar] [CrossRef]
- Chang, X.; Keller, D.; O’Donoghue, S.; Led, J.J. NMR studies of the aggregation of glucagon-like peptide-1: Formation of a symmetric helical dimer. FEBS Lett. 2002, 515, 165–170. [Google Scholar] [CrossRef]
- Callaghan, P.T.; MacGowan, D.; Packer, K.J.; Zelaya, F.O. High-resolution q-space imaging in porous structures. J. Magn. Reson. 1990, 90, 177–182. [Google Scholar] [CrossRef]
- Callaghan, P.; MacGowan, D.; Packer, K.; Zelaya, F. Influence of field gradient strength in NMR studies of diffusion in porous media. Magn. Reson. Imaging 1991, 9, 663–671. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part 1. Basic theory. Concepts in Magnetic Resonance. Concepts Magn. Reson. 1997, 9, 299–336. [Google Scholar] [CrossRef]
- Price, W.S. Pulsed-field gradient nuclear magnetic resonance as a tool for studying translational diffusion: Part II. Experimental aspects. Concepts Magn. Reson. 1998, 10, 197–237. [Google Scholar] [CrossRef]
- Forsén, S.; Hoffman, R.A. Study of Moderately Rapid Chemical Exchange Reactions by Means of Nuclear Magnetic Double Resonance. J. Chem. Phys. 1963, 39, 2892–2901. [Google Scholar] [CrossRef]
- Yoo, B.; Raam, M.S.; Rosenblum, R.M.; Pagel, M.D. Enzyme-responsive PARACEST MRI contrast agents: A new biomedical imaging approach for studies of the proteasome. Contrast Media Mol. Imaging 2007, 2, 189–198. [Google Scholar] [CrossRef] [PubMed]
- Lock, L.L.; Li, Y.; Mao, X.; Chen, H.; Staedtke, V.; Bai, R.; Ma, W.; Lin, R.; Li, Y.; Liu, G.; et al. One-Component Supramolecular Filament Hydrogels as Theranostic Label-Free Magnetic Resonance Imaging Agents. ACS Nano 2017, 11, 797–805. [Google Scholar] [CrossRef] [PubMed]
- Bar-Shir, A.; Gilad, A.A.; Chan, K.W.; Liu, G.; Van Zijl, P.C.M.; Bulte, J.W.; McMahon, M.T. Metal Ion Sensing Using Ion Chemical Exchange Saturation Transfer 19F Magnetic Resonance Imaging. J. Am. Chem. Soc. 2013, 135, 12164–12167. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Iron, M.; Bar-Shir, A. Amplifying undetectable NMR signals to study host–guest interactions and exchange. Chem. Sci. 2016, 7, 6905–6909. [Google Scholar] [CrossRef] [PubMed]
- Avram, L.; Wishard, A.D.; Gibb, B.C.; Bar-Shir, A. Quantifying Guest Exchange in Supramolecular Systems. Angew. Chem. Int. Ed. Engl. 2017, 56, 15314–15318. [Google Scholar] [CrossRef]
- Sekhar, A.; Kay, L.E. NMR paves the way for atomic level descriptions of sparsely populated, transiently formed biomolecular conformers. Proc. Natl. Acad. Sci. 2013, 110, 12867–12874. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Bouvignies, G.; Kay, L.E. Studying “Invisible” Excited Protein States in Slow Exchange with a Major State Conformation. J. Am. Chem. Soc. 2012, 134, 8148–8161. [Google Scholar] [CrossRef]
- Vallurupalli, P.; Kay, L.E. Probing Slow Chemical Exchange at Carbonyl Sites in Proteins by Chemical Exchange Saturation Transfer NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 4156–4159. [Google Scholar] [CrossRef]
- Bouvignies, G.; Vallurupalli, P.; Kay, L.E. Visualizing Side Chains of Invisible Protein Conformers by Solution NMR. J. Mol. Boil. 2014, 426, 763–774. [Google Scholar] [CrossRef]
- Zeng, H.; Xu, J.; Yadav, N.N.; McMahon, M.T.; Harden, B.; Frueh, M.; Van Zijl, P.C.M. 15N Heteronuclear Chemical Exchange Saturation Transfer MRI. J. Am. Chem. Soc. 2016, 138, 11136–11139. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, D.V.; Randtke, E.A.; Pagel, M.D. A CatalyCEST MRI Contrast Agent That Detects the Enzyme-Catalyzed Creation of a Covalent Bond. J. Am. Chem. Soc. 2013, 135, 6396–6398. [Google Scholar] [CrossRef] [PubMed]
- Sinharay, S.; Fernandez-Cuervo, G.; Acfalle, J.P.; Pagel, M.D. Detection of Sulfatase Enzyme Activity with a CatalyCEST MRI Contrast Agent. Chemistry 2016, 22, 6491–6495. [Google Scholar] [CrossRef] [PubMed]
- Sinharay, S.; Randtke, E.A.; Jones, K.M.; Howison, C.M.; Chambers, S.K.; Kobayashi, H.; Pagel, M.D. Noninvasive detection of enzyme activity in tumor models of human ovarian cancer using catalyCEST MRI. Magn. Reson. Med. 2017, 77, 2005–2014. [Google Scholar] [CrossRef] [PubMed]
- Behrend, R.; Meyer, E.; Rusche, F.I. Ueber Condensationsproducte aus Glycoluril und Formaldehyd. Eur. J. Org. Chem. 1905, 339, 1–37. [Google Scholar] [CrossRef]
- Huang, W.-H.; Zavalij, P.; Isaacs, L.D. Cucurbit[n]uril Formation Proceeds by Step-Growth Cyclo-oligomerization. J. Am. Chem. Soc. 2008, 130, 8446–8454. [Google Scholar] [CrossRef] [PubMed]
- Isaacs, L.D.; Park, S.-K.; Liu, S.; Ko, Y.H.; Selvapalam, N.; Kim, Y.; Kim, H.; Zavalij, P.; Kim, G.-H.; Lee, H.-S.; et al. The Inverted Cucurbit[n]uril Family. J. Am. Chem. Soc. 2005, 127, 18000–18001. [Google Scholar] [CrossRef]
- Huang, W.-H.; Liu, S.; Zavalij, P.Y.; Isaacs, L. Nor-Seco-Cucurbit[10]uril Exhibits Homotropic Allosterism. J. Am. Chem. Soc. 2006, 128, 14744–14745. [Google Scholar] [CrossRef]
- Huang, W.-H.; Zavalij, P.Y.; Isaacs, L. Chiral Recognition inside a Chiral Cucurbituril. Angew. Chem. Int. Ed. Engl. 2007, 46, 7425–7427. [Google Scholar] [CrossRef]
- Biltz, H.; Gonder, L. Über das Niobsulfid. Ber. der Dtsch. Chem. Ges. 1907, 40, 4963–4972. [Google Scholar] [CrossRef]
- Petersen, H. Syntheses of Cyclic Ureas by α-Ureidoalkylation. Synthesis 1973, 1973, 243–292. [Google Scholar] [CrossRef]
