
Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair

 ,
, 
 ,
,
Abstract
1. Introduction
2. Pre-PNA: TFOs and Structure-Induced Recombination
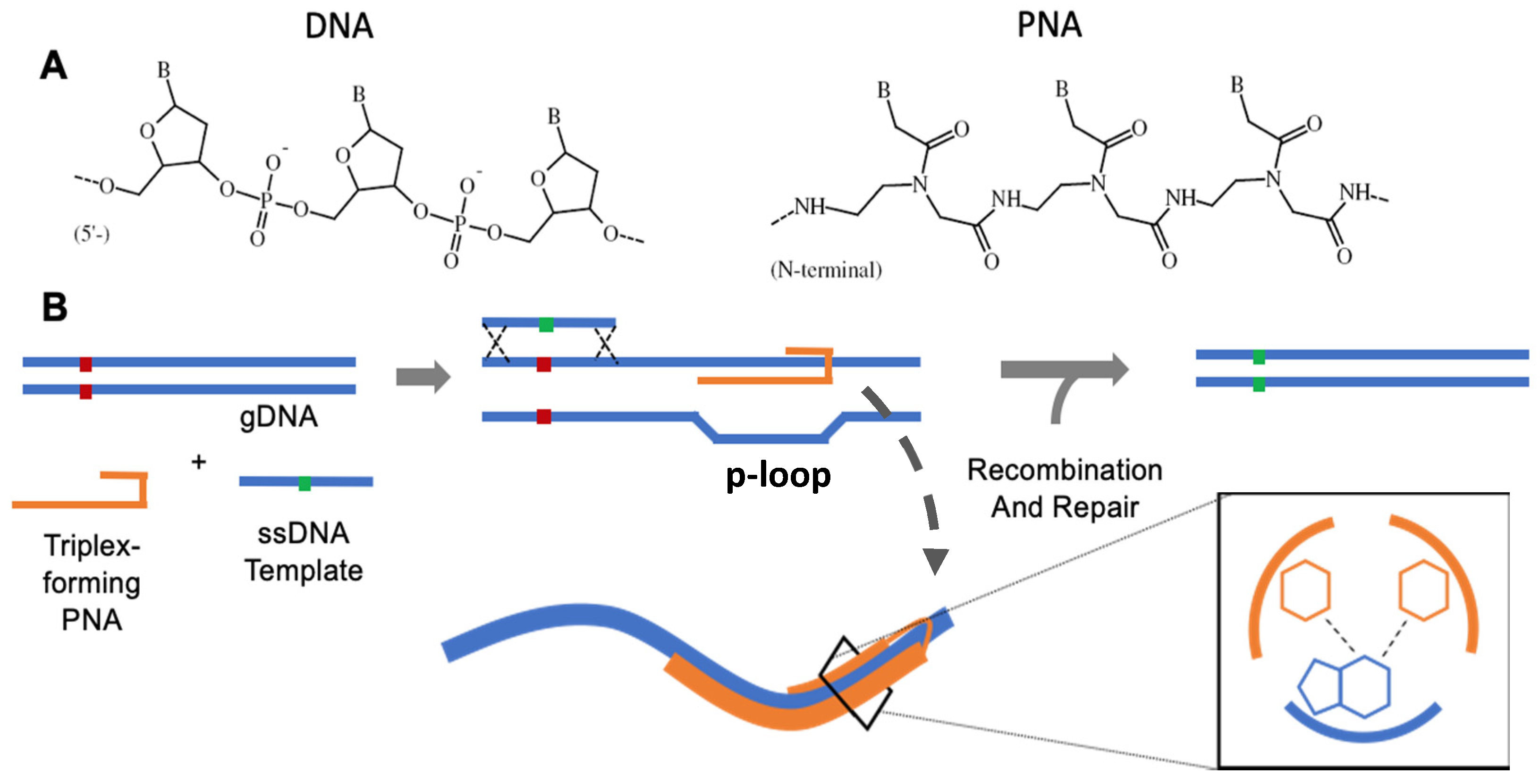
3. PNA Chemistry
4. Triplex-Forming PNAs for Gene Editing
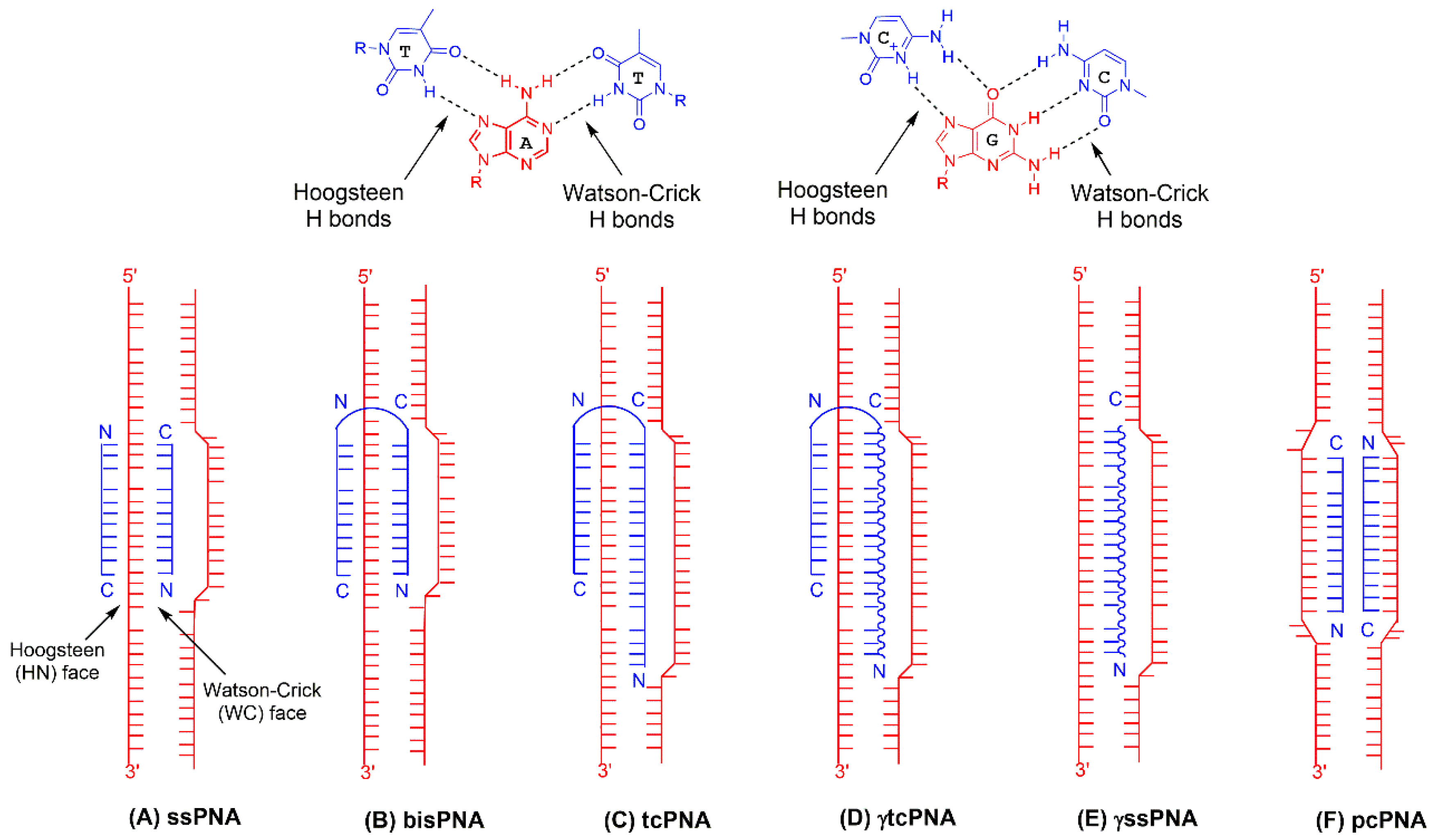
4.1. Bis-PNAs and Early Applications
4.2. tcPNAs and First In Vivo Studies
4.3. PNA Gamma(γ) Modification and Further In Vivo and In Utero Application
4.4. Ongoing Work and Democratization
5. Delivery
5.1. Peptide-Mediated Delivery of Peptide Nucleic Acids
5.2. PLGA Nanoparticle-Mediated Delivery of PNAs
5.3. PBAE/PLGA/MPG Nanoparticle-Mediated Delivery of PNAs
6. Mechanisms of PNA-Mediated Gene Editing
6.1. PNA Triplex Repair and Recombination
6.2. TFO Triplex Repair and Recombination
6.3. Endogenous Triplex Repair and Recombination
6.4. ssDNA Donor Recombination and Rad51
7. Other PNAs for Gene Editing
7.1. pcPNAs for Gene Editing
7.2. ssPNAs for Gene Editing
8. Perspectives and Limitations
Author Contributions
Funding
Conflicts of Interest
References
- Watson, J.D.; Crick, F.H.C. Molecular Structure of Nucleic Acids: A Structure for Deoxyribose Nucleic Acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Majima, T. Conformational changes of non-B DNA. Chem. Soc. Rev. 2011, 40, 5893–5909. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, G.; Del Mundo, I.M.; McKinney, J.A.; Lu, X.; Bacolla, A.; Boulware, S.B.; Zhang, C.; Zhang, H.; Ren, P.; et al. Distinct Mechanisms of Nuclease-Directed DNA-Structure-Induced Genetic Instability in Cancer Genomes. Cell Rep. 2018, 22, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Boyer, A.-S.; Grgurevic, S.; Cazaux, C.; Hoffmann, J.-S. The Human Specialized DNA Polymerases and Non-B DNA: Vital Relationships to Preserve Genome Integrity. J. Mol. Biol. 2013, 425, 4767–4781. [Google Scholar] [CrossRef] [PubMed]
- Frank-Kamenetskii, M.D.; Mirkin, S.M. Triplex Dna Structures. Annu. Rev. Biochem. 1995, 64, 65–95. [Google Scholar] [CrossRef] [PubMed]
- Havre, P.A.; Gunther, E.J.; Gasparro, F.P.; Glazer, P.M. Targeted mutagenesis of DNA using triple helix-forming oligonucleotides linked to psoralen. Proc. Natl. Acad. Sci. USA 1993, 90, 7879–7883. [Google Scholar] [CrossRef]
- Chan, P.P.; Lin, M.; Faruqi, A.F.; Powell, J.; Seidman, M.M.; Glazer, P.M. Targeted correction of an episomal gene in mammalian cells by a short DNA fragment tethered to a triplex-forming oligonucleotide. J. Biol. Chem. 1999, 274, 11541–11548. [Google Scholar] [CrossRef]
- Nielsen, P.E.; Egholm, M.; Berg, R.H.; Buchardt, O. Sequence-selective recognition of DNA by strand displacement with a thymine-substituted polyamide. Science 1991, 254, 1497–1500. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Nielsen, P.E.; Berg, R.H. Peptide nucleic acids (PNA). Oligonucleotide analogs with an achiral peptide backbone. J. Am. Chem. Soc. 1992, 114, 1895–1897. [Google Scholar] [CrossRef]
- Kim, S.K.; Nielsen, P.E.; Egholm, M.; Buchardt, O.; Berg, R.H.; Norden, B. Right-handed triplex formed between peptide nucleic acid PNA-T8 and poly(dA) shown by linear and circular dichroism spectroscopy. J. Am. Chem. Soc. 1993, 115, 6477–6481. [Google Scholar] [CrossRef]
