First Cobalt(II) Spin Crossover Compound with N4S2-Donorset



Abstract
1. Introduction
2. Results and Discussions
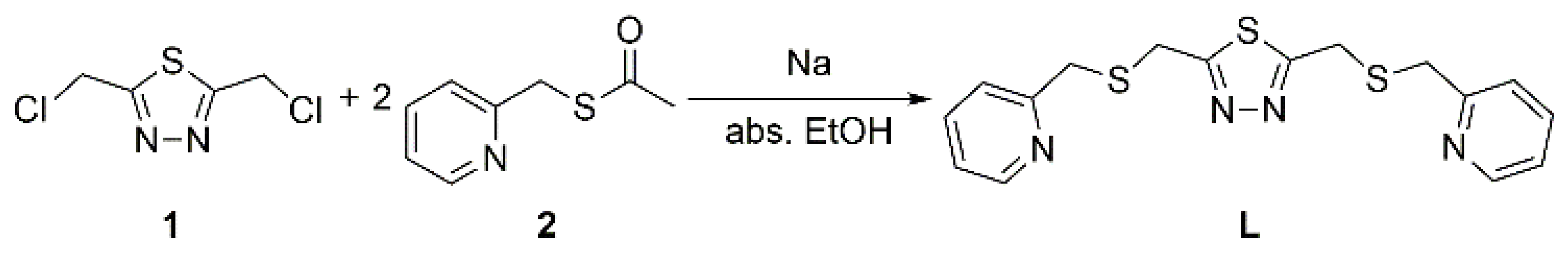
2.1. Synthesis
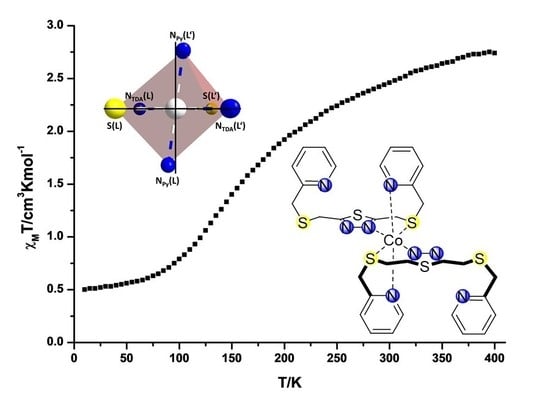
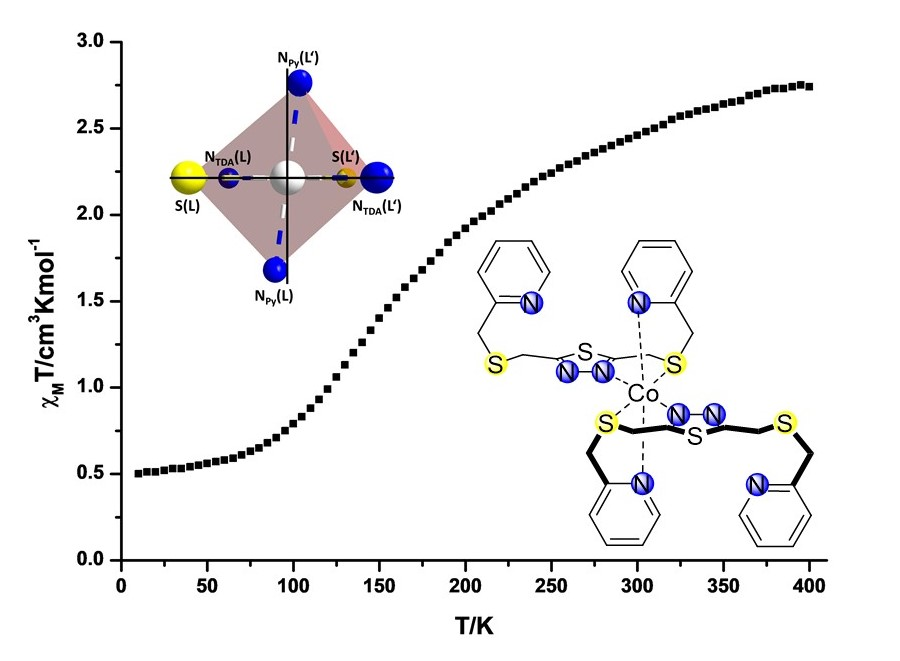
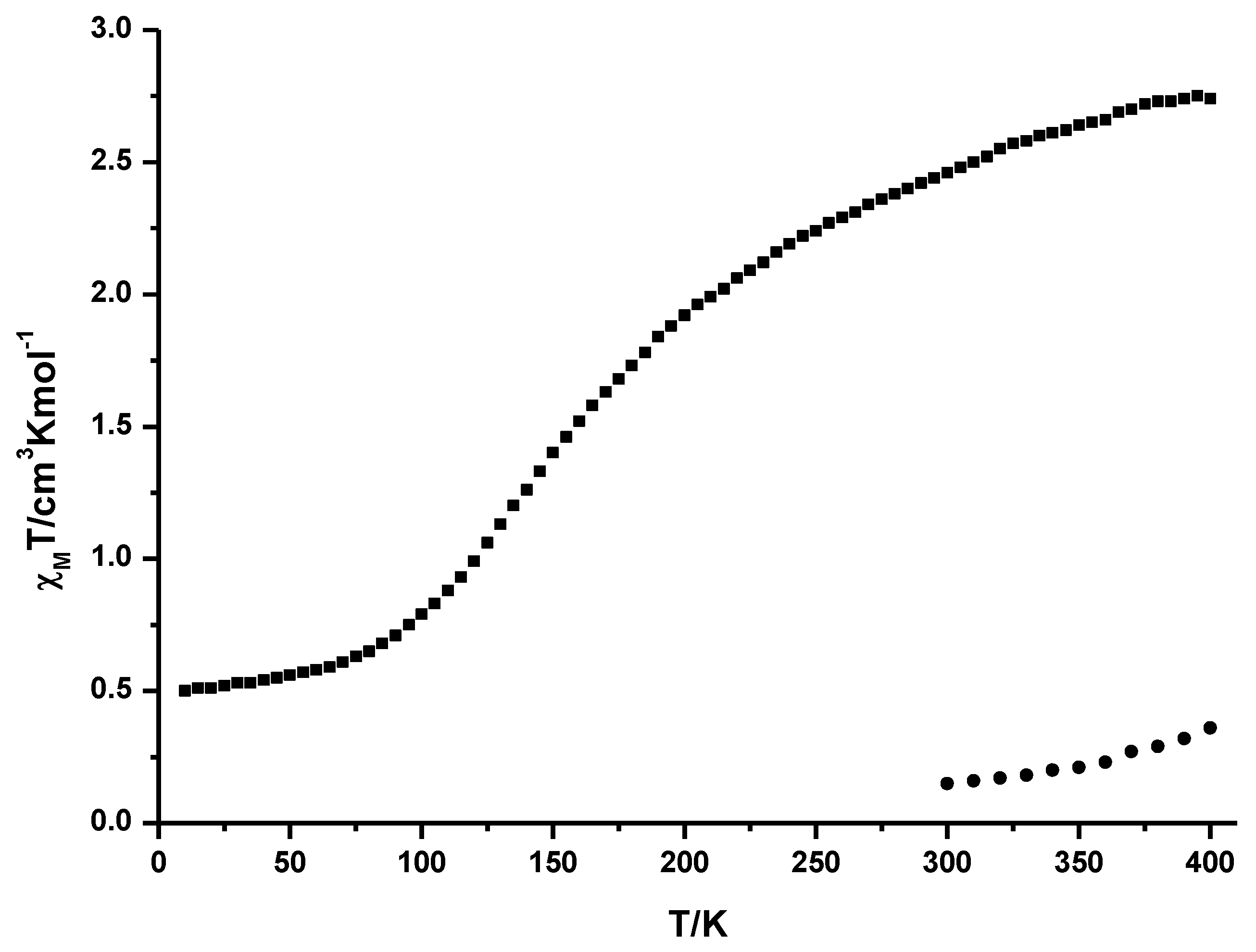
2.2. Variable Temperature Magnetic Susceptibility Measurements
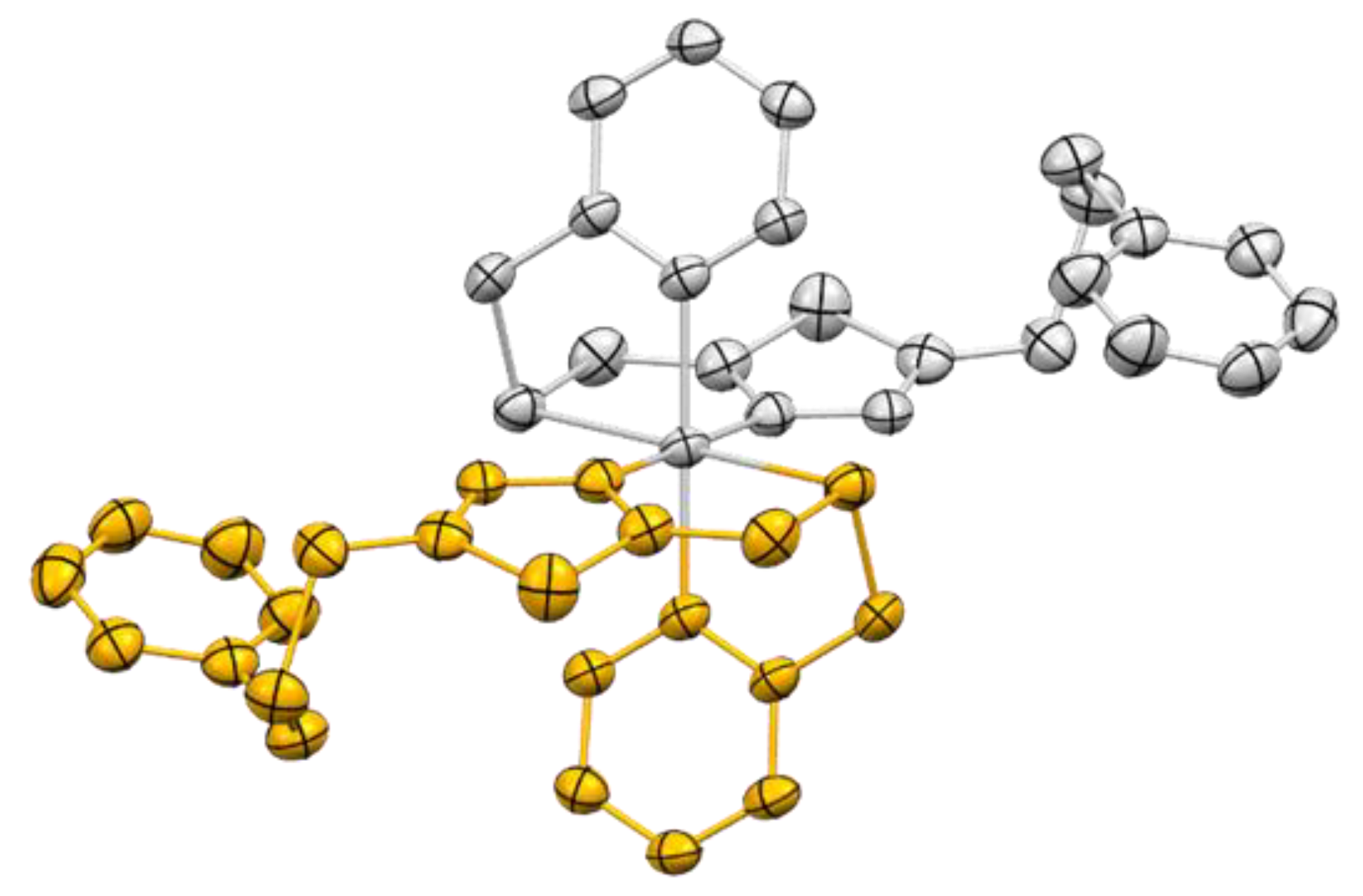
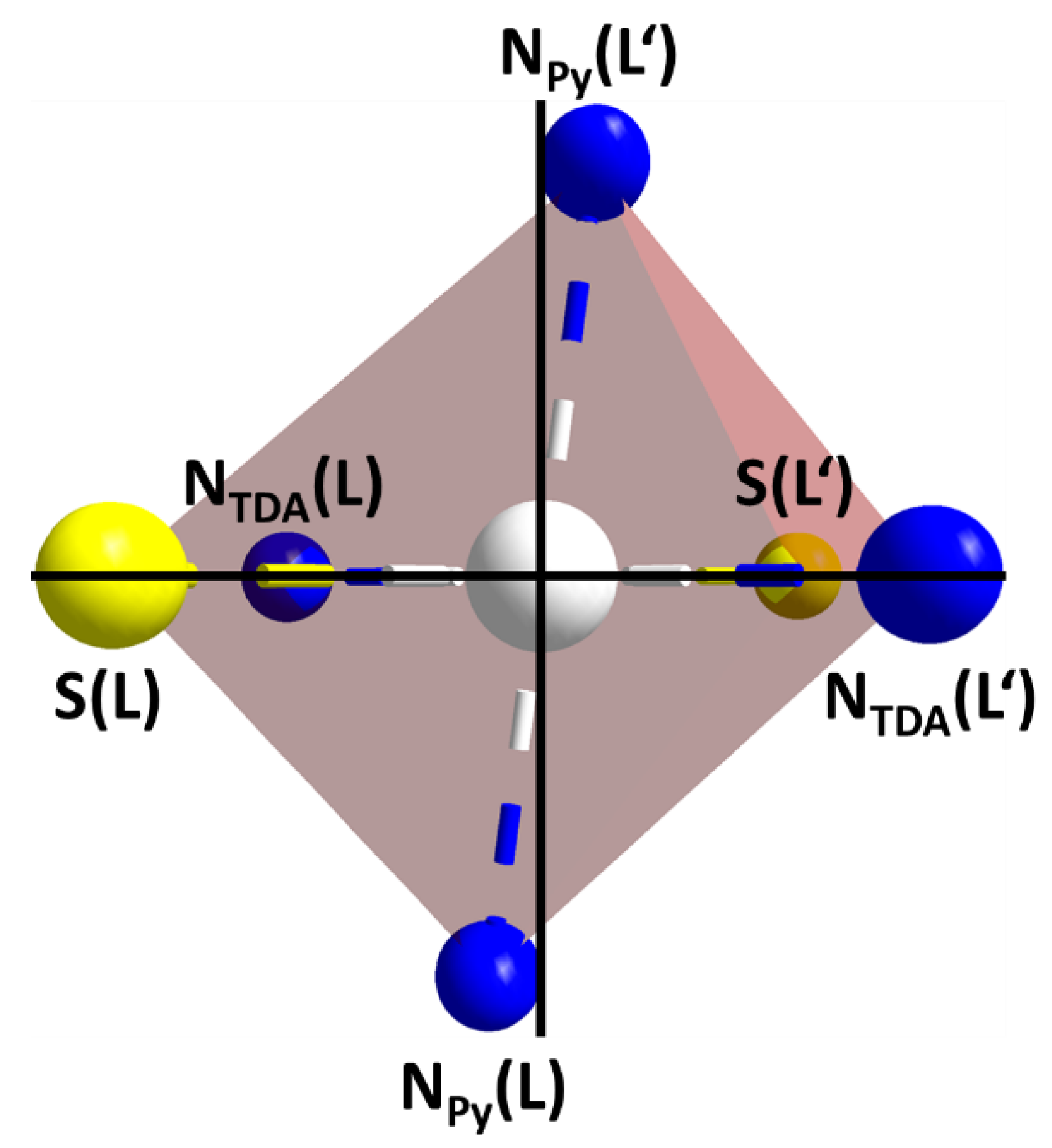
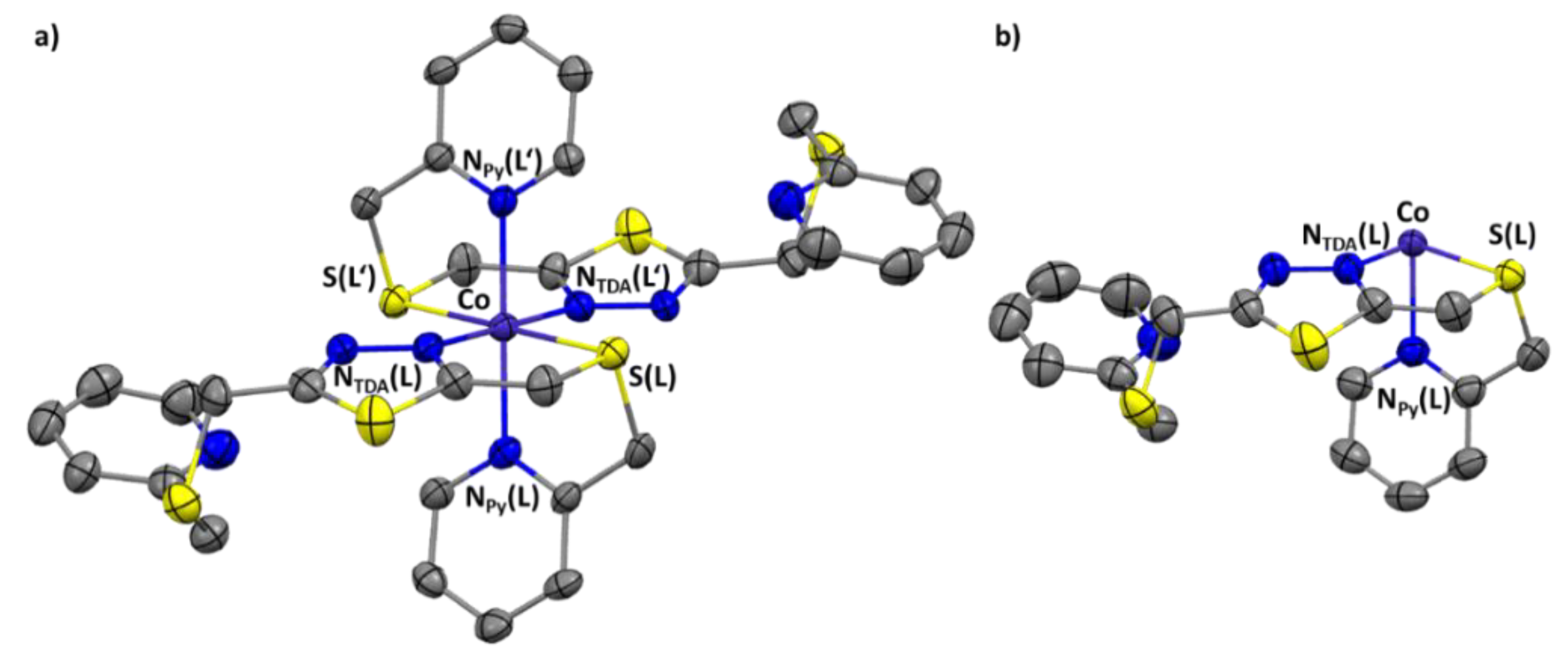
2.3. Crystal Structures
3. Materials and Methods
3.1. General Methods and Materials
3.2. Ligand Synthesis
2,5-Bis[(2-pyridylmethyl)thio]methyl-1,3,4-thiadiazole (L)
3.3. Complex Synthesis
3.3.1. [FeII(L)2](ClO4)2 (C1)
3.3.2. [CoII(L)2](ClO4)2 (C2)
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gütlich, P.; Goodwin, H.A. (Eds.) Spin Crossover in Transition Metal Compounds I; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 233, ISBN 978-3-540-40394-4. [Google Scholar]
- Gütlich, P.; Goodwin, H.A. Spin Crossover in Transition Metal Compounds II; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 234, ISBN 978-3-540-40396-8. [Google Scholar]
- Gütlich, P.; Goodwin, H.A. Spin Crossover in Transition Metal Compounds III; Topics in Current Chemistry; Springer: Berlin/Heidelberg, Germany, 2004; Volume 235, ISBN 978-3-540-40395-1. [Google Scholar]
- Halcrow, M.A. (Ed.) Spin-Crossover Materials; John Wiley & Sons Ltd.: Oxford, UK, 2013; ISBN 9781118519301. [Google Scholar]