- Grillon, E.; Gallo, R.; Pierrot, M.; Boileau, J.; Wimmer, E. Isolation and X-ray structure of the intermediate dihydroxyimidazolidine(DHI) in the synthesis of glycoluril from glyoxal and urea. Tetrahedron Lett. 1988, 29, 1015–1016. [Google Scholar] [CrossRef]
- Chakraborty, A.; Wu, A.; Witt, D.; Lagona, J.; Fettinger, J.C.; Isaacs, L.D. Diastereoselective Formation of Glycoluril Dimers: Isomerization Mechanism and Implications for Cucurbit[n]uril Synthesis. J. Am. Chem. Soc. 2002, 124, 8297–8306. [Google Scholar] [CrossRef]
- Wu, A.; Chakraborty, A.; Witt, D.; Lagona, J.; Damkaci, F.; Ofori, M.A.; Chiles, J.K.; Fettinger, J.C.; Isaacs, L.D. Methylene-Bridged Glycoluril Dimers: Synthetic Methods. J. Org. Chem. 2002, 67, 5817–5830. [Google Scholar] [CrossRef] [PubMed]
- Witt, D.; Lagona, J.; Damkaci, F.; Fettinger, J.C.; Isaacs, L.D. Diastereoselective Formation of Methylene-Bridged Glycoluril Dimers. Org. Lett. 2000, 2, 755–758. [Google Scholar] [CrossRef] [PubMed]
- Day, A.; Arnold, A.; Blanch, R.J.; Snushall, B. Controlling factors in the synthesis of cucurbituril and its homologues. J. Org. Chem. 2001, 66, 8094–8100. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Jung, I.-S.; Kim, S.-Y.; Lee, E.; Kang, J.-K.; Sakamoto, S.; Yamaguchi, K.; Kim, K. New Cucurbituril Homologues: Syntheses, Isolation, Characterization, and X-ray Crystal Structures of Cucurbit[n]uril (n= 5, 7, and 8). J. Am. Chem. Soc. 2000, 122, 540–541. [Google Scholar] [CrossRef]
- Maslii, A.N.; Grishaeva, T.N.; Kuznetsov, A.M.; Bakovets, V.V. Quantum chemical investigation of structural and thermodynamic peculiarities of the formation of cucurbit[n]urils. J. Struct. Chem. 2007, 48, 552–557. [Google Scholar] [CrossRef]
- Bakovets, V.V. A thermodynamic analysis of the mechanism of formation of homologs of the cucurbit[n]uril family. Russ. J. Phys. Chem. A 2007, 81, 1586–1590. [Google Scholar] [CrossRef]
- Oh, K.S.; Yoon, J.; Kim, K.S. Structural Stabilities and Self-Assembly of Cucurbit[n]uril (n = 4−7) and Decamethylcucurbit[n]uril (n = 4−6): A Theoretical Study. J. Phys. Chem. B 2001, 105, 9726–9731. [Google Scholar] [CrossRef]
- Cheng, X.-J.; Liang, L.-L.; Chen, K.; Ji, N.-N.; Xiao, X.; Zhang, J.-X.; Zhang, Y.-Q.; Xue, S.-F.; Zhu, Q.-J.; Ni, X.-L.; et al. Twisted Cucurbit[14]uril. Angew. Chem. Int. Ed. Engl. 2013, 52, 7252–7255. [Google Scholar] [CrossRef] [PubMed]
- Lewin, V.; Rivollier, J.; Coudert, S.; Buisson, D.-A.; Baumann, D.; Rousseau, B.; Legrand, F.-X.; Kouřilová, H.; Berthault, P.; Dognon, J.-P.; et al. Synthesis of Cucurbit[6]uril Derivatives and Insights into Their Solubility in Water. Eur. J. Org. Chem. 2013, 2013, 3857–3865. [Google Scholar] [CrossRef]
- Liu, S.; Kim, K.; Isaacs, L.D. Mechanism of the Conversion of Inverted CB[6] to CB[6]. J. Org. Chem. 2007, 72, 6840–6847. [Google Scholar] [CrossRef] [PubMed]
- Lagona, J.; Mukhopadhyay, P.; Chakrabarti, S.; Isaacs, L. The cucurbit[n]uril family. Angew. Chem. Int. Ed. Engl. 2005, 44, 4844–4870. [Google Scholar] [CrossRef] [PubMed]
- Masson, E.; Ling, X.; Joseph, R.; Kyeremeh-Mensah, L.; Lu, X. Cucurbituril chemistry: A tale of supramolecular success. RSC Adv. 2012, 2, 1213–1247. [Google Scholar] [CrossRef]
- Zhao, J.; Kim, H.-J.; Oh, J.; Kim, S.-Y.; Lee, J.W.; Sakamoto, S.; Yamaguchi, K.; Kim, K. Cucurbit[n]uril Derivatives Soluble in Water and Organic Solvents. Angew. Chem. Int. Ed. Engl. 2001, 40, 4233–4235. [Google Scholar] [CrossRef]
- Sasmal, S.; Sinha, M.K.; Keinan, E. Facile Purification of Rare Cucurbiturils by Affinity Chromatography. Org. Lett. 2004, 6, 1225–1228. [Google Scholar] [CrossRef]
- Lu, L.-B.; Zhang, Y.-Q.; Zhu, Q.-J.; Xue, S.-F.; Tao, Z. Synthesis and X-ray Structure of the Inclusion Complex of Dodecamethylcucurbit[6]uril with 1,4-Dihydroxybenzene. Molecules 2007, 12, 716–722. [Google Scholar] [CrossRef]
- Wu, F.; Wu, L.-H.; Xiao, X.; Zhang, Y.-Q.; Xue, S.-F.; Tao, Z.; Day, A.I. Locating the Cyclopentano Cousins of the Cucurbit[n]uril Family. J. Org. Chem. 2012, 77, 606–611. [Google Scholar] [CrossRef]
- Isobe, H.; Sato, S.; Nakamura, E. Synthesis of Disubstituted Cucurbit[6]uril and Its Rotaxane Derivative. Org. Lett. 2002, 4, 1287–1289. [Google Scholar] [CrossRef]
- Day, A.; Arnold, A.; Blanch, R.J. A Method for Synthesizing Partially Substituted Cucurbit[n]uril. Molecules 2003, 8, 74–84. [Google Scholar] [CrossRef]
- Zhao, Y. Synthesis of a symmetrical tetrasubstituted cucurbit[6]uril and its host-guest inclusion complex with 2,2′-bipyridine. Chin. Sci. Bull. 2004, 49, 1111. [Google Scholar] [CrossRef]
- Li-Mei, Z.; Qian-Jiang, Z.H.U.; Jian-Nan, Z.H.U.; Yun-Qian, Z.; Zhu, T.A.O.; Sai-Feng, X.U.E.; Zhan-Bing, W.E.I.; La-Sheng, L. Synthesis and Crystal Structure of a Novel Self-assembled (1,4-discyclohexyl cucurbituril) Sodium(Ⅰ) Complex. Chin. J. Inorg. Chem. 2005, 21, 1583–1588. [Google Scholar]
- Li, Z.-F.; Liang, L.-L.; Wu, F.; Zhou, F.-G.; Ni, X.-L.; Feng, X.; Xiao, X.; Zhang, Y.-Q.; Xue, S.-F.; Zhu, Q.-J.; et al. An approach to networks based on coordination of alkyl-substituted cucurbit[5]urils and potassium ions. CrystEngComm 2013, 15, 1994. [Google Scholar] [CrossRef]