- Egholm, M.; Buchardt, O.; Christensen, L.; Behrens, C.; Freier, S.M.; Driver, D.A.; Berg, R.H.; Kim, S.K.; Norden, B.; Nielsen, P.E. PNA hybridizes to complementary oligonucleotides obeying the Watson-Crick hydrogen-bonding rules. Nature 1993, 365, 566–568. [Google Scholar] [CrossRef] [PubMed]
- Demidov, V.V.; Potaman, V.N.; Frank-Kamenetskil, M.D.; Egholm, M.; Buchard, O.; Sönnichsen, S.H.; Nlelsen, P.E. Stability of peptide nucleic acids in human serum and cellular extracts. Biochem. Pharmacol. 1994, 48, 1310–1313. [Google Scholar] [CrossRef]
- Wang, G.; Seidman, M.M.; Glazer, P.M. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science 1996, 271, 802–805. [Google Scholar] [CrossRef] [PubMed]
- Faruqi, A.F.; Datta, H.J.; Carroll, D.; Seidman, M.M.; Glazer, P.M. Triple-Helix Formation Induces Recombination in Mammalian Cells via a Nucleotide Excision Repair-Dependent Pathway. Mol. Cell. Biol. 2000, 20, 990–1000. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rogers, F.A.; Vasquez, K.M.; Egholm, M.; Glazer, P.M. Site-directed recombination via bifunctional PNA–DNA conjugates. Proc. Natl. Acad. Sci. USA 2002, 99, 16695–16700. [Google Scholar] [CrossRef]
- McNeer, N.A.; Schleifman, E.B.; Cuthbert, A.; Brehm, M.; Jackson, A.; Cheng, C.; Anandalingam, K.; Kumar, P.; Shultz, L.D.; Greiner, D.L.; et al. Systemic delivery of triplex-forming PNA and donor DNA by nanoparticles mediates site-specific genome editing of human hematopoietic cells in vivo. Gene 2013, 20, 658–669. [Google Scholar] [CrossRef]
- Bahal, R.; Quijano, E.; McNeer, N.A.; Liu, Y.; Bhunia, D.C.; López-Giráldez, F.; Fields, R.J.; Saltzman, W.M.; Ly, D.H.; Glazer, P.M. Single-Stranded γPNAs for In Vivo Site-Specific Genome Editing via Watson-Crick Recognition. Curr. Gene 2014, 14, 331–342. [Google Scholar] [CrossRef]
- Fields, R.J.; Quijano, E.; McNeer, N.A.; Caputo, C.; Bahal, R.; Anandalingam, K.; Egan, M.E.; Glazer, P.M.; Saltzman, W.M. Modified Poly(lactic-co-glycolic acid) Nanoparticles for Enhanced Cellular Uptake and Gene Editing in the Lung. Adv. Healthc. Mater. 2015, 4, 361–366. [Google Scholar] [CrossRef]
- McNeer, N.A.; Anandalingam, K.; Fields, R.J.; Caputo, C.; Kopic, S.; Gupta, A.; Quijano, E.; Polikoff, L.; Kong, Y.; Bahal, R.; et al. Correction of F508del CFTR in airway epithelium using nanoparticles delivering triplex-forming PNAs. Nat. Commun. 2015, 6, 6952. [Google Scholar] [CrossRef]
- Bahal, R.; Ali McNeer, N.; Quijano, E.; Liu, Y.; Sulkowski, P.; Turchick, A.; Lu, Y.-C.; Bhunia, D.C.; Manna, A.; Greiner, D.L.; et al. In vivo correction of anaemia in β-thalassemic mice by γPNA-mediated gene editing with nanoparticle delivery. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef]
- Schleifman, E.B.; McNeer, N.A.; Jackson, A.; Yamtich, J.; Brehm, M.A.; Shultz, L.D.; Greiner, D.L.; Kumar, P.; Saltzman, W.M.; Glazer, P.M. Site-specific Genome Editing in PBMCs With PLGA Nanoparticle-delivered PNAs Confers HIV-1 Resistance in Humanized Mice. Mol. Nucleic Acids 2013, 2, e135. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Bahal, R.; Farrelly, J.S.; Quijano, E.; Bianchi, A.H.; Luks, V.L.; Putman, R.; López-Giráldez, F.; Coşkun, S.; Song, E.; et al. In utero nanoparticle delivery for site-specific genome editing. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Havre, P.A.; Glazer, P.M. Targeted mutagenesis of simian virus 40 DNA mediated by a triple helix-forming oligonucleotide. J. Virol. 1993, 67, 7324–7331. [Google Scholar] [CrossRef] [PubMed]