- Kahn, O. Spin-Transition Polymers: From Molecular Materials toward Memory Devices. Science 1998, 279, 44–48. [Google Scholar] [CrossRef]
- Gütlich, P.; Hauser, A.; Spiering, H. Thermisch und optisch schaltbare Eisen(II)-Komplexe. Angew. Chem. 1994, 106, 2109–2141. [Google Scholar] [CrossRef]
- Dhers, S.; Feltham, H.L.C.; Brooker, S. A toolbox of building blocks, linkers and crystallisation methods used to generate single-chain magnets. Coord. Chem. Rev. 2015, 296, 24–44. [Google Scholar] [CrossRef]
- Brooker, S. Spin crossover with thermal hysteresis: Practicalities and lessons learnt. Chem. Soc. Rev. 2015, 44, 2880–2892. [Google Scholar] [CrossRef]
- Barrios, L.A.; Peyrecave-Lleixà, E.; Craig, G.A.; Roubeau, O.; Teat, S.J.; Aromí, G. Unusual Crystal Packing in a Family of [Fe{2,6-bis(pyrazol-3-yl)pyridine} 2 ] 2+ Compounds and the Effect on the Occurrence of Spin Crossover and Its Cooperative Character. Eur. J. Inorg. Chem. 2014, 2014, 6013–6021. [Google Scholar] [CrossRef]
- Halcrow, M.A. Structure:function relationships in molecular spin-crossover complexes. Chem. Soc. Rev. 2011, 40, 4119–4142. [Google Scholar] [CrossRef]
- Gütlich, P.; Goodwin, H.A. Spin Crossover—An Overall Perspective. In Spin Crossover in Transition Metal Compounds I; Springer: Berlin/Heidelberg, Germany, 2004; Volume 1, pp. 1–47. [Google Scholar]