- Macartney, D.H. Encapsulation of Drug Molecules by Cucurbiturils: Effects on their Chemical Properties in Aqueous Solution. Isr. J. Chem. 2011, 51, 600–615. [Google Scholar] [CrossRef]
- Shchepotina, E.G.; Pashkina, E.A.; Yakushenko, E.V.; Kozlov, V.A. Cucurbiturils as containers for medicinal compounds. Nanotechnologies Russ. 2011, 6, 773–779. [Google Scholar] [CrossRef]
- Barrow, S.J.; Kasera, S.; Rowland, M.J.; Del Barrio, J.; Scherman, O.A. Cucurbituril-Based Molecular Recognition. Chem. Rev. 2015, 115, 12320–12406. [Google Scholar] [CrossRef]
- Sanku, R.K.K.; Karakus, O.O.; Ilies, M.; Ilies, M.A. Inclusion Complexes in Drug Delivery and Drug Targeting: Formation, Characterization, and Biological Applications. In Targeted Nanosystems for Therapeutic Applications: New Concepts, Dynamic Properties, Efficiency, and Toxicity; Sakurai, K., Ilies, M.A., Eds.; ACS Publications: Washington, DC, USA, 2019. [Google Scholar]
- Nau, W.M.; Florea, M.; Assaf, K.I. Deep Inside Cucurbiturils: Physical Properties and Volumes of their Inner Cavity Determine the Hydrophobic Driving Force for Host-Guest Complexation. Isr. J. Chem. 2011, 51, 559–577. [Google Scholar] [CrossRef]
- Assaf, K.I.; Nau, W.M. Cucurbiturils: From synthesis to high-affinity binding and catalysis. Chem. Soc. Rev. 2015, 44, 394–418. [Google Scholar] [CrossRef]
- Assaf, K.I.; Florea, M.; Antony, J.; Henriksen, N.M.; Yin, J.; Hansen, A.; Qu, Z.-W.; Sure, R.; Klapstein, D.; Gilson, M.K.; et al. HYDROPHOBE Challenge: A Joint Experimental and Computational Study on the Host–Guest Binding of Hydrocarbons to Cucurbiturils, Allowing Explicit Evaluation of Guest Hydration Free-Energy Contributions. J. Phys. Chem. B 2017, 121, 11144–11162. [Google Scholar] [CrossRef]
- Lee, T.-C.; Kalenius, E.; Lazar, A.I.; Assaf, K.I.; Kuhnert, N.; Grün, C.H.; Jänis, J.; Scherman, O.A.; Nau, W.M. Chemistry inside molecular containers in the gas phase. Nat. Chem. 2013, 5, 376–382. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Assaf, K.I.; Nau, W.M. Applications of Cucurbiturils in Medicinal Chemistry and Chemical Biology. Front. Chem. 2019, 7, 619. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Zhang, H.; Yue, L.; Li, S.; Cheng, Q.; Wang, R. Facile Preparation of Cucurbit[6]uril-Based Polymer Nanocapsules for Targeted Photodynamic Therapy. ACS Appl. Mater. Interfaces 2019, 11, 22925–22931. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Chen, Y.; Yu, Q.; Li, F.-Q.; Liu, Y. A cucurbituril/polysaccharide/carbazole ternary supramolecular assembly for targeted cell imaging. Chem. Commun. 2019, 55, 4343–4346. [Google Scholar] [CrossRef]
- Kim, K.L.; Sung, G.; Sim, J.; Murray, J.; Li, M.; Lee, A.; Shrinidhi, A.; Park, K.M.; Kim, K. Supramolecular latching system based on ultrastable synthetic binding pairs as versatile tools for protein imaging. Nat. Commun. 2018, 9, 1712. [Google Scholar] [CrossRef]
- Ko, Y.H.; Kim, E.; Hwang, I.; Kim, K. Supramolecular assemblies built with host-stabilized charge-transfer interactions. Chem. Commun. 2007, 13, 1305–1315. [Google Scholar] [CrossRef]
- Gao, R.H.; Chen, L.X.; Chen, K.; Tao, Z.; Xiao, X. Development of hydroxylated cucurbit[ n ]urils, their derivatives and potential applications. Co-ord. Chem. Rev. 2017, 348, 1–24. [Google Scholar] [CrossRef]
- Ghale, G.; Kuhnert, N.; Nau, W.M. Monitoring stepwise proteolytic degradation of peptides by supramolecular domino tandem assays and mass spectrometry for trypsin and leucine aminopeptidase. Nat. Prod. Commun. 2012, 7, 343–348. [Google Scholar] [CrossRef]
- Chinai, J.M.; Taylor, A.B.; Ryno, L.M.; Dybdal-Hargreaves, N.; Morris, C.A.; Hart, P.J.; Urbach, A.R. Molecular Recognition of Insulin by a Synthetic Receptor. J. Am. Chem. Soc. 2011, 133, 8810–8813. [Google Scholar] [CrossRef]
- Freeman, W.A.; Mock, W.L.; Shih, N.Y. Cucurbituril. J. Am. Chem. Soc. 1981, 103, 7367–7368. [Google Scholar] [CrossRef]
- Kim, B.S.; Ko, Y.H.; Kim, Y.; Lee, H.J.; Selvapalam, N.; Lee, H.C.; Kim, K. Water soluble cucurbit[6]uril derivative as a potential Xe carrier for 129Xe NMR-based biosensors. Chem. Commun. 2008, 24, 2756–2758. [Google Scholar] [CrossRef] [PubMed]
- Berthault, P.; Huber, G.; Desvaux, H. Biosensing using laser-polarized xenon NMR/MRI. Prog. Nucl. Magn. Reson. Spectrosc. 2009, 55, 35–60. [Google Scholar] [CrossRef]
- Buschmann, H.-J.; Wego, A.; Zielesny, A.; Schollmeyer, E. Structure, Stability, Electronic Properties and NMR-Shielding of the Cucurbit[6]uril–Spermine-Complex. J. Incl. Phenom. Macrocycl. Chem. 2006, 54, 241–246. [Google Scholar] [CrossRef]
- Pinjari, R.; Gejji, S.P. Electronic Structure, Molecular Electrostatic Potential, and NMR Chemical Shifts in Cucurbit[n]urils (n= 5−8), Ferrocene, and Their Complexes. J. Phys. Chem. A 2008, 112, 12679–12686. [Google Scholar] [CrossRef] [PubMed]
- Kennan, R.P.; Pollack, G.L. Pressure dependence of the solubility of nitrogen, argon, krypton, and xenon in water. J. Chem. Phys. 1990, 93, 2724–2735. [Google Scholar] [CrossRef]
- Walker, T.; Happer, W. Spin-exchange optical pumping of noble-gas nuclei. Rev. Mod. Phys. 1997, 69, 629–642. [Google Scholar] [CrossRef]
- Contributors to Wikimedia projects Hyperpolarization (physics)—Wikipedia. Available online: https://en.wikipedia.org/w/index.php?title=Hyperpolarization_(physics)&oldid=931008243 (accessed on 20 December 2019).