- Kohwi, Y.; Panchenko, Y. Transcription-dependent recombination induced by triple-helix formation. Genes Dev. 1993, 7, 1766–1778. [Google Scholar] [CrossRef]
- Faruqi, A.F.; Seidman, M.M.; Segal, D.J.; Carroll, D.; Glazer, P.M. Recombination induced by triple-helix-targeted DNA damage in mammalian cells. Mol. Cell. Biol. 1996, 16, 6820–6828. [Google Scholar] [CrossRef]
- Datta, H.J.; Chan, P.P.; Vasquez, K.M.; Gupta, R.C.; Glazer, P.M. Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51. J. Biol. Chem. 2001, 276, 18018–18023. [Google Scholar] [CrossRef]
- Wang, G.; Levy, D.D.; Seidman, M.M.; Glazer, P.M. Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol. Cell. Biol. 1995, 15, 1759–1768. [Google Scholar] [CrossRef]
- Chin, J.Y.; Glazer, P.M. Repair of DNA lesions associated with triplex-forming oligonucleotides. Mol. Carcinog. 2009, 48, 389–399. [Google Scholar] [CrossRef]
- Sahu, B.; Sacui, I.; Rapireddy, S.; Zanotti, K.J.; Bahal, R.; Armitage, B.A.; Ly, D.H. Synthesis and characterization of conformationally preorganized, (R)-diethylene glycol-containing γ-peptide nucleic acids with superior hybridization properties and water solubility. J. Org. Chem. 2011, 76, 5614–5627. [Google Scholar] [CrossRef]
- Nielsen, P.E. Peptide Nucleic Acids: Protocols and Applications; Garland Science: New York, NY, USA, 2004; ISBN 978-0-9545232-4-4. [Google Scholar]
- Nielsen, P.E.; Egholm, M.; Buchardt, O. Evidence for (PNA)2/DNA triplex structure upon binding of PNA to dsDNA by strand displacement. J. Mol. Recognit. 1994, 7, 165–170. [Google Scholar] [CrossRef]
- Peffer, N.J.; Hanvey, J.C.; Bisi, J.E.; Thomson, S.A.; Hassman, C.F.; Noble, S.A.; Babiss, L.E. Strand-invasion of duplex DNA by peptide nucleic acid oligomers. Proc. Natl. Acad. Sci. USA 1993, 90, 10648–10652. [Google Scholar] [CrossRef] [PubMed]
- Ananthanawat, C.; Hoven, V.P.; Vilaivan, T.; Su, X. Surface plasmon resonance study of PNA interactions with double-stranded DNA. Biosens. Bioelectron. 2011, 26, 1918–1923. [Google Scholar] [CrossRef] [PubMed]
- Ricciardi, A.S.; Quijano, E.; Putman, R.; Saltzman, W.M.; Glazer, P.M. Peptide Nucleic Acids as a Tool for Site-Specific Gene Editing. Molecules 2018, 23, 632. [Google Scholar] [CrossRef] [PubMed]
- Griffith, M.C.; Risen, L.M.; Greig, M.J.; Lesnik, E.A.; Sprankle, K.G.; Griffey, R.H.; Kiely, J.S.; Freier, S.M. Single and Bis Peptide Nucleic Acids as Triplexing Agents: Binding and Stoichiometry. J. Am. Chem. Soc. 1995, 117, 831–832. [Google Scholar] [CrossRef]
- Egholm, M.; Christensen, L.; Dueholm, K.L.; Buchardt, O.; Coull, J.; Nielsen, P.E. Efficient pH-independent sequence-specific DNA binding by pseudoisocytosine-containing bis-PNA. Nucleic Acids Res. 1995, 23, 217–222. [Google Scholar] [CrossRef]
- Betts, L.; Josey, J.A.; Veal, J.M.; Jordan, S.R. A nucleic acid triple helix formed by a peptide nucleic acid-DNA complex. Science 1995, 270, 1838–1841. [Google Scholar] [CrossRef]
