
Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections


Abstract
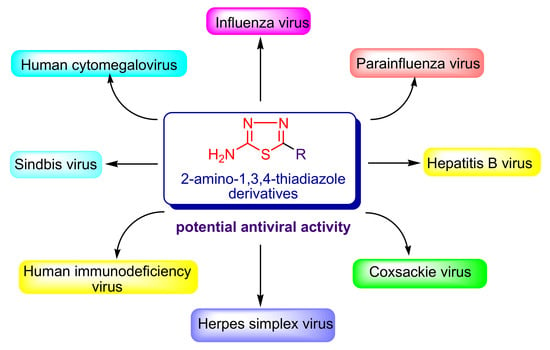
1. Human Viral Infections
2. Nitrogen-Containing Heterocycles and Thiadiazole Ring in Biology and Medicinal Chemistry
3. The Activity of 2-Amino-1,3,4-Thiadiazole Derivatives Against Human Viral Pathogens
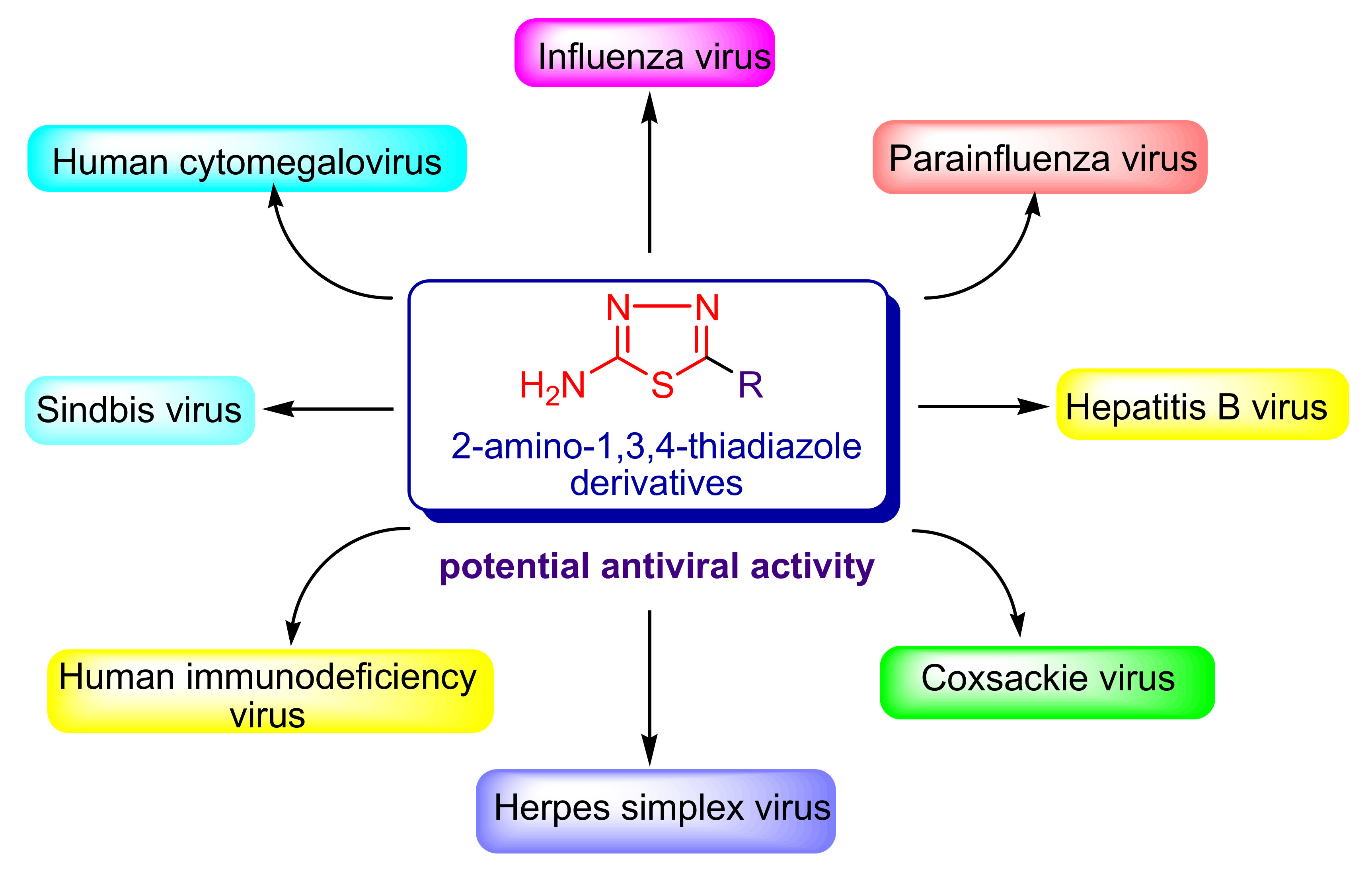
3.1. Human Immunodeficiency Virus (HIV)





3.2. Human Cytomegalovirus (HCMV)


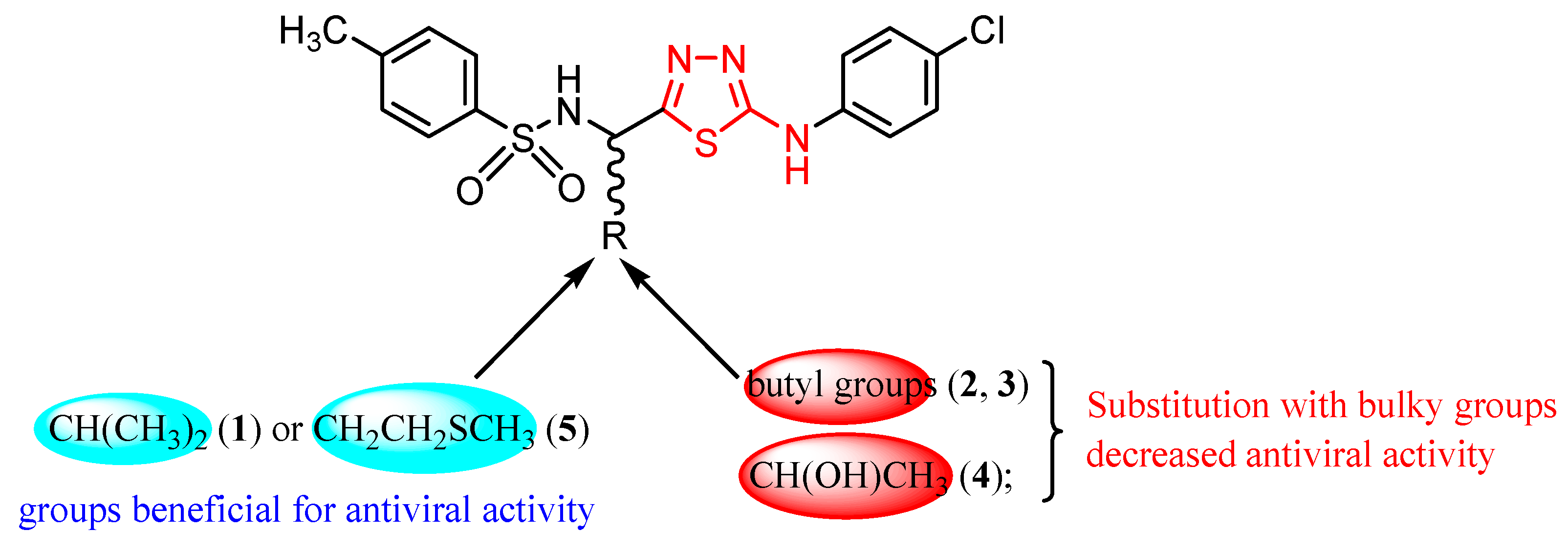
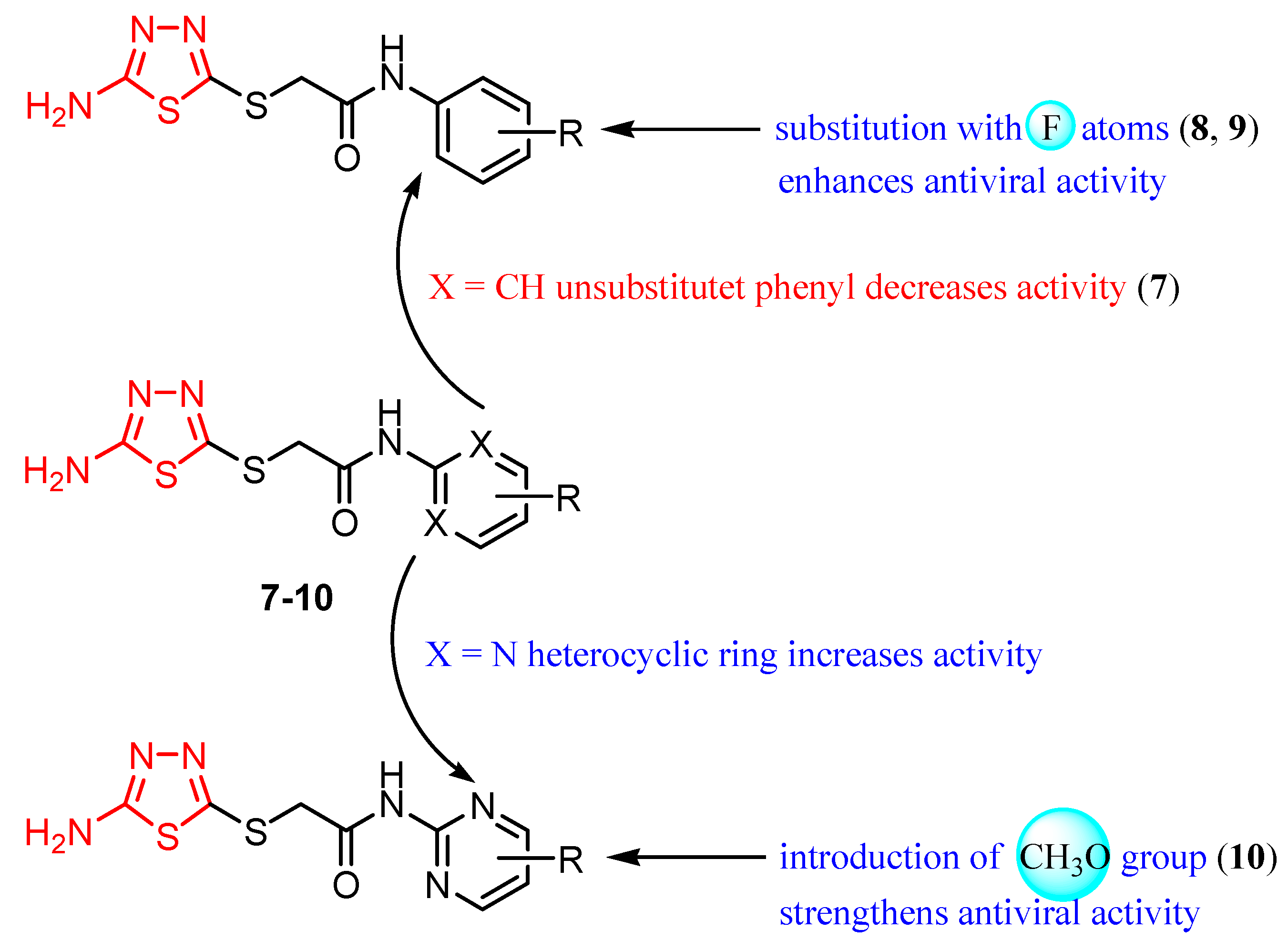
3.3. Respiratory Viruses

3.4. Hepatitis Viruses

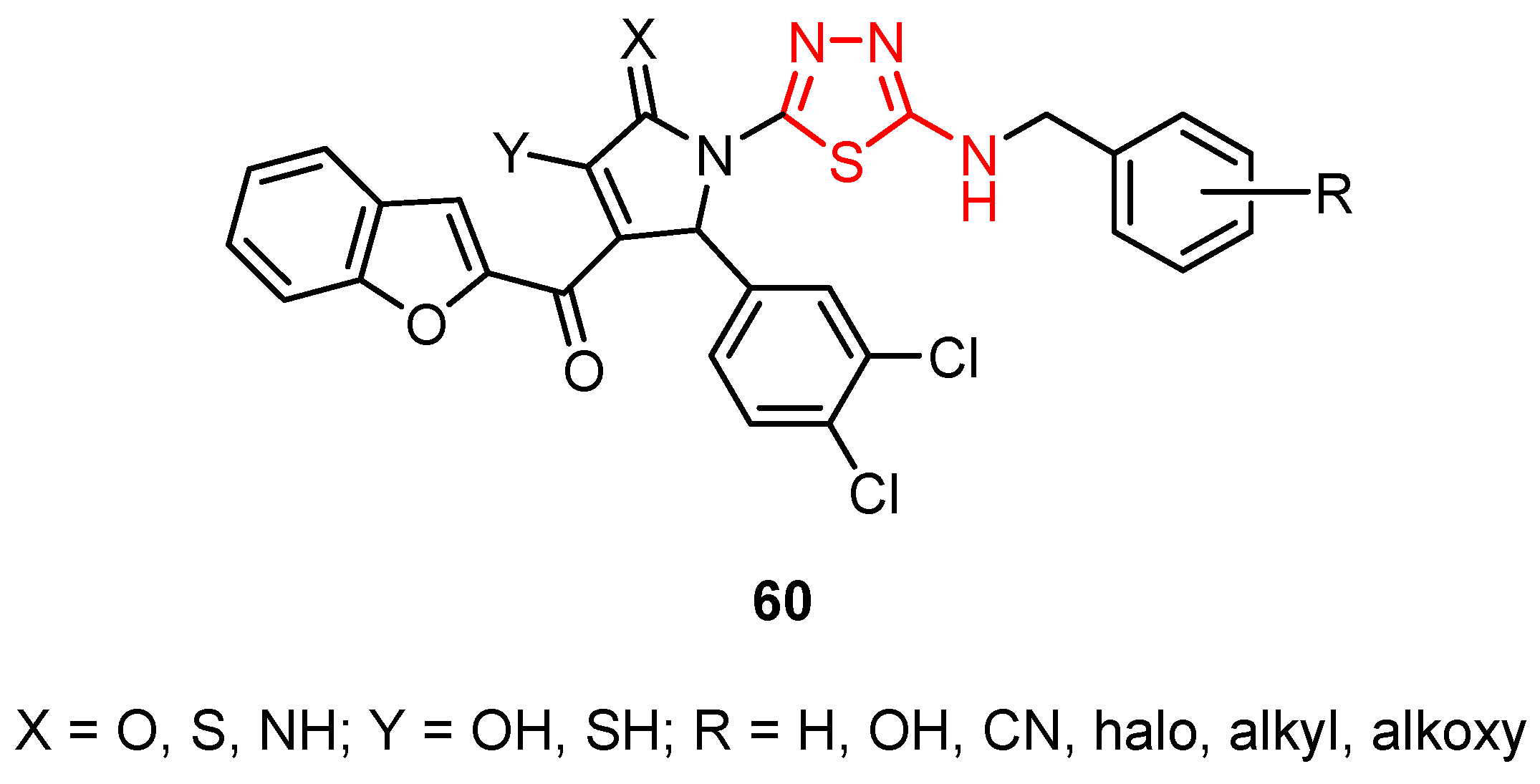
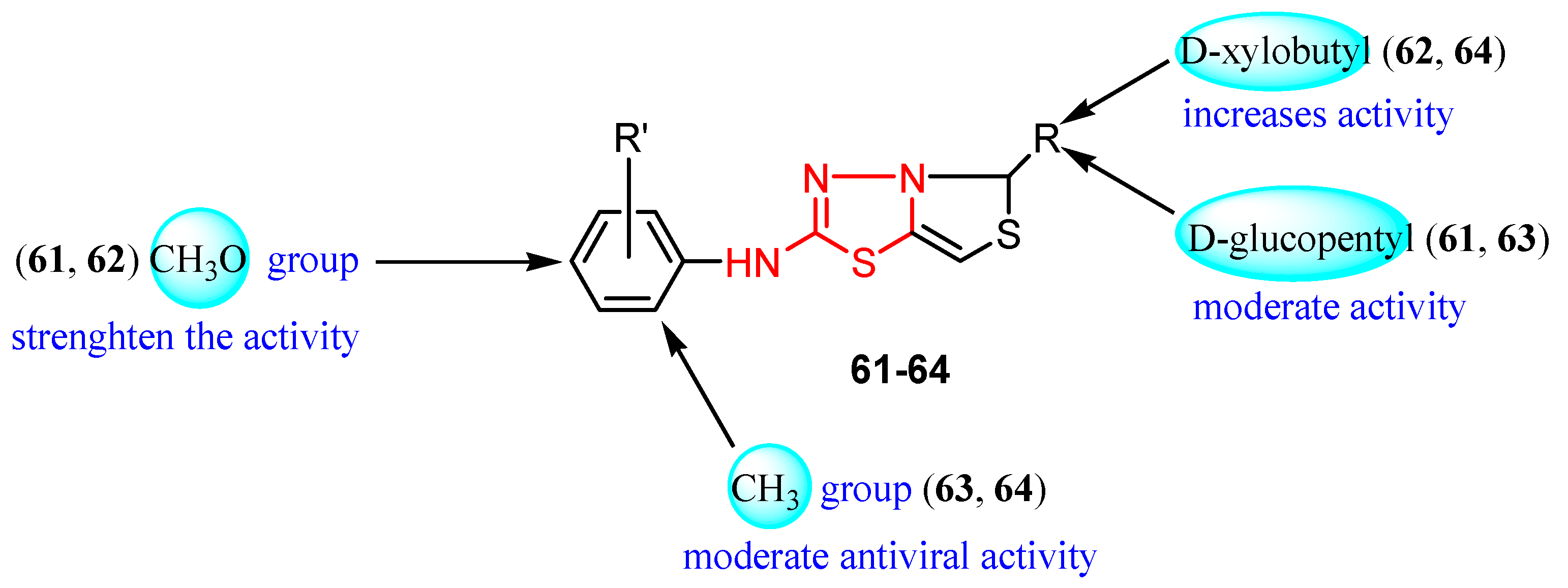
3.5. Miscellaneous Viruses



4. Conclusions
Funding
Conflicts of Interest
References
- Beale, J.M., Jr. Antiviral agents. In Wilson and Gisvold’s Textbook of Organic Medicinal and Pharmaceutical Chemistry, 12th ed.; Beale, J.M., Jr., Block, J.H., Eds.; Lippincott Williams and Wilkins: Baltimore, MD, USA, 2011; pp. 330–336. [Google Scholar]
- Rohwer, F.; Barrot, K. Viral information. Biol. Philos. 2013, 28, 283–297. [Google Scholar] [CrossRef]
- Woster, P.M. Antiviral agents and protease inhibitors. In Foye’s Principles of Medicinal Chemistry, 7th ed.; Lemke, T.L., Williams, D.A., Roche, V.F., Zito, S.W., Eds.; Lippincott Williams and Wilkins, Wolters Kluwer: Baltimore, MD, USA, 2013; pp. 1274–1302. [Google Scholar]
- Berzofsky, J.A.; Ahlers, J.D.; Janik, J.; Morris, J.; Oh, S.K.; Terabe, M.; Belyakov, I.M. Progress on new vaccine strategies against chronic viral infections. J. Clin. Invest. 2004, 114, 450–462. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Human papillomavirus vaccines: WHO position paper, May 2017, Weekly epidemiological record. Available online: http://www.who.int/wer (accessed on 11 January 2020).