- Witte, C.; Kunth, M.; Rossella, F.; Schröder, L. Observing and preventing rubidium runaway in a direct-infusion xenon-spin hyperpolarizer optimized for high-resolution hyper-CEST (chemical exchange saturation transfer using hyperpolarized nuclei) NMR. J. Chem. Phys. 2014, 140, 84203. [Google Scholar] [CrossRef]
- Nikolaou, P.; Coffey, A.M.; Walkup, L.L.; Gust, B.M.; Whiting, N.; Newton, H.; Barcus, S.; Muradyan, I.; Dabaghyan, M.; Moroz, G.D.; et al. Near-unity nuclear polarization with an open-source 129Xe hyperpolarizer for NMR and MRI. Proc. Natl. Acad. Sci. 2013, 110, 14150–14155. [Google Scholar] [CrossRef]
- Lakshmanan, A.; Lu, G.; Farhadi, A.; Nety, S.P.; Kunth, M.; Lee-Gosselin, A.; Maresca, D.; Bourdeau, R.W.; Yin, M.; Yan, J.; et al. Preparation of biogenic gas vesicle nanostructures for use as contrast agents for ultrasound and MRI. Nat. Protoc. 2017, 12, 2050–2080. [Google Scholar] [CrossRef]
- Rose, H.M.; Witte, C.; Rossella, F.; Klippel, S.; Freund, C.; Schröder, L. Development of an antibody-based, modular biosensor for 129Xe NMR molecular imaging of cells at nanomolar concentrations. Proc. Natl. Acad. Sci. 2014, 111, 11697–11702. [Google Scholar] [CrossRef]
- Huber, G.; Legrand, F.-X.; Lewin, V.; Baumann, D.; Heck, M.-P.; Berthault, P. Interaction of Xenon with Cucurbit[5]uril in Water. ChemPhysChem 2011, 12, 1053–1055. [Google Scholar] [CrossRef] [PubMed]
- Kunth, M.; Witte, C.; Schröder, L. Continuous-wave saturation considerations for efficient xenon depolarization. NMR Biomed. 2015, 28, 601–606. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.-J.; Cleve, E.; Jansen, K.; Wego, A.; Schollmeyer, E. Complex Formation between Cucurbit[n]urils and Alkali, Alkaline Earth and Ammonium Ions in Aqueous Solution. J. Inclusion Phenom. Macrocycl. Chem. 2001, 40, 117–120. [Google Scholar] [CrossRef]
- Isaacs, L.D. Cucurbit[n]urils: From mechanism to structure and function. Chem. Commun. 2009, 6, 619–629. [Google Scholar] [CrossRef]
- Buschmann, H.-J.; Jansen, K.; Meschke, C.; Schollmeyer, E. Thermodynamic Data for Complex Formation Between Cucurbituril and Alkali and Alkaline Earth Cations in Aqueous Formic Acid Solution. J. Solut. Chem. 1998, 27, 135–140. [Google Scholar] [CrossRef]
- Marquez, C.; Hudgins, R.R.; Nau, W.M. Mechanism of Host−Guest Complexation by Cucurbituril. J. Am. Chem. Soc. 2004, 126, 5806–5816. [Google Scholar] [CrossRef]
- Bardelang, D.; Udachin, K.A.; Anedda, R.; Moudrakovski, I.L.; Leek, D.M.; Ripmeester, J.A.; Ratcliffe, C.I. Single-crystal to single-crystal phase transition of cucurbit[5]uril hydrochloride hydrates: Large water-filled channels transforming to layers of unusual stability. Chem. Commun. 2008, 40, 4927–4929. [Google Scholar] [CrossRef]
- Kumar, C.P.; Pradeep Kumar, C.; Wu, F.; Woodward, C.E.; Day, A.I. The influence of equatorial substitution and K ion concentration: An encapsulation study of CH4, CH3F, CH3Cl, CH2F2and CF4, in Q[5], CyP5Q[5] and a CyP5Q[5]-carboxylate derivative. Supramol. Chem. 2014, 26, 670–676. [Google Scholar] [CrossRef]
- El Haouaj, M.; Luhmer, M.; Ko, Y.H.; Kim, K.; Bartik, K. NMR study of the reversible complexation of xenon by cucurbituril. J. Chem. Soc. Perkin Trans. 2 2001, 2, 804–807. [Google Scholar] [CrossRef][Green Version]
- Huber, G.; Brotin, T.; Dubois, L.; Desvaux, H.; Dutasta, J.-P.; Berthault, P. Water Soluble Cryptophanes Showing Unprecedented Affinity for Xenon: Candidates as NMR-Based Biosensors. J. Am. Chem. Soc. 2006, 128, 6239–6246. [Google Scholar] [CrossRef]
- McKim, S.; Hinton, J. 129Xe NMR Spectroscopic Investigation of the Interaction of Xenon with Ions in Aqueous Solution. J. Magn. Reson. Ser. A 1993, 104, 268–272. [Google Scholar] [CrossRef]
- El Haouaj, M.; Ko, Y.H.; Luhmer, M.; Kim, K.; Bartik, K. NMR Investigation of the complexation of neutral guests by cucurbituril. J. Chem. Soc. Perkin Trans. 2 2001, 2, 2104–2107. [Google Scholar] [CrossRef]
- Schnurr, M.; Döpfert, J.; Schröder, L.; Hennig, A.; Sloniec-Myszk, J. Supramolecular Assays for Mapping Enzyme Activity by Displacement-Triggered Change in Hyperpolarized 129 Xe Magnetization Transfer NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2015, 54, 13444–13447. [Google Scholar] [CrossRef] [PubMed]
- Truxal, A.E.; Cao, L.; Isaacs, L.; Wemmer, D.E.; Pines, A. Directly Functionalized Cucurbit[7]uril as a Biosensor for the Selective Detection of Protein Interactions by 129 Xe hyperCEST NMR. Chemistry 2019, 25, 6108–6112. [Google Scholar] [CrossRef]
- Schröder, L.; Lowery, T.J.; Hilty, C.; Wemmer, D.E.; Pines, A. Molecular Imaging Using a Targeted Magnetic Resonance Hyperpolarized Biosensor. Science 2006, 314, 446–449. [Google Scholar] [CrossRef]
- Klippel, S.; Freund, C.; Schröder, L. Multichannel MRI Labeling of Mammalian Cells by Switchable Nanocarriers for Hyperpolarized Xenon. Nano Lett. 2014, 14, 5721–5726. [Google Scholar] [CrossRef]
- Spence, M.M.; Rubin, S.M.; Dimitrov, I.; Ruiz, E.J.; Wemmer, D.E.; Pines, A.; Yao, S.Q.; Tian, F.; Schultz, P.G. Functionalized xenon as a biosensor. Proc. Natl. Acad. Sci. 2001, 98, 10654–10657. [Google Scholar] [CrossRef]