- Hansen, G.I.; Bentin, T.; Larsen, H.J.; Nielsen, P.E. Structural isomers of bis-PNA bound to a target in duplex DNA11Edited by I. Tinoco. J. Mol. Biol. 2001, 307, 67–74. [Google Scholar] [CrossRef]
- Pack, G.R.; Wong, L.; Lamm, G. PKa of cytosine on the third strand of triplex DNA: Preliminary Poisson–Boltzmann calculations. Int. J. Quantum Chem. 1998, 70, 1177–1184. [Google Scholar] [CrossRef]
- Chin, J.Y.; Kuan, J.Y.; Lonkar, P.S.; Krause, D.S.; Seidman, M.M.; Peterson, K.R.; Nielsen, P.E.; Kole, R.; Glazer, P.M. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc. Natl. Acad. Sci. USA 2008, 105, 13514–13519. [Google Scholar] [CrossRef]
- McNeer, N.A.; Chin, J.Y.; Schleifman, E.B.; Fields, R.J.; Glazer, P.M.; Saltzman, W.M. Nanoparticles deliver triplex-forming PNAs for site-specific genomic recombination in CD34+ human hematopoietic progenitors. Mol. Ther. 2011, 19, 172–180. [Google Scholar] [CrossRef]
- Chin, J.Y.; Reza, F.; Glazer, P.M. Triplex-forming Peptide Nucleic Acids Induce Heritable Elevations in Gamma-globin Expression in Hematopoietic Progenitor Cells. Mol. Ther. 2013, 21, 580–587. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bentin, T.; Larsen, H.J.; Nielsen, P.E. Combined Triplex/Duplex Invasion of Double-Stranded DNA by “Tail-Clamp” Peptide Nucleic Acid. Biochemistry 2003, 42, 13987–13995. [Google Scholar] [CrossRef] [PubMed]
- Kaihatsu, K.; Shah, R.H.; Zhao, X.; Corey, D.R. Extending recognition by peptide nucleic acids (PNAs): Binding to duplex DNA and inhibition of transcription by tail-clamp PNA-peptide conjugates. Biochemistry 2003, 42, 13996–14003. [Google Scholar] [CrossRef] [PubMed]
- Schleifman, E.B.; Bindra, R.; Leif, J.; Del Campo, J.; Rogers, F.A.; Uchil, P.; Kutsch, O.; Shultz, L.D.; Kumar, P.; Greiner, D.L.; et al. Targeted disruption of the CCR5 gene in human hematopoietic stem cells stimulated by peptide nucleic acids. Chem. Biol. 2011, 18, 1189–1198. [Google Scholar] [CrossRef] [PubMed]
- Dragulescu-Andrasi, A.; Rapireddy, S.; Frezza, B.M.; Gayathri, C.; Gil, R.R.; Ly, D.H. A Simple γ-Backbone Modification Preorganizes Peptide Nucleic Acid into a Helical Structure. J. Am. Chem. Soc. 2006, 128, 10258–10267. [Google Scholar] [CrossRef]
- He, G.; Rapireddy, S.; Bahal, R.; Sahu, B.; Ly, D.H. Strand invasion of extended, mixed-sequence B-DNA by gammaPNAs. J. Am. Chem. Soc. 2009, 131, 12088–12090. [Google Scholar] [CrossRef]
- Bahal, R.; Sahu, B.; Rapireddy, S.; Lee, C.-M.; Ly, D.H. Sequence-unrestricted, Watson-Crick recognition of double helical B-DNA by (R)-miniPEG-γPNAs. Chembiochem 2012, 13, 56–60. [Google Scholar] [CrossRef]
- Yeh, J.I.; Shivachev, B.; Rapireddy, S.; Crawford, M.J.; Gil, R.R.; Du, S.; Madrid, M.; Ly, D.H. Crystal structure of chiral gammaPNA with complementary DNA strand: Insights into the stability and specificity of recognition and conformational preorganization. J. Am. Chem. Soc. 2010, 132, 10717–10727. [Google Scholar] [CrossRef]
- Demidov, V.V.; Yavnilovich, M.V.; Belotserkovskii, B.P.; Frank-Kamenetskii, M.D.; Nielsen, P.E. Kinetics and mechanism of polyamide (“peptide”) nucleic acid binding to duplex DNA. Proc. Natl. Acad. Sci. USA 1995, 92, 2637–2641. [Google Scholar] [CrossRef]