- El-Essawy, F.A.; El-Sayed, W.A.; El-Kafrawy, S.A.; Morshedy, A.S.; Abdel-Rahman, A.H. Anti-Hepatitis B virus activity of new 1,2,4-triazol-2-yl- and1,3,4-oxadiazol-2-yl-2-pyridinone derivatives. Z. Naturforsch. 2008, 63c, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Cichero, E. Fight against H1N1 Influenza A virus: recent insights towards the development of druggable compounds. Curr. Med. Chem. 2016, 23, 1802–1817. [Google Scholar] [CrossRef] [PubMed]
- Diaba, F.; Montiel, J.A.; Serban, G.; Bonjoch, J. Synthesis of normorphans through an efficient intramolecular carbamoylation of ketones. Org. Lett. 2015, 17, 3860–3863. [Google Scholar] [CrossRef]
- Serban, G.; Abe, H.; Takeuchi, Y. Synthetic studies of substituted pyridine aldehydes as intermediates for the synthesis of toddaquinoline, its derivatives and other natural products. Heterocycles 2011, 83, 1989–2000. [Google Scholar] [CrossRef]
- Serban, G.; Abe, H.; Takeuchi, Y.; Harayama, T. A new approach to the benzopyridoxepine core by metal mediated intramolecular biaryl ether formation. Heterocycles 2008, 75, 2949–2958. [Google Scholar] [CrossRef]
- Serban, G.; Shigeta, Y.; Nishioka, H.; Abe, H.; Takeuchi, Y.; Harayama, T. Studies toward the synthesis of toddaquinoline by intramolecular cyclization. Heterocycles 2007, 71, 1623–1630. [Google Scholar] [CrossRef]
- Gupta, R. Biological significance of nitrogen containing heterocyclic compounds—a mini review. IJCA 2015, 8, 18–23. [Google Scholar]
- Joule, J.A. Natural products containing nitrogen heterocycles—some highlights 1990–2015. In Advances in Heterocyclic Chemistry: Heterocyclic Chemistry in the 21st Century—A Tribute to Alan Katritzky, 1st ed.; Scriven, E.F.V., Ramsden, C.A., Eds.; Academic Press Elsevier: Cambridge, UK, 2016; Volume 119, pp. 81–106. [Google Scholar]
- Takashima, K.; Hayakawa, D.; Gouda, H.; Toyooka, N. Formal syntheses of (-)-Lepadiformines A, C and (-)-Fasicularin. J. Org. Chem. 2019, 84, 5222–5229. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the structural diversity, substitution patterns, and frequency of nitrogen heterocycles among U.S. FDA approved pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Shukla, P.K.; Verma, A.; Mishra, P. Significance of nitrogen heterocyclic nuclei in the search of pharmacological active compounds. In New Perspective in Agricultural and Human Health; Shukla, R.P., Mishra, R.S., Tripathi, A.D., Yadav, A.K., Tiwari, M., Mishra, R.R., Eds.; Bharti Publication: New Delhi, India, 2017; pp. 100–126. [Google Scholar]
- Rajput, A.P.; Kankhare, A.R. Synthetic utility of azaheterocycles: A short review. Int. J. Pharm. Sci. Invent. 2017, 6, 19–25. [Google Scholar]
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [PubMed]
- Razonable, R.R. Antiviral drugs for viruses other than human immunodeficiency virus. Mayo Clin. Proc. 2011, 86, 1009–1026. [Google Scholar] [CrossRef] [PubMed]
- Serban, G.; Stanasel, O.; Serban, E.; Bota, S. 2-Amino-1,3,4-thiadiazole as a potential scaffold for promising antimicrobial agents. Drug Des. Devel. Ther. 2018, 12, 1545–1566. [Google Scholar] [CrossRef]
- Schenone, S.; Brullo, C.; Bruno, O.; Bondavalli, F.; Ranise, A.; Filippelli, W.; Rinaldi, B.; Capuano, A.; Falcone, G. New 1,3,4-thiadiazole derivatives endowed with analgesic and anti-inflammatory activities. Bioorg Med Chem. 2006, 14, 1698–1705. [Google Scholar] [CrossRef]
- Labanauskas, L.; Kalcas, V.; Udrenaite, E.; Gaidelis, P.; Brukstus, A.; Dauksas, A. Synthesis of 3-(3,4-dimethoxyphenyl)-1H-1,2,4-triazole-5-thiol and 2-amino-5-(3,4-dimethoxyphenyl)-1,3,4-thiadiazole derivatives exhibiting anti-inflammatory activity. Pharmazie 2001, 56, 617–619. [Google Scholar]
- Clerici, F.; Pocar, D.; Guido, M.; Loche, A.; Perlini, V.; Brufani, M. Synthesis of 2-amino-5-sulfanyl-1,3,4-thiadiazole derivatives and evaluation of their antidepressant and anxiolytic activity. J Med Chem. 2001, 44, 931–936. [Google Scholar] [CrossRef]
- Basso, A.; Liu, M.; Dai, C.; Gray, K.; Nale, L.; Tevar, S.; Lee, S.; Liang, L.; Ponery, A.; Yaremko, B.; et al. SCH 2047069, a novel oral kinesin spindle protein inhibitor, shows single-agent antitumor activity and enhances the efficacy of chemotherapeutics. Mol. Cancer Ther. 2019, 9, 2993–3002. [Google Scholar] [CrossRef]
- Dawood, K.M.; Farghaly, T.A. Thiadiazole inhibitors: A patent review. Expert Opin. Ther. Pat. 2017, 27, 477–505. [Google Scholar] [CrossRef]
- Haider, S.; Alam, M.S.; Hamid, H. 1,3,4-Thiadiazoles: a potent multi targeted pharmacological scaffold. Eur. J. Med. Chem. 2015, 92, 156–177. [Google Scholar] [CrossRef] [PubMed]
- Serban, G. Future prospects in the treatment of parasitic diseases: 2-amino-1,3,4-thiadiazoles in leishmaniasis. Molecules 2019, 24, 1557. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Geng, J.; Liu, Y.; Yu, S.; Zhao, G. Thiadiazole—A promising structure in Medicinal Chemistry. ChemMedChem 2013, 8, 27–41. [Google Scholar] [CrossRef] [PubMed]
- Senff-Ribeiro, A.; Echevarria, A.; Silva, E.F.; Franco, C.R.C.; Veiga, S.S.; Oliveira, M.B.M. Cytotoxic effect of a new 1,3,4-thiadiazolium mesoionic compound (MI-D) on cell lines of human mellanoma. Br. J. Cancer 2004, 91, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Wermuth, C.G. Are pyridazines privileged structures? Med.Chem.Commun. 2011, 2, 935–941. [Google Scholar] [CrossRef]
- Biziere, K.; Worms, P.; Kan, J.P.; Mandel, P.; Garattini, S.; Roncucci, R. Minaprine, a new drug with antidepressant properties. Drugs Exp. Clin. Res. 1985, 11, 831–840. [Google Scholar] [PubMed]
- Ueno, S.; Bracamontes, J.; Zorumski, C.; Weiss, D.S.; Steinbach, J.H. Bicuculline and gabazine are allosteric inhibitors of channel opening of the GABA A receptor. J. Neurosci. 1997, 17, 625–634. [Google Scholar] [CrossRef] [PubMed]
- Holla, B.S.; Poorjary, K.N.; Rao, B.S.; Shivananda, M.K. New bis-aminomercaptotriazoles and bis-triazolothiadiazoles as possible anticancer agents. Eur. J. Med. Chem. 2002, 37, 511–517. [Google Scholar] [CrossRef]
