
Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning System-Natural Product Analysis

{kind=link}
{kind=link}
Abstract
1. Introduction
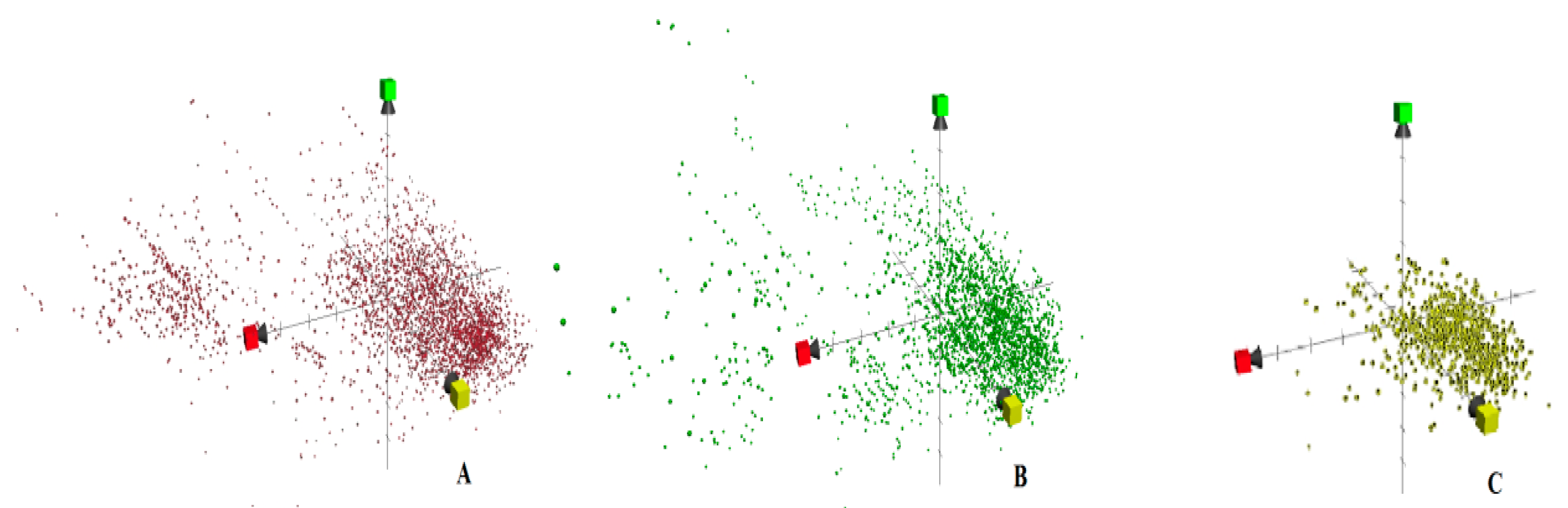
2. Results and Discussion
3. Material and Methods
3.1. Data Collection and Sources
3.1.1. Screening Schemes
3.1.2. In-House Active Set
3.1.3. Database
3.2. Compound Classification:
3.2.1. Classification According to Activity
3.2.2. Classification According to Predicted Permeability
3.2.3. Classification According to Toxicity Profile
3.3. Chemical Global Positioning System- Natural Product (ChemGPS-NP)
Descriptors
3.4. Data Analysis and Presentation:
3.4.1. Calculation of Euclidean Distances
3.4.2. Calculation of the Tanimoto Coefficient
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
Abbreviation
References
- Cambau, E.; Drancourt, M. Steps towards the discovery of Mycobacterium tuberculosis by Robert Koch, 1882. Clin. Microbio. Infec. 2014, 20, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Dheda, K.; Migliori, G.B. The global rise of extensively drug resistant tuberculosis: is the time to bring back sanatoria now overdue? Lancet 2012, 379, 773–775. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report 2012; World Health Organization: Geneva, Switzerland, 2013; Available online: http://www.who.int/tb/publications/global_report/en/ (accessed on 1 February 2020).
- Knox, C.; Law, V.; Jewison, T.; Liu, P.; Ly, S.; Frolkis, A.; Pon, A.; Banco, K.; Mak, C.; Neveu, V.; et al. DrugBank 3.0: A comprehensive resource for ‘Omics’ research on drugs. Nucleic Acids Res. 2014, 39, 1035–1041. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Treatment of Tuberculosis Guidelines, 4th ed.; WHO: Geneva, Switzerland, 2010; Available online: http://whqlibdoc.who.int/publications/2010/9789241547833_eng.pdf (accessed on 1 February 2020).
- Global Alliance for TB Drug Development. Ongoing Projects without a Lead Compound Series. Available online: https://www.newtbdrugs.org/pipeline/clinical (accessed on 30 June 2018).