- Roberts, T.D.; Little, M.A.; Kershaw Cook, L.J.; Halcrow, M.A. Iron(ii) complexes of 2,6-di(1H-pyrazol-3-yl)-pyridine derivatives with hydrogen bonding and sterically bulky substituents. Dalt. Trans. 2014, 43, 7577–7588. [Google Scholar] [CrossRef]
- Bauer, W.; Lochenie, C.; Weber, B. Synthesis and characterization of 1D iron( ii ) spin crossover coordination polymers with hysteresis. Dalt. Trans. 2014, 43, 1990–1999. [Google Scholar] [CrossRef]
- Kahn, O.; Kröber, J.; Jay, C. Spin Transition Molecular Materials for displays and data recording. Adv. Mater. 1992, 4, 718–728. [Google Scholar] [CrossRef]
- Kröber, J.; Codjovi, E.; Kahn, O.; Grolière, F.; Jay, C. A Spin Transition System with a Thermal Hysteresis at Room Temperature. J. Am. Chem. Soc. 1993, 115, 9810–9811. [Google Scholar] [CrossRef]
- Real, J.A.; Gaspar, A.B.; Niel, V.; Muñoz, M.C. Communication between iron(II) building blocks in cooperative spin transition phenomena. Coord. Chem. Rev. 2003, 236, 121–141. [Google Scholar] [CrossRef]
- Real, J.A.; Gaspar, A.B.; Muñoz, M.C. Thermal, pressure and light switchable spin-crossover materials. Dalt. Trans. 2005, 2062–2079. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.; Rentschler, E. The First 1,3,4-Oxadiazole Based Dinuclear Iron(II) Complexes Showing Spin Crossover Behavior with Hysteresis. Eur. J. Inorg. Chem. 2016, 2016, 1955–1960. [Google Scholar] [CrossRef]
- Herold, C.F.; Carrella, L.M.; Rentschler, E. A Family of Dinuclear Iron(II) SCO Compounds Based on a 1,3,4-Thiadiazole Bridging Ligand. Eur. J. Inorg. Chem. 2015, 2015, 3632–3636. [Google Scholar] [CrossRef]