- Yousif, E.; Majeed, A.; Al-Sammarrae, K.; Salih, N.; Salimon, J.; Abdullah, B. Metal complexes of Schiff base: preparation, characterization and antibacterial activity. Arabian J. Chem. 2013, 5. [Google Scholar] [CrossRef]
- Juszczak, M.; Matysiak, J.; Brzana, W.; Niewiadomy, A.; Rzeski, W. Evaluation of antiproliferative activity of 2-(monohalogenophenylamino)-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazoles. Arzneim. Forsch. Drug Res. 2008, 58, 353–357. [Google Scholar] [CrossRef]
- Matysiak, J. Evaluation of antiproliferative effect in vitro of some 2-amino-5-(2,4-dihydroxyphenyl)-1,3,4-thiadiazole derivatives. Chem. Pharm. Bull. 2006, 54, 988–991. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Yoosefian, M.; Chermahini, Z.J.; Raissi, H.; Mola, A.; Sadeghi, M. A theoretical study on the structure of 2-amino-1,3,4-thiadiazole and its 5-substituted derivatives in the gas phase, water, THF and DMSO solutions. J. Mol. Liq. 2015, 203, 137–142. [Google Scholar] [CrossRef]
- Serban, G. 2-Amino-1,3,4-thiadiazoles as prospective agents in trypanosomiasis and other parasitoses. Acta Pharm. 2020, 70, 259–290. [Google Scholar] [CrossRef] [PubMed]
- Serban, G. 5-Arylamino-1,3,4-thiadiazol-2-yl acetic acid esters as intermediates for the synthesis of new bisheterocyclic compounds. Farmacia 2015, 63, 146–149. [Google Scholar]
- Serban, G.; Coman, M.; Curea, E. Synthesis of some heterocyclic nitro–coumarins by Knoevenagel condensation. Farmacia 2005, 53, 78–84. [Google Scholar]
- Serban, G.; Matinca, D.; Bradea, O.; Gherman, L.; Coman, M.; Curea, E. The study of the biological activity of some heterocyclic coumarins. Farmacia 2005, 53, 91–99. [Google Scholar]
- Serban, G.; Suciu, A.; Coman, M.; Curea, E. Synthesis and physical-chemical study of some 3-(5-arylamino-1,3,4-thiadiazol-2-yl)cou–marins. Farmacia 2002, 50, 50–54. [Google Scholar]
- Horvath, T.; Serban, G.; Cuc, S. Synthesis of new 2-phenylamino-5-[(α-acylamino)-p-X-stiryl] -1,3,4-thiadiazole compounds. Farmacia 2014, 62, 422–427. [Google Scholar]
- Serban, G.; Coman, M.; Curea, E.; Proinov, L. Synthesis and description of some heterocyclic coumarins. Farmacia 2001, 49, 45–52. [Google Scholar]
- Global AIDS Update 2016. Joint United Nations Programme on HIV/AIDS, Geneva, Switzerland. Available online: https://www.unaids.org/sites/default/files/media_asset/global-AIDS-update-2016en.pdf (accessed on 17 July 2018).
- Sharma, P.C.; Sinhmar, A.; Sharma, A.; Rajak, H.; Pathak, D.P. Medicinal significance of benzothiazole scaffold: An insight view. J. Enzyme Inhib. Med. Chem. 2013, 28, 240–266. [Google Scholar] [CrossRef]
- Xiaohe, Z.; Yu, Q.; Hong, Y.; Xiuqing, S.; Rugang, Z. Synthesis, biological evaluation and molecular modeling studies of N-aryl-2-arylthioacetamides as non-nucleoside HIV-1 reverse transcriptase inhibitors. Chem. Biol. Drug Des. 2010, 76, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Harms, A. Non-nucleoside reverse transcriptase inhibitors. In The art of drug synthesis; Johnson, D.S., Li, J.J., Eds.; Wiley Interscience, John Wiley and Sons, Inc., Hoboken: New Jersey, NJ, USA, 2007; p. 84. [Google Scholar]
- Tatar, E.; Kucukguzel, S.G.; Karakus, S.; De Clercq, E.; Andrei, G.; Snoeck, R.; Pannecouque, C.; OktemOkullu, S.; Unubol, N.; Kocagoz, T.; et al. Synthesis and biological evaluation of some new 1,3,4-thiadiazole and 1,2,4-triazole derivatives from L-methionine as antituberculosis and antiviral agents. Marmara Pharm. J. 2015, 19, 88–102. [Google Scholar] [CrossRef]
- Wit, F.W.N.M.; Lange, J.M.A.; Volberding, P.A. New HIV Drug Development. In Global HIV/AIDS Medicine; Volberding, P.A., Sande, M.A., Greene, W.C., Lange, J.M.A., Eds.; Elsevier: Philadelphia, PA, USA, 2008; pp. 123–134. [Google Scholar]
- Schiller, D.S.; Youssef-Bessler, M. Etravirine: A second-generation nonnucleoside reverse transcriptase inhibitor (NNRTI) active against NNRTI-resistant strains of HIV. Clin. Ther. 2009, 31, 692–704. [Google Scholar] [CrossRef] [PubMed]
- Miller, C.D.; Crain, J.; Tran, B.; Patel, N. Rilpivirine: A new addition to the anti-HIV-1 armamentarium. Drugs Today 2011, 47, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Doravirine combination pill looks good for initial HIV treatment. Available online: https://www.aidsmap.com/news/jul-2017/doravirine-combination-pill-looks-good-initial-hiv-treatment; http://www.aidsmap.com/Doravirine-combination-pill-looks-good-for-initial-HIV-treatment/pagee3160520 (accessed on 5 June 2018).
- Gatell, J.M.; Raffi, F.; Plettenberg, A.; Smith, D.; Portilla, J.; Hoffmann, C.; Arasteh, K.; Thompson, M.; Xu, X.; Teppler, H. Doravirine 100 mg QD vs Efavirenz + TDF/FTC in ART-Naive HIV+ Patients: Week 48 Results. In Proceedings of the Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 22–25 February 2016. abstract number 470. [Google Scholar]
- FDA Accepts New Drug Applications for Merck’s Doravirine, the Company’s Investigational Non-Nucleoside Reverse Transcriptase Inhibitor (NNRTI), for Treatment of HIV-1 Infection. Available online: http://investors.merck.com/news/press-release-details/2018/FDA-Accepts-New-Drug-Applications-for-Mercks-Doravirine-the-Companys-Investigational-Non-Nucleoside-Reverse-Transcriptase-Inhibitor-NNRTI-for-Treatment-of-HIV-1-Infection/default.aspx (accessed on 5 June 2018).
- FDA approves Merck’s Pifeltro (doravirine). Available online: https://www.drugs.com/newdrugs/ fda-approves-merck-s-pifeltro-doravirine-hiv-1-appropriate-patients-4815.html (accessed on 6 November 2019).
- Merck’s Pifeltro (doravirine) and Delstrigo (doravirine/lamivudine/tenofovir disoproxil fumarate) receive FDA approval for use in appropriate adults living with HIV-1 who are virologically suppressed. 20 September 2019. Available online: https://investors.merck.com/news/press-release-details/2019/Mercks-PIFELTRO-doravirine-and-DELSTRIGO-doravirinelamivudinetenofovir-disoproxil-fumarate-Receive-US-FDA-Approval-for-Use-in-Appropriate-Adults-Living-with-HIV-1-Who-Are-Virologically-Suppressed/default.aspx (accessed on 6 November 2019).