- Knauert, M.P.; Lloyd, J.A.; Rogers, F.A.; Datta, H.J.; Bennett, M.L.; Weeks, D.L.; Glazer, P.M. Distance and Affinity Dependence of Triplex-Induced Recombination. Biochemistry 2005, 44, 3856–3864. [Google Scholar] [CrossRef]
- Manesia, J.K.; Xu, Z.; Broekaert, D.; Boon, R.; Van Vliet, A.; Eelen, G.; Vanwelden, T.; Stegen, S.; Van Gastel, N.; Pascual-Montano, A.; et al. Highly proliferative primitive fetal liver hematopoietic stem cells are fueled by oxidative metabolic pathways. Stem Cell Res. 2015, 15, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Pietras, E.M.; Warr, M.R.; Passegué, E. Cell cycle regulation in hematopoietic stem cells. J. Cell Biol. 2011, 195, 709–720. [Google Scholar] [CrossRef] [PubMed]
- Piotrowski-Daspit, A.S.; Barone, C.; Kauffman, A.C.; Lin, C.Y.; Nguyen, R.; Gupta, A.; Glazer, P.M.; Saltzman, W.M.; Egan, M.E. Gene Correction of Cystic Fibrosis Mutations In Vitro and In Vivo Mediated by PNA Nanoparticles. In Proceedings of the American Society of Gene and Cell Therapy 22nd Annual Meeting, Washington, DC, USA, 29 April–2 May 2019. Abstract Number 81. [Google Scholar]
- Piotrowski-Daspit, A.S.; Kauffman, A.C.; Lin, C.Y.; Liu, Y.; Glazer, P.M.; Saltzman, W.M. Gene Correction of Beta-Thalassemia Ex Vivo and In Vivo Mediated by PNA Nanoparticles. In Proceedings of the American Society of Gene and Cell Therapy 22nd Annual Meeting, Washington, DC, USA, 29 April–2 May 2019. Abstract Number 976. [Google Scholar]
- Ricciardi, A.; Barone, C.; Putnam, R.; Quijano, E.; Nguyen, R.; Gupta, A.; Mandl, H.; Freedman-Weiss, M.; Lopez-Giraldez, F.; Stitelman, D.; et al. Systemic in Utero Gene Editing as a Treatment for Cystic Fibrosis. In Proceedings of the American Society of Gene and Cell Therapy 22nd Annual Meeting, Washington, DC, USA, 29 April–2 May 2019. Abstract Number 83. [Google Scholar]
- Quijano, E.; Bahal, R.; Liu, Y.; Gupta, A.; Oyaghire, S.; Ricciardi, A.S.; Mandl, H.K.; Suh, H.W.; Lezon-Geyda, K.; Economos, N.; et al. Nanoparticle-Mediated Correction of the Sickle Cell Mutation. In Proceedings of the American Society of Gene and Cell Therapy 22nd Annual Meeting, Washington, DC, USA, 29 April–2 May 2019. Abstract Number 594. [Google Scholar]
- Economos, N.; Bahal, R.; Quijano, E.; Saltzman, W.M.; Glazer, P. Mechanisms of Non-Enzymatic PNA-Mediated Gene Editing. In Proceedings of the American Society of Gene and Cell Therapy 22nd Annual Meeting, Washington, DC, USA, 29 April–2 May 2019. Abstract Number 631. [Google Scholar]
- Félix, A.J.; Ciudad, C.J.; Noé, V. Correction of the aprt Gene Using Repair-Polypurine Reverse Hoogsteen Hairpins in Mammalian Cells. Mol. Ther. Nucleic Acids 2020, 19, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Gaddis, S.S.; Wu, Q.; Thames, H.D.; DiGiovanni, J.; Walborg, E.F.; MacLeod, M.C.; Vasquez, K.M. A web-based search engine for triplex-forming oligonucleotide target sequences. Oligonucleotides 2006, 16, 196–201. [Google Scholar] [CrossRef]
- Wittung, P.; Kajanus, J.; Edwards, K.; Haaima, G.; Nielsen, P.E.; Nordén, B.; Malmström, B.G. Phospholipid membrane permeability of peptide nucleic acid. FEBS Lett. 1995, 375, 27–29. [Google Scholar] [CrossRef]