- Fürmeyer, F.; Carrella, L.M.; Ksenofontov, V.; Möller, A.; Rentschler, E. Phase trapping in multistep spin crossover compound. Inorg. Chem. 2020, in press. [Google Scholar]
- Hogue, R.W.; Feltham, H.L.C.; Miller, R.G.; Brooker, S. Spin Crossover in Dinuclear N 4 S 2 Iron(II) Thioether–Triazole Complexes: Access to [HS-HS], [HS-LS], and [LS-LS] States. Inorg. Chem. 2016, 55, 4152–4165. [Google Scholar] [CrossRef]
- Hayami, S.; Komatsu, Y.; Shimizu, T.; Kamihata, H.; Lee, Y.H. Spin-crossover in cobalt(II) compounds containing terpyridine and its derivatives. Coord. Chem. Rev. 2011, 255, 1981–1990. [Google Scholar] [CrossRef]
- Grillo, A.V.; Gahan, R.L.; Hanson, R.G.; Stranger, R.; Hambley, W.T.; Murray, S.K.; Moubaraki, B.; Cashion, D.J. Iron(III) and iron(II) complexes of 1-thia-4{,}7-diazacyclononane ([9]aneN2S) and 1{,}4-dithia-7-azacyclononane ([9]aneNS2). X-Ray structural analyses{,} magnetic susceptibility{,} Mössbauer{,} EPR and electronic spectroscopy †. J. Chem. Soc., Dalt. Trans. 1998, 2341–2348. [Google Scholar] [CrossRef]
- England, J.; Gondhia, R.; Bigorra-Lopez, L.; Petersen, A.R.; White, A.J.P.; Britovsek, G.J.P. Towards robust alkane oxidation catalysts: Electronic variations in non-heme iron(ii) complexes and their effect in catalytic alkane oxidation. Dalt. Trans. 2009, 27, 5319–5334. [Google Scholar] [CrossRef][Green Version]