- Margolis, D.A.; Eron, J.J.; DeJesus, E.; White, S.; Wannamaker, P.; Stancil, B.; Johnson, M. Unexpected finding of delayed-onset seizures in HIV-positive, treatment-experienced subjects in the Phase IIb evaluation of fosdevirine (GSK2248761). Antivir. Ther. 2014, 19, 69–78. [Google Scholar] [CrossRef]
- Vernazza, P.; Wang, C.; Pozniak, A.; Weil, E.; Pulik, P.; Cooper, D.A.; Kaplan, R.; Lazzarin, A.; Valdez, H.; Goodrich, J.; et al. Efficacy and safety of lersivirine (UK-453,061) versus efavirenz in antiretroviral treatment-naive HIV-1 infected patients: week 48 primary analysis results from an ongoing, multicenter, randomized, double-blind, Phase IIb trial. J. Acquir. Immune Defic. Syndr. 2013, 62, 171–179. [Google Scholar] [CrossRef]
- Platten, M.; Fatkenheuer, G. Lersavirine—a new drug for HIV infection therapy. Expert. Opin. Investig. Drugs 2013, 22, 1687–1694. [Google Scholar] [CrossRef]
- Sacks, D.; Ledwaba, J.; Morris, L.; Hunt, G.M. Rapid detection of common HIV-1 drug resistance mutations by use of high-resolution melting analysis and unlabeled probes. J. Clin. Microbiol. 2017, 55, 122–133. [Google Scholar] [CrossRef][Green Version]
- Yahi, N.; Tamalet, C.; Tourres, C.; Tivoli, N.; Ariasi, F.; Volot, F.; Gastaut, J.A.; Gallais, H.; Moreau, J.; Fantini, J. Mutation patterns of the reverse transcriptase and protease genes in Human Immunodeficiency Virus type 1-infected patients undergoing combination therapy: survey of 787 sequences. J. Clin. Microbiol. 1999, 37, 4099–4106. [Google Scholar] [CrossRef]
- Hu, Y.; Li, C.Y.; Wang, X.M.; Yang, Y.H.; Zhu, H.L. 1,3,4-Thiadiazole: synthesis, reactions and applications in medicinal, agricultural, and materials chemistry. Chem. Rev. 2014, 114, 5572–5610. [Google Scholar] [CrossRef]
- Szulczyk, D.; Tomaszewski, P.; Jozwiak, M.; Koziol, A.E.; Lis, T.; Collu, D.; Iuliano, F.; Struga, M. Synthesis and biological activities of ethyl 2-(2-pyridylacetate) derivatives containing thiourea, 1,2,4-triazole, thiadiazole and oxadiazole moieties. Molecules 2017, 22, 409. [Google Scholar] [CrossRef] [PubMed]
- Akhtar, T.; Hameed, S.; Al-Masoudi, N.A.; Khan, K.M. Synthesis and anti-HIV activity of new chiral 1,2,4-triazoles and 1,3,4-thiadiazoles. Heteroatom Chem. 2007, 18, 316–322. [Google Scholar] [CrossRef]
- Hamad, N.S.; Al-Haidery, N.H.; Al-Masoudi, I.A.; Sabri, M.; Sabri, L.; Al-Masoudi, N.A. Amino acid derivatives, part 4: synthesis and anti-HIV activity of new naphthalene derivatives. Arch. Pharm. Chem. Life Sci. 2010, 343, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wu, B.; Kuhen, K.L.; Bursulaya, B.; Nguyen, T.N.; Nguyen, D.G.; He, Y. Synthesis and biological evaluations of sulfanyltriazoles as novel HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2006, 16, 4174–4177. [Google Scholar] [CrossRef] [PubMed]
- Muraglia, E.; Kinzel, O.D.; Laufer, R.; Miller, M.D.; Moyer, G.; Munshi, V.; Orvieto, F.; Palumbi, M.C.; Pescatore, G.; Rowley, M.; et al. Tetrazole thioacetanilides: potent non-nucleoside inhibitors of WT HIV reverse transcriptase and its K103N mutant. Bioorg. Med. Chem. Lett. 2006, 16, 2748–2752. [Google Scholar] [CrossRef] [PubMed]
- Zhan, P.; Liu, X.; Cao, Y.; Wang, Y.; Pannecouque, C.; De Clercq, E. 1,2,3-Thiadiazole thioacetanilides as a novel class of potent HIV-1 non-nucleoside reverse transcriptase inhibitors. Bioorg. Med. Chem. Lett. 2008, 18, 5368–5371. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Xu, W.; Koh, Y.H.; Shim, J.H.; Girardet, J.L.; Yeh, L.T.; Hamatake, R.K.; Hong, Z. A novel non-nucleoside analogue that inhibits human immunodeficiency virus type 1 isolates resistant to current non-nucleoside reverse transcriptase inhibitors. Antimicrob. Agents Chemother. 2007, 51, 429–437. [Google Scholar] [CrossRef]
- Huang, Y.; Zhao, L.; Jia, B.; Wu, L.; Li, Y.; Curthoys, N.; Zheng, J.C. Glutaminase dysregulation in HIV-1-infected human microglia mediates neurotoxicity: relevant to HIV-1 associated neurocognitive disorders. J. Neurosci. 2011, 31, 15195–15204. [Google Scholar] [CrossRef]
- Zhao, J.; Lopez, A.L.; Erichsen, D.; Herek, S.; Cotter, R.L.; Curthoys, N.P.; Zheng, J. Mitochondrial glutaminase enhances extracellular glutamate production in HIV-1-infected macrophages: Linkage to HIV-1 associated dementia. J. Neurochem. 2004, 88, 169–180. [Google Scholar] [CrossRef]
- Stumvoll, M.; Perriello, G.; Meyer, C.; Gerich, J. Role of glutamine in human carbohydrate metabolism in kidney and other tissues. Kidney Int. 1999, 55, 778–792. [Google Scholar] [CrossRef]
- Smith, R.J. Glutamine metabolism and its physiological importance. J. Parenter. Enteral. Nutr. 1990, 14, 40S–44S. [Google Scholar] [CrossRef] [PubMed]
- Bromley, S.D.; Bennett, M.K.; Gross, M.I.; Li, J.; Chen, L.; Goyal, B.; Laidig, G.; Stanton, T.F.; Sjogren, E.B. Treatment of viral infections with inhibitors of glutaminase. PCT Int. Appl. PCT/US2014/061746; WO 2015/061432 A1. 2015. [Google Scholar]
- Newsholme, P.; Lima, M.M.R.; Procopio, J.; Pithon-Curi, T.C.; Doi, S.Q.; Bazotte, R.B.; Curi, R. Glutamine and glutamate as vital metabolites. Braz. J. Med. Biol. Res. 2003, 36, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Behar, K.L.; Rothman, D.L. In vivo NMR studies of glutamate-GABA-glutamine cycling in rodent and human cortex: the central role of glutamine. J. Nutr. 2001, 131, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Erdmann, N.; Zhao, J.; Lopez, A.L.; Herek, S.; Curthoys, N.; Hexum, T.D.; Tsukamoto, T.; Ferraris, D.; Zheng, J. Glutamate production by HIV-1 infected human macrophage is blocked by the inhibition of glutaminase. J. Neurochem. 2007, 102, 539–549. [Google Scholar] [CrossRef]
- Babayo, A.; Thairu, Y.; Nasir, I.A.; Baba, M.M. Serological evidence and sociodemographic risk factors of recent cytomegalovirus infection in pregnant women attending a tertiary hospital in Maiduguri, Nigeria. J. Med. Microbiol. Infec. Dis. 2014, 2, 49–55. [Google Scholar]
- Forbes, B.A. Acquisition of cytomegalovirus infection: An update. Clin. Microbiol. Rev. 1989, 2, 204–216. [Google Scholar] [CrossRef]
- Spruance, S.L. Viral infections. In The Merck Manual of Medical Information: Second Home Edition; Beers, M.H., Ed.; Merck Research Laboratories, Merck and Co., Inc.: Whitehouse Station, NJ, USA, 2003; p. 1164. [Google Scholar]
- Centers for Disease Control and Prevention. Cytomegalovirus (CMV) and Congenital CMV Infection. Available online: https://www.cdc.gov/cmv/overview.html (accessed on 11 July 2018).