- Schröder, L. Xenon Biosensor HyperCEST MRI. In Hyperpolarized and Inert Gas MRI.; Elsevier: Amsterdam, the Netherlands, 2017; pp. 263–277. [Google Scholar]
- Jayapaul, J.; Schröder, L. Nanoparticle-Based Contrast Agents for 129Xe HyperCEST NMR and MRI Applications. Contrast Media Mol. Imaging 2019, 2019, 9498173. [Google Scholar] [CrossRef]
- Kunth, M.; Witte, C.; Hennig, A.; Schröder, L. Identification, classification, and signal amplification capabilities of high-turnover gas binding hosts in ultra-sensitive NMR. Chem. Sci. 2015, 6, 6069–6075. [Google Scholar] [CrossRef]
- Avram, L.; Bar-Shir, A. 19F-GEST NMR: Studying dynamic interactions in host–guest systems. Org. Chem. Front. 2019, 6, 1503–1512. [Google Scholar] [CrossRef]
- Kaizerman-Kane, D.; Hadar, M.; Tal, N.; Dobrovetsky, R.; Zafrani, Y.; Cohen, Y.; Dobtovetsky, R. pH-Responsive Pillar[6]arene-based Water-Soluble Supramolecular Hexagonal Boxes. Angew. Chem. Int. Ed. Engl. 2019, 58, 5302–5306. [Google Scholar] [CrossRef] [PubMed]
- Cragg, P.J. Pillar[ n ]arenes at the Chemistry-Biology Interface. Isr. J. Chem. 2018, 58, 1194–1208. [Google Scholar] [CrossRef]
- Adiri, T.; Marciano, D.; Cohen, Y. Potential 129Xe-NMR biosensors based on secondary and tertiary complexes of a water-soluble pillar[5]arene derivative. Chem. Commun. 2013, 49, 7082. [Google Scholar] [CrossRef] [PubMed]
- Kunth, M.; Döpfert, J.; Witte, C.; Rossella, F.; Schröder, L. Optimized Use of Reversible Binding for Fast and Selective NMR Localization of Caged Xenon. Angew. Chem. Int. Ed. Engl. 2012, 51, 8217–8220. [Google Scholar] [CrossRef]
- Döpfert, J.; Witte, C.; Schröder, L. Fast Gradient-Encoded CEST Spectroscopy of Hyperpolarized Xenon. ChemPhysChem 2014, 15, 261–264. [Google Scholar] [CrossRef]
- Xu, X.; Lee, J.-S.; Jerschow, A. Ultrafast scanning of exchangeable sites by NMR spectroscopy. Angew. Chem. Int. Ed. Engl. 2013, 52, 8281–8284. [Google Scholar] [CrossRef]
- Döpfert, J.; Zaiss, M.; Witte, C.; Schröder, L. Ultrafast CEST imaging. J. Magn. Reson. 2014, 243, 47–53. [Google Scholar] [CrossRef]
- Boutin, C.; Léonce, E.; Brotin, T.; Jerschow, A.; Berthault, P. Ultrafast Z-Spectroscopy for 129Xe NMR-Based Sensors. J. Phys. Chem. Lett. 2013, 4, 4172–4176. [Google Scholar] [CrossRef][Green Version]
- Döpfert, J.; Witte, C.; Schröder, L. Slice-selective gradient-encoded CEST spectroscopy for monitoring dynamic parameters and high-throughput sample characterization. J. Magn. Reson. 2013, 237, 34–39. [Google Scholar] [CrossRef]
- Schnurr, M.; Joseph, R.; Naugolny-Keisar, A.; Kaizerman-Kane, D.; Bogdanoff, N.; Schuenke, P.; Cohen, Y.; Schröder, L. High Exchange Rate Complexes of Xe with Water-Soluble Pillar[5]arenes for Adjustable Magnetization Transfer MRI. Chemphyschem 2019, 20, 246–251. [Google Scholar] [CrossRef]
- Avram, L.; Havel, V.; Shusterman-Krush, R.; Iron, M.; Zaiss, M.; Šindelář, V.; Bar-Shir, A. Cover Feature: Dynamic Interactions in Synthetic Receptors: A Guest Exchange Saturation Transfer Study (Chem. Eur. J. 7/2019). Chemistry 2019, 25, 1604. [Google Scholar] [CrossRef]
- Zaiss, M.; Schnurr, M.; Bachert, P. Analytical solution for the depolarization of hyperpolarized nuclei by chemical exchange saturation transfer between free and encapsulated xenon (HyperCEST). J. Chem. Phys. 2012, 136, 144106. [Google Scholar] [CrossRef] [PubMed]
- Kunth, M.; Witte, C.; Schröder, L. Quantitative chemical exchange saturation transfer with hyperpolarized nuclei (qHyper-CEST): Sensing xenon-host exchange dynamics and binding affinities by NMR. J. Chem. Phys. 2014, 141, 194202. [Google Scholar] [CrossRef] [PubMed]
- Korchak, S.; Kilian, W.; Schröder, L.; Mitschang, L. Design and comparison of exchange spectroscopy approaches to cryptophane–xenon host–guest kinetics. J. Magn. Reson. 2016, 265, 139–145. [Google Scholar] [CrossRef]
- Korchak, S.; Riemer, T.; Kilian, W.; Mitschang, L. Quantitative Assessment of Xenon Exchange Kinetics with Cucurbit[6]uril in Physiological Saline. ChemPhysChem 2018, 19, 1859–1865. [Google Scholar] [CrossRef]
- Döpfert, J.; Schnurr, M.; Kunth, M.; Rose, H.M.; Hennig, A.; Schröder, L. Time-resolved monitoring of enzyme activity with ultrafast Hyper-CEST spectroscopy. Magn. Reson. Chem. 2018, 56, 679–688. [Google Scholar] [CrossRef]
- Pagel, M.D.; Daryaei, I. Double agents and secret agents: The emerging fields of exogenous chemical exchange saturation transfer and T2-exchange magnetic resonance imaging contrast agents for molecular imaging. Res. Rep. Nucl. Med. 2015, 5, 19–32. [Google Scholar] [CrossRef][Green Version]
- Fernandez-Cuervo, G.; Tucker, K.A.; Malm, S.; Jones, K.M.; Pagel, M.D. Diamagnetic Imaging Agents with a Modular Chemical Design for Quantitative Detection of β-Galactosidase and β-Glucuronidase Activities with CatalyCEST MRI. Bioconjugate Chem. 2016, 27, 2549–2557. [Google Scholar] [CrossRef]
- Li, Y.; Sheth, V.R.; Liu, G.; Pagel, M.D. A self-calibrating PARACEST MRI contrast agent that detects esterase enzyme activity. Contrast Media Mol. Imaging 2010, 6, 219–228. [Google Scholar] [CrossRef]