- Reus, C.; Ruth, K.; Tüllmann, S.; Bolte, M.; Lerner, H.-W.; Weber, B.; Holthausen, M.C.; Wagner, M. Synthesis, Molecular Structure, and Physical Properties of the Complexes [{PhB(pz)2(CH2SMe)}2M] (M = MnII, FeII; pz = pyrazol-1-yl) Containing a Novel [N,N,S]-Heteroscorpionate Ligand. Eur. J. Inorg. Chem. 2011, 2011, 1709–1718. [Google Scholar] [CrossRef]
- Lennartson, A.; Bond, A.D.; Piligkos, S.; McKenzie, C.J. Four-Site Cooperative Spin Crossover in a Mononuclear Fe II Complex. Angew. Chem. 2012, 124, 11211–11214. [Google Scholar] [CrossRef]
- Lennartson, A.; Southon, P.; Sciortino, N.F.; Kepert, C.J.; Frandsen, C.; Mørup, S.; Piligkos, S.; McKenzie, C.J. Reversible Guest Binding in a Non-Porous Fe II Coordination Polymer Host Toggles Spin Crossover. Chem.-A Eur. J. 2015, 21, 16066–16072. [Google Scholar] [CrossRef]
- Arroyave, A.; Lennartson, A.; Dragulescu-Andrasi, A.; Pedersen, K.S.; Piligkos, S.; Stoian, S.A.; Greer, S.M.; Pak, C.; Hietsoi, O.; Phan, H.; et al. Spin Crossover in Fe(II) Complexes with N 4 S 2 Coordination. Inorg. Chem. 2016, 55, 5904–5913. [Google Scholar] [CrossRef]
- Yergeshbayeva, S.; Hrudka, J.J.; Lengyel, J.; Erkasov, R.; Stoian, S.A.; Dragulescu-Andrasi, A.; Shatruk, M. Heteroleptic Fe(II) Complexes with N 4 S 2 Coordination as a Platform for Designing Spin-Crossover Materials. Inorg. Chem. 2017, 56, 11096–11103. [Google Scholar] [CrossRef]