- Tan, B.H. Cytomegalovirus treatment. Curr. Treat. Options Infect. Dis. 2014, 6, 256–270. [Google Scholar] [CrossRef]
- McGavin, J.K.; Goa, K.L. Ganciclovir: An update of its use in the prevention of cytomegalovirus infection and disease in transplant recipients. Drugs 2001, 61, 1153–1183. [Google Scholar] [CrossRef]
- Sakamoto, H.; Hirano, M.; Nose, K.; Ueno, S.; Oki, T.; Sugimoto, K.; Nishioka, T.; Kusunoki, S.; Nakamura, Y. A case of severe ganciclovir-induced encephalopathy. Case Rep. Neurol. 2013, 5, 183–186. [Google Scholar] [CrossRef]
- Thaisrivongs, S.; Turner, S.R. 1,3,4-Thiadiazoles useful for the treatment of CMV infections. US Patent 6,150,385, 2000. [Google Scholar]
- World Health Organization. Battle against Respiratory Viruses (BRaVe) initiative. Available online: http://www.who.int/influenza/patient_care/clinical/ brave/en/ (accessed on 14 June 2018).
- World Health Organization. Research needs for the battle against respiratory viruses (BRAve). Background document 2013. Available online: http://www.who.int/influenza/patient_care/clinical/BRaVe _ Research_Agenda _ 2013.pdf (accessed on 14 June 2018).
- Bellos, A.; Mulholland, K.; O’Brien, K.L.; Qazi, S.A.; Gayer, M.; Checchil, F. The burden of acute respiratory infections in crisis-affected populations: A systematic review. Confl. Health 2010, 4, 1–12. [Google Scholar] [CrossRef]
- Straliotto, S.M.; Siqueira, M.M.; Muller, R.L.; Fischer, G.B.; Cunha, M.L.T.; Nestor, S.M. Viral etiology of acute respiratory infections among children in Porto Alegre, RS, Brazil. Rev. Soc. Bras. Med. Tro. 2002, 35, 283–291. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.C.; da Silva Mendes, G.; Rojas, M.A.; Amorim, A.R.; Couceiro, J.N.; Lupi, O.; Elabras, J.; Pires, G.; Valle, S.; Santos, N. Frequency of viral etiology in symptomatic adult upper respiratory tract infections. Braz. J. Infect. Dis. 2015, 19, 30–35. [Google Scholar] [CrossRef] [PubMed]
- Fendrick, A.M.; Monto, A.S.; Nightengale, B.; Sarnes, M. The economic burden of non-influenza-related viral respiratory tract infections in the United States. Arch. Intern. Med. 2003, 163, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.S.; Li, J.J. Neuraminidase inhibitors for influenza: oseltamivir phosphate (Tamiflu) and zanamivir (Relenza). In The art of drug synthesis; Johnson, D.S., Li, J.J., Eds.; Wiley Interscience, John Wiley and Sons, Inc., Hoboken: New Jersey, NJ, USA, 2007; pp. 95–96. [Google Scholar]
- Al-Muharrmi, Z. Understanding the Influenza A H1N1 2009 pandemic. SQU Med. J. 2010, 10, 187–195. [Google Scholar]
- Tonelli, M.; Naesens, L.; Gazzarrini, S.; Santucci, M.; Cichero, E.; Tasso, B.; Moroni, A.; Costi, M.P.; Loddo, R. Host dihydrofolate reductase (DHFR)-directed cycloguanil analogues endowed with activity against influenza virus and respiratory syncytial virus. Eur. J. Med. Chem. 2017, 135, 467–478. [Google Scholar] [CrossRef]
- Francesconi, V.; Giovannini, L.; Santucci, M.; Cichero, E.; Costi, M.P.; Naesens, L.; Giordanetto, F.; Tonelli, M. Synthesis, biological evaluation and molecular modeling of novel azaspiro dihydrotriazines as influenza virus inhibitors targeting the host factor dihydrofolate reductase (DHFR). Eur. J. Med. Chem. 2018, 155, 229–243. [Google Scholar] [CrossRef]