- Osborne, R. First novel anti-tuberculosis drug in 40 years. Nat. Biotech. 2012, 31, 89–91. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, T.; Cooper, C.; Mdluli, K. Challenges and opportunities in developing novel drugs for TB. Fut. Med. Chem. 2011, 3, 1373–1400. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M. Chemical space and biology. Nature 2004, 432, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Pollock, S.N.; Coutsias, E.A.; Wester, M.J.; Oprea, T.I. Scaffold Topologies II: Analysis of Chemical Databases. J. Chem. Inf. Model. 2008, 48, 1304–1310. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Reymond, J.L.; Awale, M. Exploring chemical space for drug discovery using the chemical universe database. ACS Chem. Neurosci. 2012, 3, 649–657. [Google Scholar] [CrossRef] [PubMed]
- Brennan, P.J.; Young, B.D. Handbook of anti-tuberculosis agents. Tuberculosis 2008, 88, 85–170. [Google Scholar]
- Sacchettini, J.C.; Rubin, E.J.; Freundlich, J.S. Drugs versus bugs: In pursuit of the persistent predator Mycobacterium tuberculosis. Nat. Rev. Microbiol. 2008, 6, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Maddry, J.A.; Ananthan, S.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Maddox, C.; Rasmussen, L.; Reynolds, R.C.; Secrist, J.A.; Sosa, M.I.; et al. Antituberculosis activity of the molecular libraries screening center network library. Tuberculosis 2009, 89, 354–363. [Google Scholar] [CrossRef] [PubMed]
- Macarron, R.; Banks, M.N.; Bojanic, D.; Burns, D.J.; Cirovic, D.A.; Garyantes, T.; Green, D.V.S.; Hertzberg, R.P.; Janzen, W.P.; Paslay, J.W.; et al. Impact of high-throughput screening in biomedical research. Nat. Rev. Drug Discov. 2011, 10, 188–195. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Bradford, J.; Dole, K.; Spektor, A.; Gregory, K.; Blondeau, D.; Hohman, M.; Bunin, B.A. A Collaborative database and computational models for tuberculosis drug discovery. Mol. Biosyst. 2010, 6, 840–851. [Google Scholar] [CrossRef] [PubMed]
- Ballell, L.; Bates, R.H.; Young, R.J.; Alvarez-Gomez, D.; Alvarez-Ruiz, E.; Barroso, V.; Blanco, D.; Crespo, B.; Escribano, J.; González, R.; et al. Fueling Open-Source Drug Discovery: 177 Small-Molecule Leads against Tuberculosis. Chem. Med. Chem. 2013, 8, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Prathipati, P.; Ma, N.L.; Keller, T.H. Global Bayesian models for the prioritization of antitubercular agents. J. Chem. Inf. Model. 2008, 48, 2362–2370. [Google Scholar] [CrossRef] [PubMed]
- Merget, B.; Zilian, D.; Muller, T.; Sotriffer, C.A. MycPermCheck: The Mycobacterium tuberculosis permeability prediction tool for small molecules. Bioinformatics 2013, 29, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Larsson, J.; Gottfries, J.; Muresan, S.; Backlund, A. ChemGPS-NP: Tuned for navigation in biologically relevant chemical space. J Nat. Prod. 2007, 70, 789–794. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, L.; Johansson, E.; Kettaneh-Wold, N.; Trygg, J.; Wilkström, C.; Wold, S. Multiand Megavariate Data Analysis Part I Basic Principles and Applications, 2nd ed.; Utmetrics Inc.: Umeå, Sweden, 2006; pp. 1–425. [Google Scholar]
- Rosén, J.; Lövgren, A.; Kogej, T.; Muresan, S.; Gottfries, J.; Backlund, A. ChemGPS-NPweb—Chemical space navigation online. J. Comp. Aid. Mol. Design 2009, 23, 253–259. [Google Scholar] [CrossRef] [PubMed]
- Stanley, S.A.; Grant, S.S.; Kawate, T.; Iwase, N.; Shimizu, M.; Wivagg, C.; Silvis, M.; Kazyanskaya, E.; Aquadro, J.; Golas, A.; et al. Identification of novel inhibitors of M. Tuberculosis growth using whole cell based high-throughput screening. ACS Chem Biol. 2012, 7, 1377–1384. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available from the authors. |


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alajlani, M.M.; Backlund, A. Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning System-Natural Product Analysis. Molecules 2020, 25, 945. https://doi.org/10.3390/molecules25040945
Alajlani MM, Backlund A. Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning System-Natural Product Analysis. Molecules. 2020; 25(4):945. https://doi.org/10.3390/molecules25040945
Chicago/Turabian StyleAlajlani, Muaaz Mutaz, and Anders Backlund. 2020. "Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning System-Natural Product Analysis" Molecules 25, no. 4: 945. https://doi.org/10.3390/molecules25040945
APA StyleAlajlani, M. M., & Backlund, A. (2020). Evaluating Antimycobacterial Screening Schemes Using Chemical Global Positioning System-Natural Product Analysis. Molecules, 25(4), 945. https://doi.org/10.3390/molecules25040945