- Wei, Z.; Xie, X.; Zhao, J.; Huang, L.; Liu, X. A novel hexadentate ligand and its complexes with divalent metal ions (Zinc, Copper, and Cobalt): Synthesis, characterization, and electrochemical investigation. Inorg. Chim. Acta 2012, 387, 277–282. [Google Scholar] [CrossRef]
- Funkemeier, D.; Mattes, R. Synthesis and structural studies of copper(II), nickel(II) and cobalt(II) complexes of a 14-membered trans-N2S2 dibenzo macrocycle with two pendant pyridylmethyl groups. J. Chem. Soc. Dalton Trans. 1993, 1313–1319. [Google Scholar] [CrossRef]
- Mohamadou, A.; Jubert, C.; Barbier, J.-P. Novel cobalt(II) complexes with pyridyl/ether or pyridyl/thioether ligands. The conversion of pyridyl/thioether cobalt(II) complex to pyridyl/sulfinato cobalt(III) compound. Inorg. Chim. Acta 2006, 359, 273–282. [Google Scholar] [CrossRef]
- Magwa, N.P.; Hosten, E.; Watkins, G.M.; Tshentu, Z.R. The coordination and extractive chemistry of the later 3d transition metals with bis ((1 R -benzimidazol-2-yl)methyl)sulfide. J. Coord. Chem. 2013, 66, 114–125. [Google Scholar] [CrossRef]
- Hogue, R.W.; Schott, O.; Hanan, G.S.; Brooker, S. A Smorgasbord of 17 Cobalt Complexes Active for Photocatalytic Hydrogen Evolution. Chem. A Eur. J. 2018, 24, 9820–9832. [Google Scholar] [CrossRef]
- Wu, H.; Yang, Z.; Wang, F.; Peng, H.; Zhang, H.; Wang, C.; Wang, K. V-shaped ligand 1,3-bis(1-ethylbenzimidazol-2-yl)-2-thiapropane and manganese(II), cobalt(II) and copper(II) complexes: Synthesis, crystal structure, DNA-binding properties and antioxidant activities. J. Photochem. Photobiol. B Biol. 2015, 148, 252–261. [Google Scholar] [CrossRef]