- Fonseca, S.B.; Pereira, M.P.; Kelley, S.O. Recent advances in the use of cell-penetrating peptides for medical and biological applications. Adv. Drug Deliv. Rev. 2009, 61, 953–964. [Google Scholar] [CrossRef]
- Zhang, X.-Y.; Dinh, A.; Cronin, J.; Li, S.-C.; Reiser, J. Cellular uptake and lysosomal delivery of galactocerebrosidase tagged with the HIV Tat protein transduction domain. J. Neurochem. 2008, 104, 1055–1064. [Google Scholar] [CrossRef]
- Wender, P.A.; Mitchell, D.J.; Pattabiraman, K.; Pelkey, E.T.; Steinman, L.; Rothbard, J.B. The design, synthesis, and evaluation of molecules that enable or enhance cellular uptake: Peptoid molecular transporters. Proc. Natl. Acad. Sci. USA 2000, 97, 13003–13008. [Google Scholar] [CrossRef]
- Zhou, P.; Wang, M.; Du, L.; Fisher, G.W.; Waggoner, A.; Ly, D.H. Novel Binding and Efficient Cellular Uptake of Guanidine-Based Peptide Nucleic Acids (GPNA). J. Am. Chem. Soc. 2003, 125, 6878–6879. [Google Scholar] [CrossRef]
- Fabani, M.M.; Gait, M.J. miR-122 targeting with LNA/2′-O-methyl oligonucleotide mixmers, peptide nucleic acids (PNA), and PNA–peptide conjugates. RNA 2008, 14, 336–346. [Google Scholar] [CrossRef]
- Aldrian-Herrada, G.; Desarménien, M.G.; Orcel, H.; Boissin-Agasse, L.; Méry, J.; Brugidou, J.; Rabié, A. A peptide nucleic acid (PNA) is more rapidly internalized in cultured neurons when coupled to a retro-inverso delivery peptide. The antisense activity depresses the target mRNA and protein in magnocellular oxytocin neurons. Nucleic Acids Res. 1998, 26, 4910–4916. [Google Scholar] [CrossRef] [PubMed]
- Pooga, M.; Soomets, U.; Hällbrink, M.; Valkna, A.; Saar, K.; Rezaei, K.; Kahl, U.; Hao, J.X.; Xu, X.J.; Wiesenfeld-Hallin, Z.; et al. Cell penetrating PNA constructs regulate galanin receptor levels and modify pain transmission in vivo. Nat. Biotechnol. 1998, 16, 857–861. [Google Scholar] [CrossRef] [PubMed]
- Shiraishi, T.; Nielsen, P.E. Cellular delivery of peptide nucleic acids (PNAs). Methods Mol. Biol. 2014, 1050, 193–205. [Google Scholar] [PubMed]
- Turner, J.J.; Ivanova, G.D.; Verbeure, B.; Williams, D.; Arzumanov, A.A.; Abes, S.; Lebleu, B.; Gait, M.J. Cell-penetrating peptide conjugates of peptide nucleic acids (PNA) as inhibitors of HIV-1 Tat-dependent trans-activation in cells. Nucleic Acids Res. 2005, 33, 6837–6849. [Google Scholar] [CrossRef]
- Simmons, C.G.; Pitts, A.E.; Mayfield, L.D.; Shay, J.W.; Corey, D.R. Synthesis and membrane permeability of PNA-peptide conjugates. Bioorganic Med. Chem. Lett. 1997, 7, 3001–3006. [Google Scholar] [CrossRef]
- Rogers, F.A.; Lin, S.S.; Hegan, D.C.; Krause, D.S.; Glazer, P.M. Targeted gene modification of hematopoietic progenitor cells in mice following systemic administration of a PNA-peptide conjugate. Mol. Ther. 2012, 20, 109–118. [Google Scholar] [CrossRef]
- Adochite, R.-C.; Moshnikova, A.; Golijanin, J.; Andreev, O.A.; Katenka, N.V.; Reshetnyak, Y.K. Comparative Study of Tumor Targeting and Biodistribution of pH (Low) Insertion Peptides (pHLIP(®) Peptides) Conjugated with Different Fluorescent Dyes. Mol. Imaging Biol. 2016, 18, 686–696. [Google Scholar] [CrossRef]