- World Health Organization. Fact sheets on sustainable development goals: health target, Viral hepatitis, World Health Organization 2017, Copenhagen, Denmark, www.euro.who.int/sdgs. Available online: http://www.euro.who.int/__data/assets/pdf_file/0003/348222/Fact-sheet-SDG-viral-hepatitis-FINAL-en.pdf?ua=1 (accessed on 23 January 2020).
- World Health Organization. Hepatitis B in the WHO European Region, Fact sheet – July 2019, World Health Organization 2019. Available online: http://www.euro.who.int/__data/assets/pdf_file/0007/ 377251/Fact-Sheet-Hepatitis-B_2019-ENG.pdf?ua=1 (accessed on 23 January 2020).
- FDA. Hepatitis B and C treatments. Available online: https://www.fda.gov/patients/hepatitis-b-c/ hepatitis-b-and-c-treatments (accessed on 23 January 2020).
- Kurkela, S.; Ratti, O.; Huhtamo, E.; Uzcategui, N.; Nuorti, J.P.; Laakkonen, J.; Manni, T.; Helle, P.; Vaheri, A.; Vapalahti, O. Sindbis virus infection in resident birds, migratory birds, and humans, Finland. Emerg. Infect. Dis. 2008, 14, 41–47. [Google Scholar] [CrossRef]
- Kucukguzel, S.G.; Kucukguzel, I.; Tatar, E.; Rollas, S.; Sahin, F.; Gulluce, M.; De Clercq, E.; Kabasakal, L. Synthesis of some novel heterocyclic compounds derived from diflunisal hydrazide as potential anti-infective and anti-inflammatory agents. Eur. J. Med. Chem. 2007, 42, 893–901. [Google Scholar] [CrossRef]
- Bonina, L.; Orzalesi, G.; Merendino, R.; Arena, A.; Mastroeni, P. Structure-activity relationships of new antiviral compounds. Antimicrob. Agents Chemother. 1982, 22, 1067–1069. [Google Scholar] [CrossRef][Green Version]
- Streissle, G.; Paesseus, A.; Oediger, H. New antiviral compounds. In Advances in Virus Research; Maramorosch, K., Murphy, F.A., Shatkin, A.J., Eds.; Academic Press., Elsevier: London, UK, 1985; Volume 30, p. 115. [Google Scholar]
- Cui, T.; Chum, M.P.; Lam, Y.; Gao, Y. Compounds for use as anti-viral agents. Singapore Pat. Appl. SG 162629 A1 20100729. 2010. [Google Scholar]
- Yadav, L.D.S.; Singh, S. Synthesis of antiviral acyclic C-nucleosides incorporating thiazolo-1,3,4-oxa(thia)diazole or thiazolo-1,2,4-triazole structure as a nucleobase. Indian J. Chem. 2001, 40B, 440–442. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
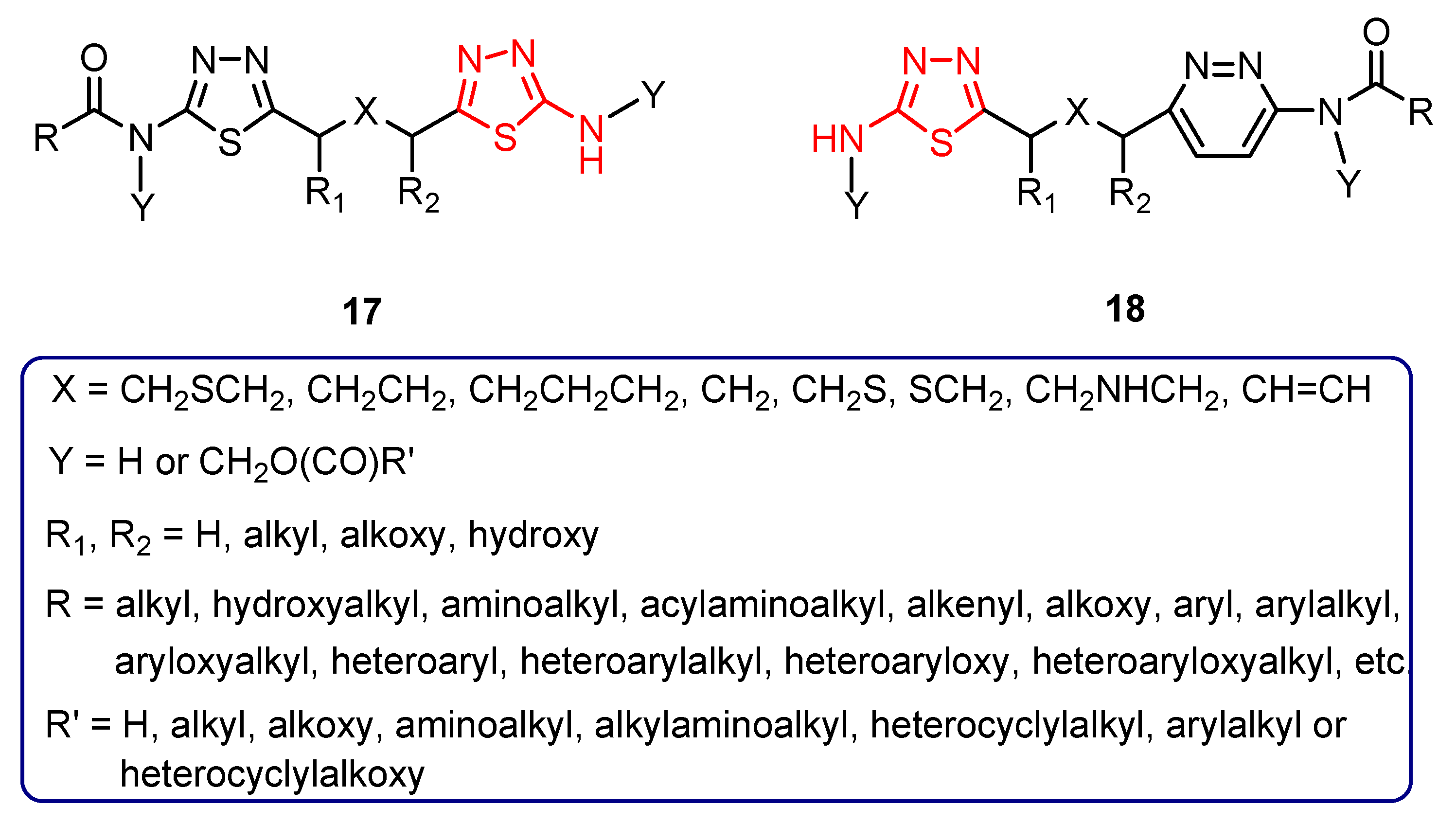{kind=link}
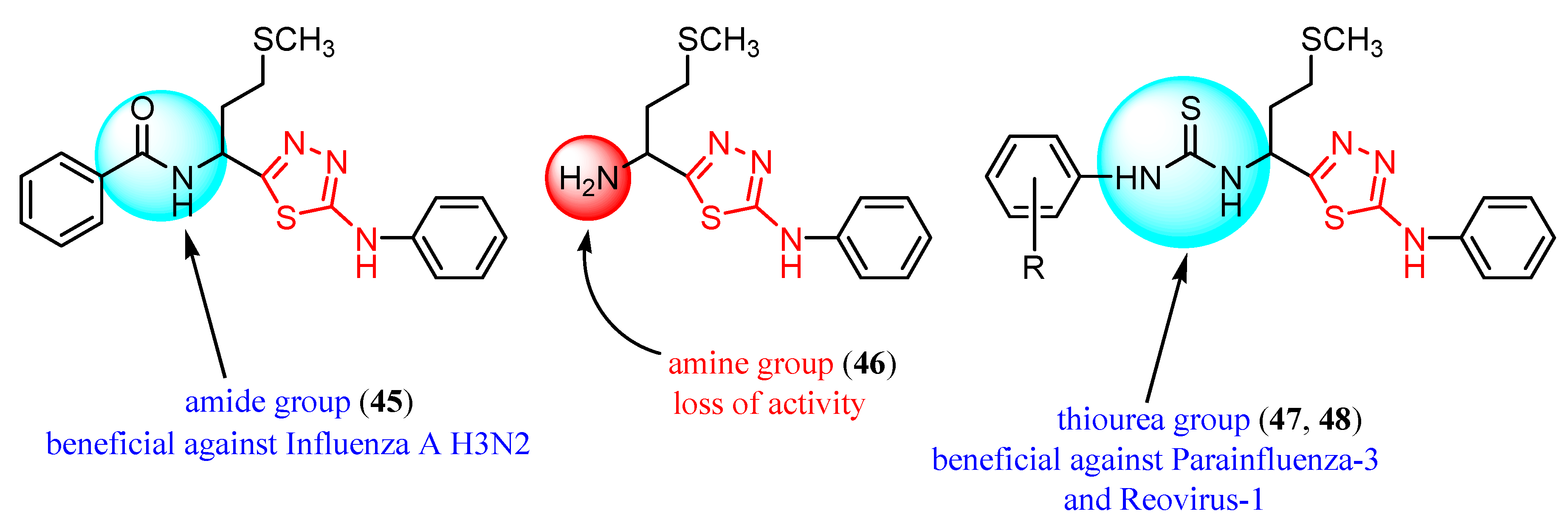{kind=link}
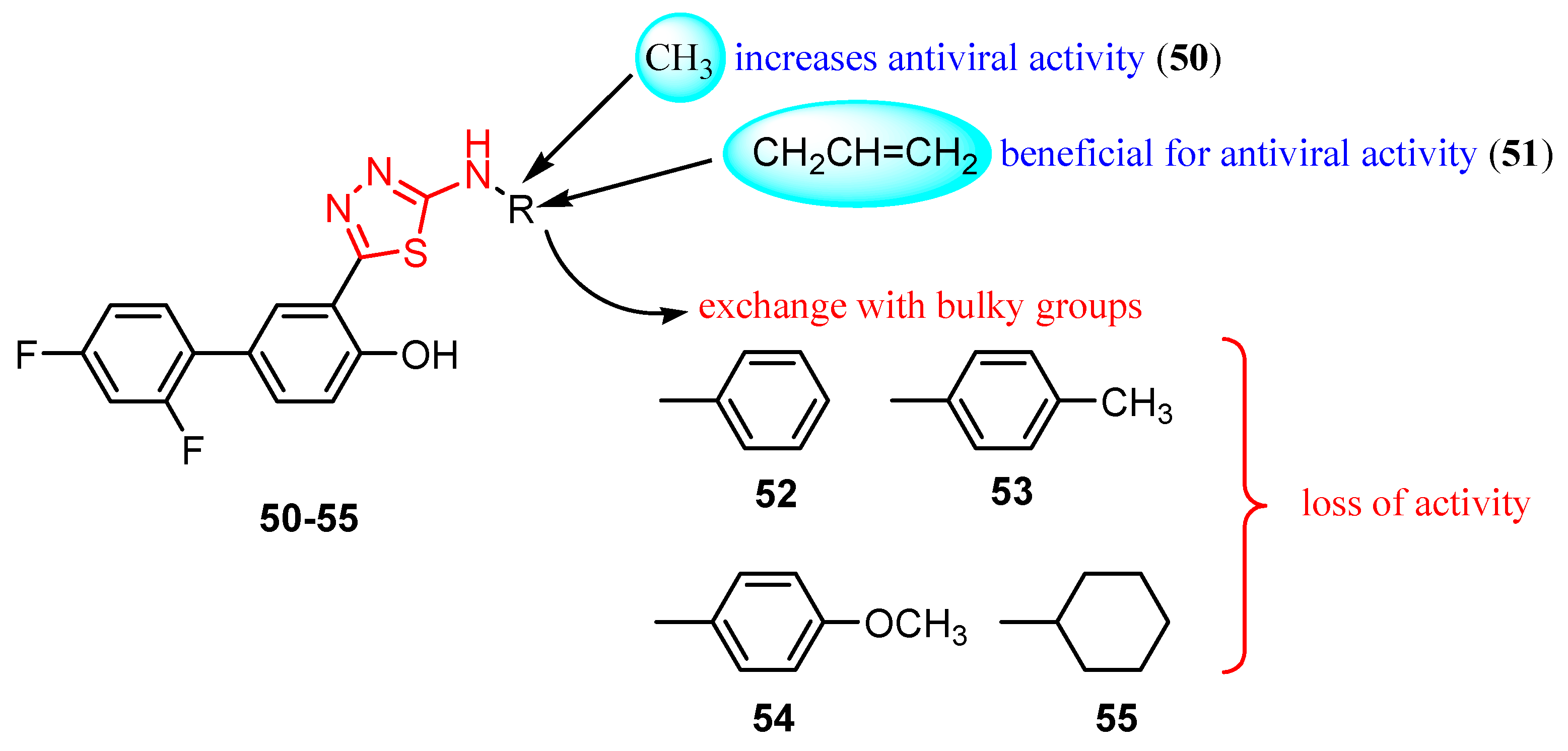{kind=link}
{kind=link}
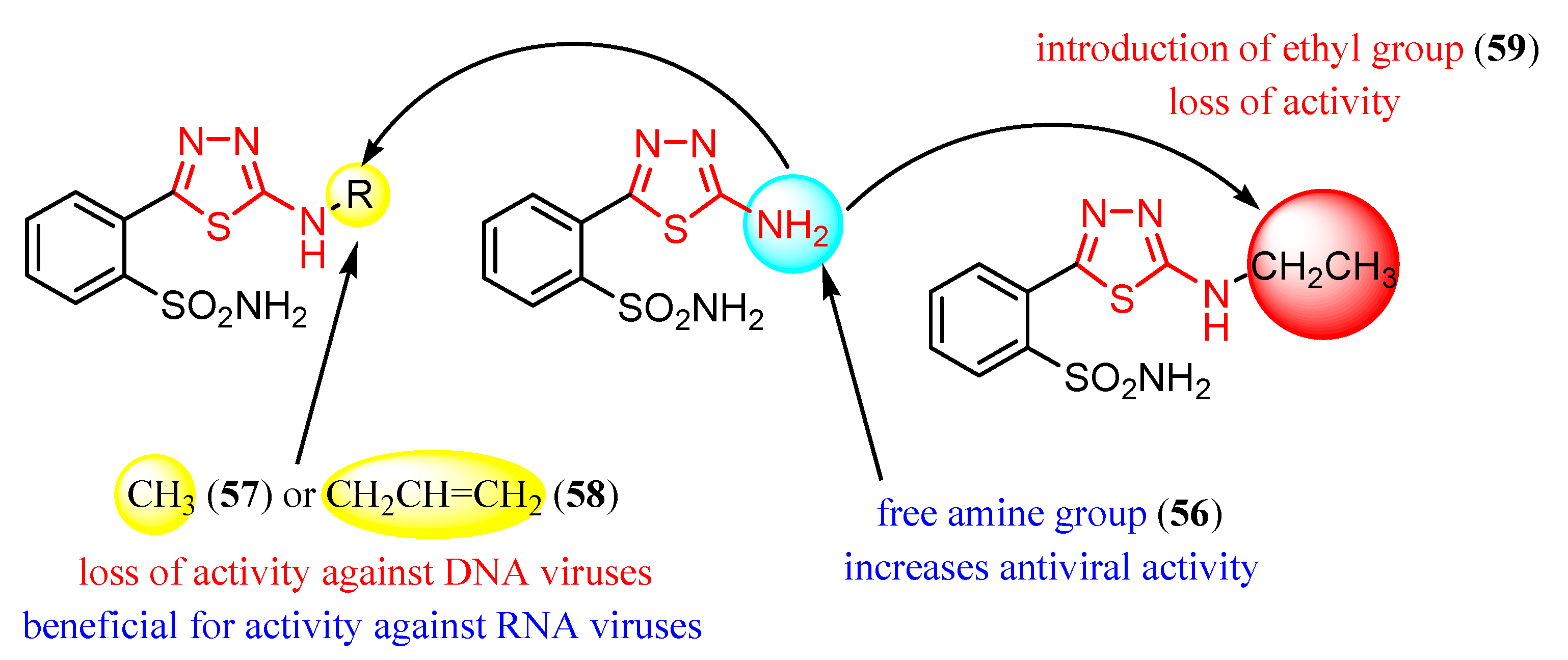{kind=link}
{kind=link}
{kind=link}
| X | R | IC50 (μM) | |
|---|---|---|---|
| 7 | CH | H | 20.83 ± 1.17 |
| 8 | CH | 2,5-F2 | 16.10 ± 0.24 |
| 9 | CH | 3,5-(CF3)2 | 14.93 ± 0.84 |
| 10 | N | 4,6-(OCH3)2 | 7.50 ± 1.06 |
| R | Inhibition (%) | R | Inhibition (%) | ||
|---|---|---|---|---|---|
| 21 | (CH2)10CH3 | 100 | 26 |  | 99.8 |
| 22 |  | 100 | 27 |  | 99.8 |
| 23 |  | 100 | 28 |  | 99.7 |
| 24 |  | 99.9 | 29 |  | 99.6 |
| 25 |  | 99.8 | 30 |  | 99.5 |
| R | Inhibition (%) | R | Inhibition (%) | ||
|---|---|---|---|---|---|
| 31 |  | 100 | 35 |  | 97.7 |
| 32 |  | 99.9 | 36 |  | 97.0 |
| 33 |  | 99 | 37 |  | 92.6 |
| 34 |  | 98.3 | 38 |  | 91.8 |
| R | Inhibition (%) | R | Inhibition (%) | ||
|---|---|---|---|---|---|
| 39 |  | 93.1 | 41 |  | 83.9 |