- Fernandez-Cuervo, G.; Sinharay, S.; Pagel, M.D. A CatalyCEST MRI Contrast Agent that Can Simultaneously Detect Two Enzyme Activities. ChemBioChem 2016, 17, 383–387. [Google Scholar] [CrossRef]
- Lee, H.H.; Choi, T.S.; Lee, S.J.C.; Lee, J.W.; Park, J.; Ko, Y.H.; Kim, W.J.; Kim, K.; Kim, H.I. Supramolecular inhibition of amyloid fibrillation by cucurbit[7]uril. Angew. Chem. Int. Ed. Engl. 2014, 53, 7461–7465. [Google Scholar] [CrossRef] [PubMed]
- Jost, J.O.; Witte, C.; Schröder, L. Macrocyclic Xenon hosts: Potential inhibitors and reporters for protein aggregation in Hyper-CEST MRI. In Proceedings of the Annual Meeting ISMRM-ESMRMB, Paris, France, 16–21 June 2018. [Google Scholar]
- Cheng, C.; McGonigal, P.; Schneebeli, S.; Li, H.; Vermeulen, N.A.; Ke, C.; Stoddart, J.F. An artificial molecular pump. Nat. Nanotechnol. 2015, 10, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Whitesides, G.M. Self-Assembly at All Scales. Science 2002, 295, 2418–2421. [Google Scholar] [CrossRef] [PubMed]
- Browne, W.R.; Feringa, B.L. Making molecular machines work. Nat. Nanotechnol. 2006, 1, 25–35. [Google Scholar] [CrossRef]
- Chambers, J.M.; Hill, P.A.; Aaron, J.A.; Han, Z.; Christianson, D.W.; Kuzma, N.N.; Dmochowski, I.J. Cryptophane Xenon-129 Nuclear Magnetic Resonance Biosensors Targeting Human Carbonic Anhydrase. J. Am. Chem. Soc. 2009, 131, 563–569. [Google Scholar] [CrossRef]
- Supuran, C.T. Carbonic anhydrases: Novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov. 2008, 7, 168–181. [Google Scholar] [CrossRef]
- Gao, J.; Qiao, S.; Whitesides, G.M. Increasing Binding Constants of Ligands to Carbonic Anhydrase by Using "Greasy Tails". J. Med. Chem. 1995, 38, 2292–2301. [Google Scholar] [CrossRef]
- Wang, Y.; Roose, B.W.; Philbin, J.P.; Doman, J.L.; Dmochowski, I.J. Programming A Molecular Relay for Ultrasensitive Biodetection through 129 Xe NMR. Angew. Chem. Int. Ed. Engl. 2016, 128, 1765–1768. [Google Scholar] [CrossRef]
- Zhang, S.; Dominguez, Z.; Assaf, K.I.; Nilam, M.; Thiele, T.; Pischel, U.; Schedler, U.; Nau, W.M.; Hennig, A. Precise supramolecular control of surface coverage densities on polymer micro- and nanoparticles. Chem. Sci. 2018, 9, 8575–8581. [Google Scholar] [CrossRef]
- Jost, J.O.; Liu, Y.-C.; Hennig, A.; Schröder, L. HyperCEST NMR Characterization of the Transition Between Two Hosts for the Two-Faced Guest AMADA-Putr. In Proceedings of the 60th Experimental Nuclear Magnetic Resonance Conference (ENC), Pacific Grove, CA, USA, 7–12 April 2019. [Google Scholar]
- Jiang, W.; Winkler, H.D.F.; Schalley, C.A. Integrative Self-Sorting: Construction of a Cascade-Stoppered Hetero[3]rotaxane. J. Am. Chem. Soc. 2008, 130, 13852–13853. [Google Scholar] [CrossRef]
- Yang, W.; Li, Y.; Liu, H.; Chi, L.; Li, Y. Design and Assembly of Rotaxane-Based Molecular Switches and Machines. Small 2012, 8, 504–516. [Google Scholar] [CrossRef] [PubMed]
- Finbloom, J.A.; Slack, C.; Bruns, C.; Jeong, K.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane-mediated suppression and activation of cucurbit[6]uril for molecular detection by 129Xe hyperCEST NMR. Chem. Commun. 2016, 52, 3119–3122. [Google Scholar] [CrossRef] [PubMed]
- Tuncel, D.; Steinke, J.H.G. The synthesis of [2], [3] and [4]rotaxanes and semirotaxanes. Chem. Commun. 2002, 5, 496–497. [Google Scholar] [CrossRef] [PubMed]
- Ke, C.; Smaldone, R.A.; Kikuchi, T.; Li, H.; Davis, A.P.; Stoddart, J.F. ChemInform Abstract: Quantitative Emergence of Hetero[4]rotaxanes by Template-Directed Click Chemistry. Angew. Chem. Int. Ed. Engl. 2013, 52, 381–387. [Google Scholar] [CrossRef]
- Hou, X.; Ke, C.; Bruns, C.; McGonigal, P.; Pettman, R.B.; Stoddart, J.F. Tunable solid-state fluorescent materials for supramolecular encryption. Nat. Commun. 2015, 6, 6884. [Google Scholar] [CrossRef]
- Slack, C.; Finbloom, J.A.; Jeong, K.; Bruns, C.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane probes for protease detection by 129 Xe hyperCEST NMR. Chem. Commun. 2017, 53, 1076–1079. [Google Scholar] [CrossRef]
- Niethammer, P.; Grabher, C.; Look, A.T.; Mitchison, T.J. A tissue-scale gradient of hydrogen peroxide mediates rapid wound detection in zebrafish. Nature 2009, 459, 996–999. [Google Scholar] [CrossRef]
- Weinstain, R.; Savariar, E.N.; Felsen, C.N.; Tsien, R.Y. In Vivo Targeting of Hydrogen Peroxide by Activatable Cell-Penetrating Peptides. J. Am. Chem. Soc. 2014, 136, 874–877. [Google Scholar] [CrossRef]
- Finbloom, J.A.; Aanei, I.L.; Bernard, J.M.; Klass, S.H.; Elledge, S.K.; Han, K.; Ozawa, T.; Nicolaides, T.; Berger, M.S.; Francis, M.B. Evaluation of Three Morphologically Distinct Virus-Like Particles as Nanocarriers for Convection-Enhanced Drug Delivery to Glioblastoma. Nanomaterials 2018, 8, 1007. [Google Scholar] [CrossRef]
- Adriani, G.; De Tullio, M.D.; Ferrari, M.; Hussain, F.; Pascazio, G.; Liu, X.; Decuzzi, P. The preferential targeting of the diseased microvasculature by disk-like particles. Biomaterials 2012, 33, 5504–5513. [Google Scholar] [CrossRef]