- Svoronos, A.A.; Bahal, R.; Pereira, M.C.; Barrera, F.N.; Deacon, J.C.; Bosenberg, M.; DiMaio, D.; Glazer, P.M.; Engelman, D.M. Tumor-Targeted, Cytoplasmic Delivery of Large, Polar Molecules Using a pH-Low Insertion Peptide. Mol. Pharm. 2020. [Google Scholar] [CrossRef]
- Reshetnyak, Y.K.; Andreev, O.A.; Lehnert, U.; Engelman, D.M. Translocation of molecules into cells by pH-dependent insertion of a transmembrane helix. Proc. Natl. Acad. Sci. USA 2006, 103, 6460–6465. [Google Scholar] [CrossRef]
- Reshetnyak, Y.K.; Segala, M.; Andreev, O.A.; Engelman, D.M. A Monomeric Membrane Peptide that Lives in Three Worlds: In Solution, Attached to, and Inserted across Lipid Bilayers. Biophys. J. 2007, 93, 2363–2372. [Google Scholar] [CrossRef]
- Cheng, C.J.; Bahal, R.; Babar, I.A.; Pincus, Z.; Barrera, F.; Liu, C.; Svoronos, A.; Braddock, D.T.; Glazer, P.M.; Engelman, D.M.; et al. MicroRNA silencing for cancer therapy targeted to the tumour microenvironment. Nature 2015, 518, 107–110. [Google Scholar] [CrossRef]
- Erazo-Oliveras, A.; Muthukrishnan, N.; Baker, R.; Wang, T.-Y.; Pellois, J.-P. Improving the endosomal escape of cell-penetrating peptides and their cargos: Strategies and challenges. Pharmaceuticals 2012, 5, 1177–1209. [Google Scholar] [CrossRef]
- Shiraishi, T.; Pankratova, S.; Nielsen, P.E. Calcium ions effectively enhance the effect of antisense peptide nucleic acids conjugated to cationic tat and oligoarginine peptides. Chem. Biol. 2005, 12, 923–929. [Google Scholar] [CrossRef]
- Gupta, A.; Bahal, R.; Gupta, M.; Glazer, P.M.; Saltzman, W.M. Nanotechnology for delivery of peptide nucleic acids (PNAs). J. Control. Release 2016, 240, 302–311. [Google Scholar] [CrossRef] [PubMed]
- Schoubben, A.; Ricci, M.; Giovagnoli, S. Meeting the unmet: From traditional to cutting-edge techniques for poly lactide and poly lactide-co-glycolide microparticle manufacturing. J. Pharm. Investig. 2019, 49, 381–404. [Google Scholar] [CrossRef]
- Semete, B.; Booysen, L.; Lemmer, Y.; Kalombo, L.; Katata, L.; Verschoor, J.; Swai, H.S. In vivo evaluation of the biodistribution and safety of PLGA nanoparticles as drug delivery systems. Nanomedicine 2010, 6, 662–671. [Google Scholar] [CrossRef] [PubMed]
- Woodrow, K.A.; Cu, Y.; Booth, C.J.; Saucier-Sawyer, J.K.; Wood, M.J.; Saltzman, W.M. Intravaginal gene silencing using biodegradable polymer nanoparticles densely loaded with small-interfering RNA. Nat. Mater. 2009, 8, 526–533. [Google Scholar] [CrossRef] [PubMed]
- Swider, E.; Koshkina, O.; Tel, J.; Cruz, L.J.; De Vries, I.J.M.; Srinivas, M. Customizing poly(lactic-co-glycolic acid) particles for biomedical applications. Acta Biomater. 2018, 73, 38–51. [Google Scholar] [CrossRef]
- Mandl, H.K.; Quijano, E.; Suh, H.W.; Sparago, E.; Oeck, S.; Grun, M.; Glazer, P.M.; Saltzman, W.M. Optimizing biodegradable nanoparticle size for tissue-specific delivery. J. Control. Release 2019, 314, 92–101. [Google Scholar] [CrossRef]
- McNeer, N.A.; Schleifman, E.B.; Glazer, P.M.; Saltzman, W.M. Polymer delivery systems for site-specific genome editing. J. Control. Release 2011, 155, 312–316. [Google Scholar] [CrossRef]
- Fields, R.J.; Cheng, C.J.; Quijano, E.; Weller, C.; Kristofik, N.; Duong, N.; Hoimes, C.; Egan, M.E.; Saltzman, W.M. Surface modified poly(β amino ester)-containing nanoparticles for plasmid DNA delivery. J. Control. Release 2012, 164, 41–48. [Google Scholar] [CrossRef] [PubMed]