| 40 |  | 92.9 | 42 |  | 83.1 |
| Influenza A H1N1 | Influenza A H3N2 | Para Influenza-3 | Reovirus-1 | Feline Coronavirus | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | CC50 (μM) | EC50 (μM) | CC50 (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | CC50 (μM) | |
| 45 | 42 | > 100 | 31.4 | > 100 | > 20 | ≥ 20 | > 20 | ≥ 20 | > 100 | > 100 |
| 46 | - | 23 | - | 23 | > 100 | >100 | > 100 | > 100 | > 100 | > 100 |
| 47 | - | 79 | - | 79 | > 20 | 100 | > 20 | 100 | > 100 | > 100 |
| 48 | - | 2.7 | - | 2.7 | > 4 | 20 | > 4 | 20 | > 4 | 11 |
| Oseltamivir carboxylate | 4.7 | > 100 | 9 | > 100 | - | - | - | - | - | - |
| Ribavirine | 8 | > 100 | 8.1 | > 100 | 50 | > 250 | > 250 | > 250 | - | - |
| Amantadine | 127 | > 500 | 1.7 | > 500 | - | - | - | - | - | - |
| HSV-1 | HSV-2 | HSV-1 (TK-KOS ACV) | Sindbis Virus | Coxsackie Virus B4 | Punto Toro Virus | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | EC50 (μM) | MCC (μM) | |
| 45 | > 20 | ≥ 20 | > 20 | ≥ 20 | > 20 | ≥ 20 | > 20 | ≥ 20 | > 20 | ≥ 20 | > 20 | ≥ 20 |
| 46 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 47 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 20 | 100 | > 20 | 100 | > 20 | 100 |
| 48 | > 20 | 100 | > 20 | 100 | > 20 | 100 | > 4 | 20 | > 4 | 20 | > 4 | 20 |
| Acyclovir | 0.9 | > 250 | 0.4 | > 250 | > 250 | > 250 | - | - | - | - | - | - |
| Ribavirine | - | - | - | - | - | - | > 250 | > 250 | > 250 | > 250 | 112 | > 250 |
| Concn (μg/mL) | Ad17 (Cells Number) | HSV-1 (Cells Number) | Poliovirus 1 (Cells Number) | Echovirus 2 (Cells Number) | Coxsackie virus B4 (Cells Number) | MNC (μg/mL) | |
|---|---|---|---|---|---|---|---|
| blank | 5 × 108 | 3 × 106 | 3 × 109 | 2 × 109 | 3 × 108 | ||
| 56 | 20 | 7 × 106 | 6 × 105 | 4 × 106 | 0 | 6 × 105 | 900 |
| 50 | 4 × 102 | 5 × 104 | 2 × 103 | 0 | 2 × 103 | ||
| 100 | 2 × 102 | 3 × 104 | 1 × 103 | 0 | 2 × 103 | ||
| 57 | 50 | 6 × 108 | 3 × 106 | 2 × 105 | - | 8 × 104 | 800 |
| 100 | 6 × 108 | 2 × 106 | 8 × 104 | - | 2 × 104 | ||
| 58 | 50 | 4 × 108 | 2 × 106 | 3 × 104 | - | 2 × 104 | 800 |
| 100 | 5 × 108 | 3 × 106 | 2 × 104 | - | 2 × 104 | ||
| 59 | 50 | 4 × 108 | 2 × 106 | 3 × 109 | 2 × 109 | 3 × 108 | 1300 |
| 100 | 5 × 108 | 3 × 106 | 3 × 109 | 2 × 109 | 3 × 108 |
| R’ | R | Viral Infection Control (%) | ||
|---|---|---|---|---|
| 1000 ppm | 100 ppm | |||
| 61 | 4-CH3O | D-glucopentyl | 69 | 26 |
| 62 | 4-CH3O | D-xylobutyl | 82 | 35 |
| 63 | 2-CH3 | D-glucopentyl | 59 | 22 |
| 64 | 2-CH3 | D-xylobutyl | 76 | 30 |
| Virazole | 100 | 100 | ||
© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serban, G. Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections. Molecules 2020, 25, 942. https://doi.org/10.3390/molecules25040942
Serban G. Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections. Molecules. 2020; 25(4):942. https://doi.org/10.3390/molecules25040942
Chicago/Turabian StyleSerban, Georgeta. 2020. "Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections" Molecules 25, no. 4: 942. https://doi.org/10.3390/molecules25040942
APA StyleSerban, G. (2020). Synthetic Compounds with 2-Amino-1,3,4-Thiadiazole Moiety Against Viral Infections. Molecules, 25(4), 942. https://doi.org/10.3390/molecules25040942