- Dixon, S.J.; Stockwell, B.R. The role of iron and reactive oxygen species in cell death. Nat. Chem. Biol. 2014, 10, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Outschoorn, U.; Lin, Z.; Trimmer, C.; Flomenberg, N.; Wang, C.; Pavlides, S.; Pestell, R.G.; Howell, A.; Sotgia, F.; Lisanti, M.P. Cancer cells metabolically “fertilize” the tumor microenvironment with hydrogen peroxide, driving the Warburg effect. Cell Cycle 2011, 10, 2504–2520. [Google Scholar] [CrossRef] [PubMed]
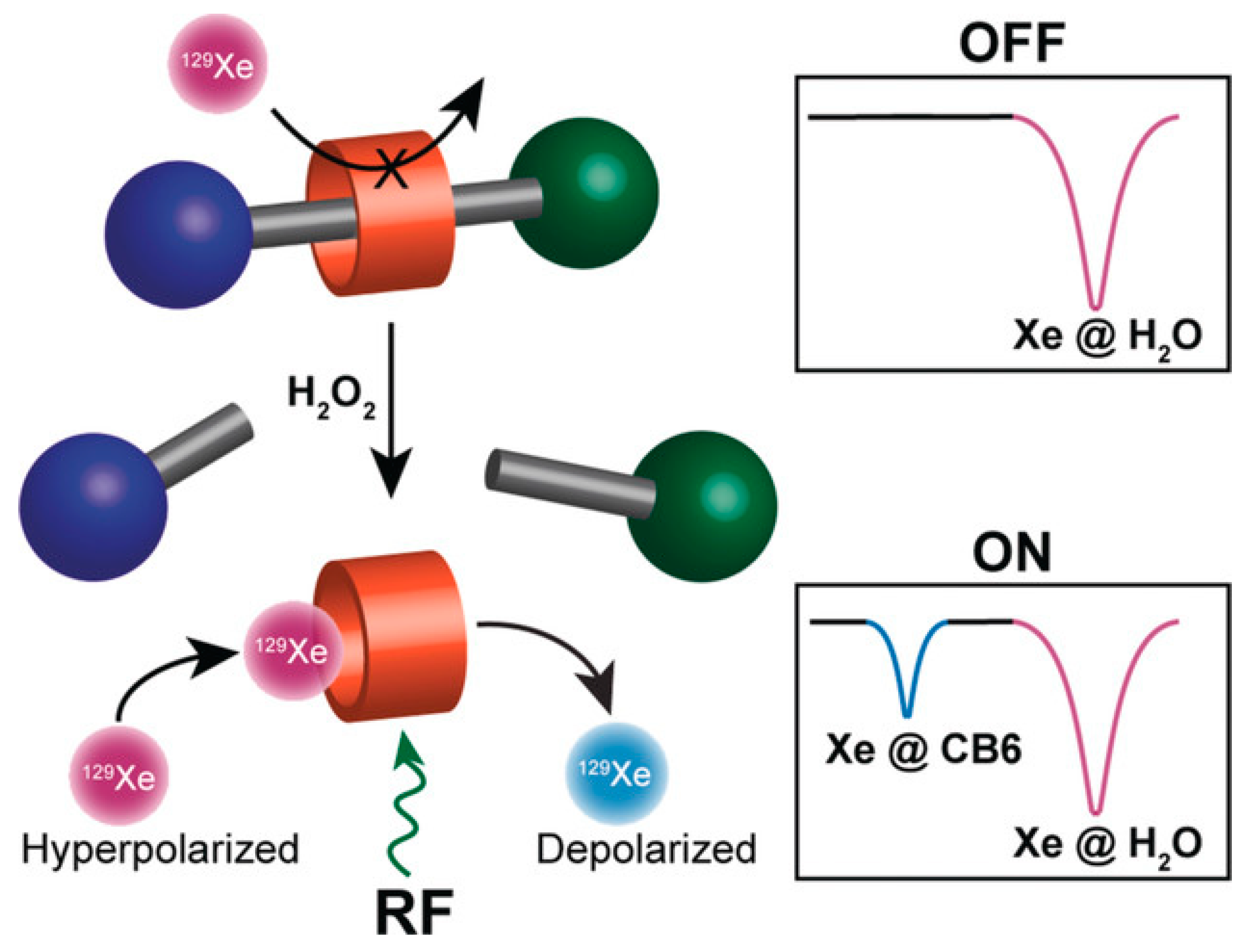
- Klass, S.H.; Truxal, A.E.; Fiala, T.A.; Kelly, J.; Nguyen, D.; Finbloom, J.A.; Wemmer, D.E.; Pines, A.; Francis, M.B. Rotaxane Probes for the Detection of Hydrogen Peroxide by 129 Xe HyperCEST NMR Spectroscopy. Angew. Chem. Int. Ed. Engl. 2019, 58, 9948–9953. [Google Scholar] [CrossRef] [PubMed]
- Finbloom, J.A.; Han, K.; Slack, C.; Furst, A.L.; Francis, M.B. Cucurbit[6]uril-Promoted Click Chemistry for Protein Modification. J. Am. Chem. Soc. 2017, 139, 9691–9697. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.-J.; Cleve, E.; Jansen, K.; Wego, A.; Schollmeyer, E. The determination of complex stabilities between different cyclodextrins and dibenzo-18-crown-6, cucurbit[6]uril, decamethylcucurbit[5]uril, cucurbit[5]uril, p-tert-butylcalix[4]arene and p-tert-butylcalix[6]arene in aqueous solutions using a spectrophotometric method. Mater. Sci. Eng. C 2001, 14, 35–39. [Google Scholar]
- Rekharsky, M.V.; Yamamura, H.; Kawai, M.; Osaka, I.; Arakawa, R.; Sato, A.; Ko, Y.H.; Selvapalam, N.; Kim, K.; Inoue, Y. Sequential Formation of a Ternary Complex among Dihexylammonium, Cucurbit[6]uril, and Cyclodextrin with Positive Cooperativity. Org. Lett. 2006, 8, 815–818. [Google Scholar] [CrossRef]
- Bügler, J.; Engbersen, J.F.J.; Reinhoudt, D.N. Novel Water-Soluble β-Cyclodextrin−Calix[4]arene Couples as Fluorescent Sensor Molecules for the Detection of Neutral Analytes. J. Org. Chem. 1998, 63, 5339–5344. [Google Scholar] [CrossRef]
- Bügler, J.; Sommerdijk, N.A.J.M.; Visser, A.J.W.G.; Van Hoek, A.; Nolte, R.; Engbersen, J.F.J.; Reinhoudt, D.N. Interconnective Host−Guest Complexation of β-Cyclodextrin−Calix[4]arene Couples. J. Am. Chem. Soc. 1999, 121, 28–33. [Google Scholar] [CrossRef]
- Tian, X.; Chen, L.X.; Yao, Y.-Q.; Chen, K.; Chen, M.-D.; Zeng, X.; Tao, Z. 4-Sulfocalix[4]arene/Cucurbit[7]uril-Based Supramolecular Assemblies through the Outer Surface Interactions of Cucurbit[n]uril. ACS Omega 2018, 3, 6665–6672. [Google Scholar] [CrossRef]
- Ni, X.-L.; Xiao, X.; Cong, H.; Zhu, Q.-J.; Xue, S.-F.; Tao, Z. Self-Assemblies Based on the “Outer-Surface Interactions” of Cucurbit[n]urils: New Opportunities for Supramolecular Architectures and Materials. Accounts Chem. Res. 2014, 47, 1386–1395. [Google Scholar] [CrossRef]
- Shen, F.-F.; Zhao, J.-L.; Chen, K.; Hua, Z.-Y.; Chen, M.-D.; Zhang, Y.-Q.; Zhu, Q.-J.; Tao, Z. Supramolecular coordination assemblies of a symmetrical octamethyl-substituted cucurbituril with alkali metal ions based on the outer-surface interactions of cucurbit[n]urils. CrystEngComm 2017, 19, 2464–2474. [Google Scholar] [CrossRef]
- Shen, F.-F.; Zhao, J.-L.; Chen, K.; Xu, J.; Wang, Y.; Hua, Z.-Y.; Wu, L.; Chen, M.-D.; Zhang, Y.-Q.; Tao, Z. Coordination and supramolecular assemblies of mono-hydroxylated octamethylcucurbit[6]uril with alkali and alkaline earth metal ions in the presence of polychloride cadmium anions. CrystEngComm 2017, 19, 4017–4024. [Google Scholar] [CrossRef]
- Jabadurai Jayapaul, L.S. In 129Xe Hyper-CEST for sensing Supramolecular Complexes. In Proceedings of the XeMAT, Technical University Dresden, Dresden, German, 13–17 September 2015; pp. 82–83. [Google Scholar]
- Flinn, A.; Hough, G.C.; Fraser Stoddart, J.; Williams, D.J. Decamethylcucurbit[5]uril. Angew. Chem. Int. Ed. Engl. 1992, 31, 1475–1477. [Google Scholar] [CrossRef]
- Kim, K.; Selvapalam, N.; Ko, Y.H.; Park, K.M.; Kim, D.; Kim, J. Functionalized cucurbiturils and their applications. Chem. Soc. Rev. 2007, 36, 267–279. [Google Scholar] [CrossRef]
- Lucas, D.; Minami, T.; Iannuzzi, G.; Cao, L.; Wittenberg, J.B.; Anzenbacher, P.; Isaacs, L.D. Templated Synthesis of Glycoluril Hexamer and Monofunctionalized Cucurbit[6]uril Derivatives. J. Am. Chem. Soc. 2011, 133, 17966–17976. [Google Scholar] [CrossRef]
- Cao, L.; Isaacs, L.D. Daisy Chain Assembly Formed from a Cucurbit[6]uril Derivative. Org. Lett. 2012, 14, 3072–3075. [Google Scholar] [CrossRef]
- Vinciguerra, B.; Cao, L.; Cannon, J.R.; Zavalij, P.; Fenselau, C.; Isaacs, L.D. Synthesis and Self-Assembly Processes of Monofunctionalized Cucurbit[7]uril. J. Am. Chem. Soc. 2012, 134, 13133–13140. [Google Scholar] [CrossRef]
- Cao, L.; Hettiarachchi, G.; Briken, V.; Isaacs, L. Cucurbit[7]uril containers for targeted delivery of oxaliplatin to cancer cells. Angew. Chem. Int. Ed. Engl. 2013, 52, 12033–12037. [Google Scholar] [CrossRef]
- Zhao, N.; Lloyd, G.; Scherman, O.A. Monofunctionalised cucurbit[6]uril synthesis using imidazolium host–guest complexation. Chem. Commun. 2012, 48, 3070. [Google Scholar] [CrossRef]