- Pandiyan, T.; Hernández, J.G.; Medina, N.T.; Bernés, S. Geometrical isomers of bis(benzimidazol-2-ylethyl)sulfide)cobalt(II) diperchlorates: Synthesis, structure, spectra and redox behavior of pink-[Co(bbes)2](ClO4)2 and blue-[Co(bbes)2](ClO4)2. Inorg. Chim. Acta 2004, 357, 2570–2578. [Google Scholar] [CrossRef]
- Singh, A.K.; Mukherjee, R. Cobalt(ii) and cobalt(iii) complexes of thioether-containing hexadentate pyrazine amide ligands: C–S bond cleavage and cyclometallation reaction. J. Chem. Soc. Dalt. Trans. 2008, 260–270. [Google Scholar] [CrossRef] [PubMed]
- Nandi, S.; Bannerjee, D.; Datta, P.; Lu, T.H.; Slawin, A.M.Z.; Sinha, C. Cobalt-thioalkylazoimidazole complexes: Structures, spectra and redox properties. Polyhedron 2009, 28, 3519–3525. [Google Scholar] [CrossRef]
- Thomas, L.; Hsiung, T.; Breen, J.; Worrell, J.H. Kinetics of substitution and isomerization of nitrite ion on aqua(7-methyl-4, 10-dithia-1, 7, 13- triazatridecane)cobalt(III) and the structure of the product. J. Coord. Chem. 1992, 26, 15–34. [Google Scholar]
- Eaborn, C. Purification of Laboratory Chemicals. J. Organomet. Chem. 1981. [Google Scholar] [CrossRef]
- Cobas, J.C.; Sardina, F.J. Nuclear magnetic resonance data processing. MestRe-C: A software package for desktop computers. Concepts Magn. Reson. 2003, 19A, 80–96. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
Sample Availability: Not available. |