- Jon, S.Y.; Selvapalam, N.; Oh, D.H.; Kang, J.-K.; Kim, S.-Y.; Jeon, Y.J.; Lee, J.W.; Kim, K. Facile Synthesis of Cucurbit[n]uril Derivatives via Direct Functionalization: Expanding Utilization of Cucurbit[n]uril. J. Am. Chem. Soc. 2003, 125, 10186–10187. [Google Scholar] [CrossRef]
- Ayhan, M.M.; Karoui, H.; Hardy, M.; Rockenbauer, A.; Charles, L.; Rosas, R.; Udachin, K.; Tordo, P.; Bardelang, D.; Ouari, O. Comprehensive Synthesis of Monohydroxy-Cucurbit[n]urils (n = 5, 6, 7, 8): High Purity and High Conversions. J. Am. Chem. Soc. 2015, 137, 10238–10245. [Google Scholar] [CrossRef] [PubMed]
- Ahn, Y.; Jang, Y.; Selvapalam, N.; Yun, G.; Kim, K. Back Cover: Supramolecular Velcro for Reversible Underwater Adhesion. Angew. Chem. Int. Ed. Engl. 2013, 52, 3282. [Google Scholar] [CrossRef]
- Zeng, Q.; Guo, Q.; Yuan, Y.; Yang, Y.; Zhang, B.; Ren, L.; Zhang, X.; Luo, Q.; Liu, M.; Bouchard, L.-S.; et al. Mitochondria Targeted and Intracellular Biothiol Triggered Hyperpolarized 129Xe Magnetofluorescent Biosensor. Anal. Chem. 2017, 89, 2288–2295. [Google Scholar] [CrossRef] [PubMed]
- Hane, F.T.; Li, T.; Smylie, P.; Pellizzari, R.M.; Plata, J.A.; DeBoef, B.; Albert, M.S. In vivo detection of cucurbit[6]uril, a hyperpolarized xenon contrast agent for a xenon magnetic resonance imaging biosensor. Sci. Rep. 2017, 7, 41027. [Google Scholar] [CrossRef]
- Uzunova, V.D.; Cullinane, C.; Brix, K.; Nau, W.M.; Day, A. Toxicity of cucurbit[7]uril and cucurbit[8]uril: An exploratory in vitro and in vivo study. Org. Biomol. Chem. 2010, 8, 2037–2042. [Google Scholar] [CrossRef]
- Hettiarachchi, G.; Nguyen, D.; Wu, J.; Lucas, D.; Ma, D.; Isaacs, L.D.; Briken, V. Toxicology and Drug Delivery by Cucurbit[n]uril Type Molecular Containers. PLoS ONE 2010, 5, 10514. [Google Scholar] [CrossRef]
- Colloc’h, N.; Sopkova-de Oliveira Santos, J.; Retailleau, P.; Vivarès, D.; Bonneté, F.; Langlois d’Estainto, B.; Gallois, B.; Brisson, A.; Risso, J.-J.; Lemaire, M.; et al. Protein crystallography under xenon and nitrous oxide pressure: Comparison with in vivo pharmacology studies and implications for the mechanism of inhaled anesthetic action. Biophys. J. 2007, 92, 217–224. [Google Scholar] [CrossRef]
- Jordan, B.D.; Wright, E.L. Xenon as an anesthetic agent. AANA J. 2010, 78, 387–392. [Google Scholar]
- Banks, P.; Franks, N.P.; Dickinson, R. Competitive Inhibition at the Glycine Site of the N-Methyl-d-Aspartate Receptor Mediates Xenon Neuroprotection against Hypoxia–Ischemia. Anesthesiology 2010, 112, 614–622. [Google Scholar] [CrossRef]
- Muradyan, I.; Patz, S. Hyperpolarized 129Xenon MRI of the Lung. In MRI of the Lung; Springer: Cham, Switzerland, 2017; pp. 99–124. [Google Scholar]
- Driehuys, B.; Cofer, G.P.; Pollaro, J.; Mackel, J.B.; Hedlund, L.; Johnson, G. Imaging alveolar–capillary gas transfer using hyperpolarized 129Xe MRI. Proc. Natl. Acad. Sci. 2006, 103, 18278–18283. [Google Scholar] [CrossRef]
- Shapiro, M.G.; Ramirez, R.M.; Sperling, L.J.; Sun, G.; Sun, J.; Pines, A.; Schaffer, D.V.; Bajaj, V.S. Genetically encoded reporters for hyperpolarized xenon magnetic resonance imaging. Nat. Chem. 2014, 6, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Matsumoto, S.; Norquay, G.; Griffiths, P.D.; Wild, J. High resolution spectroscopy and chemical shift imaging of hyperpolarized129Xe dissolved in the human brain in vivo at 1.5 tesla. Magn. Reson. Med. 2016, 75, 2227–2234. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.R.; Stewart, N.J.; Griffiths, P.D.; Norquay, G.; Wild, J. Imaging Human Brain Perfusion with Inhaled Hyperpolarized 129Xe MR Imaging. Radiology 2018, 286, 659–665. [Google Scholar] [CrossRef] [PubMed]
- Rao, M.; Norquay, G.; Stewart, N.J.; Hoggard, N.; Griffiths, P.D.; Wild, J. Assessment of brain perfusion using hyperpolarized 129 Xe MRI in a subject with established stroke. J. Magn. Reson. Imaging 2019, 50, 1002–1004. [Google Scholar] [CrossRef] [PubMed]
- Chacon-Caldera, J.; Maunder, A.; Rao, M.; Norquay, G.; Rodgers, O.I.; Clemence, M.; Puddu, C.; Schad, L.R.; Wild, J.M. Dissolved hyperpolarized xenon-129 MRI in human kidneys. Magn. Reson. Med. 2020, 83, 262–270. [Google Scholar] [CrossRef]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jayapaul, J.; Schröder, L. Probing Reversible Guest Binding with Hyperpolarized 129Xe-NMR: Characteristics and Applications for Cucurbit[n]urils. Molecules 2020, 25, 957. https://doi.org/10.3390/molecules25040957
Jayapaul J, Schröder L. Probing Reversible Guest Binding with Hyperpolarized 129Xe-NMR: Characteristics and Applications for Cucurbit[n]urils. Molecules. 2020; 25(4):957. https://doi.org/10.3390/molecules25040957
Chicago/Turabian StyleJayapaul, Jabadurai, and Leif Schröder. 2020. "Probing Reversible Guest Binding with Hyperpolarized 129Xe-NMR: Characteristics and Applications for Cucurbit[n]urils" Molecules 25, no. 4: 957. https://doi.org/10.3390/molecules25040957
APA StyleJayapaul, J., & Schröder, L. (2020). Probing Reversible Guest Binding with Hyperpolarized 129Xe-NMR: Characteristics and Applications for Cucurbit[n]urils. Molecules, 25(4), 957. https://doi.org/10.3390/molecules25040957