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Selected Parameters [a] | C1 (@173 K) | C2 (@120 K) | C2 (@250 K) |
|---|---|---|---|
| M-NTDA(L) | 1.967(1) | 1.979(4) | 2.057(2) |
| M-NPy(L) | 2.014(1) | 2.019(4) | 2.075(2) |
| M-NTDA(L′) | 1.964(1) | 1.981(4) | |
| M-NPy(L′) | 2.016(1) | 2.027(4) | |
| M-S(L) | 2.263(1) | 2.472(2) | 2.479(1) |
| M-S(L′) | 2.264(1) | 2.470(2) | |
| cis NTDA-M-NPy | 84.1–95.9 | 84.6–95.6 | 85.1, 94.1 |
| cis NTDA-M-S | 85.3–94.8 | 82.9–97.4 | 83.8, 96.2 |
| cis NPy-M-S | 84.8–95.2 | 83.2–96.7 | 82.8, 97.2 |
| av. trans X-M-X | 179.8 | 179.6 | 180.0 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fürmeyer, F.; Münzberg, D.; Carrella, L.M.; Rentschler, E. First Cobalt(II) Spin Crossover Compound with N4S2-Donorset. Molecules 2020, 25, 855. https://doi.org/10.3390/molecules25040855
Fürmeyer F, Münzberg D, Carrella LM, Rentschler E. First Cobalt(II) Spin Crossover Compound with N4S2-Donorset. Molecules. 2020; 25(4):855. https://doi.org/10.3390/molecules25040855
Chicago/Turabian StyleFürmeyer, Fabian, Danny Münzberg, Luca M. Carrella, and Eva Rentschler. 2020. "First Cobalt(II) Spin Crossover Compound with N4S2-Donorset" Molecules 25, no. 4: 855. https://doi.org/10.3390/molecules25040855
APA StyleFürmeyer, F., Münzberg, D., Carrella, L. M., & Rentschler, E. (2020). First Cobalt(II) Spin Crossover Compound with N4S2-Donorset. Molecules, 25(4), 855. https://doi.org/10.3390/molecules25040855