- Petit, C.; Sancar, A. Nucleotide excision repair: From E. coli to man. Biochimie 1999, 81, 15–25. [Google Scholar] [CrossRef]
- Cubbon, A.; Ivancic-Bace, I.; Bolt, E.L. CRISPR-Cas immunity, DNA repair and genome stability. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed]
- Richardson, C.D.; Kazane, K.R.; Feng, S.J.; Zelin, E.; Bray, N.L.; Schäfer, A.J.; Floor, S.N.; Corn, J.E. CRISPR–Cas9 genome editing in human cells occurs via the Fanconi anemia pathway. Nat. Genet. 2018, 50, 1132–1139. [Google Scholar] [CrossRef]
- Rees, H.A.; Liu, D.R. Base editing: Precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet. 2018, 19, 770–788. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Homology-directed repair of DNA nicks via pathways distinct from canonical double-strand break repair. Proc. Natl. Acad. Sci. USA 2014, 111, E924–E932. [Google Scholar] [CrossRef] [PubMed]
- Davis, L.; Maizels, N. Two Distinct Pathways Support Gene Correction by Single-Stranded Donors at DNA Nicks. Cell Rep. 2016, 17, 1872–1881. [Google Scholar] [CrossRef]
- Lohse, J.; Dahl, O.; Nielsen, P.E. Double duplex invasion by peptide nucleic acid: A general principle for sequence-specific targeting of double-stranded DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 11804–11808. [Google Scholar] [CrossRef]
- Breslauer, K.J.; Remeta, D.P.; Chou, W.Y.; Ferrante, R.; Curry, J.; Zaunczkowski, D.; Snyder, J.G.; Marky, L.A. Enthalpy-entropy compensations in drug-DNA binding studies. Proc. Natl. Acad. Sci. USA 1987, 84, 8922–8926. [Google Scholar] [CrossRef]
- Lonkar, P.; Kim, K.-H.; Kuan, J.Y.; Chin, J.Y.; Rogers, F.A.; Knauert, M.P.; Kole, R.; Nielsen, P.E.; Glazer, P.M. Targeted correction of a thalassemia-associated beta-globin mutation induced by pseudo-complementary peptide nucleic acids. Nucleic Acids Res. 2009, 37, 3635–3644. [Google Scholar] [CrossRef][Green Version]
- Kayali, R.; Bury, F.; Ballard, M.; Bertoni, C. Site-directed gene repair of the dystrophin gene mediated by PNA-ssODNs. Hum. Mol. Genet. 2010, 19, 3266–3281. [Google Scholar] [CrossRef] [PubMed]
- Nik-Ahd, F.; Bertoni, C. Ex vivo gene editing of the dystrophin gene in muscle stem cells mediated by peptide nucleic acid single stranded oligodeoxynucleotides induces stable expression of dystrophin in a mouse model for Duchenne muscular dystrophy. Stem Cells 2014, 32, 1817–1830. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are not available. |




{kind=link}
{kind=link}
{kind=link}
| Guidelines for Triplex PNA Design for Gene Editing |
|---|
|
|
|
|
|
|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Economos, N.G.; Oyaghire, S.; Quijano, E.; Ricciardi, A.S.; Saltzman, W.M.; Glazer, P.M. Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules 2020, 25, 735. https://doi.org/10.3390/molecules25030735
Economos NG, Oyaghire S, Quijano E, Ricciardi AS, Saltzman WM, Glazer PM. Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules. 2020; 25(3):735. https://doi.org/10.3390/molecules25030735
Chicago/Turabian StyleEconomos, Nicholas G., Stanley Oyaghire, Elias Quijano, Adele S. Ricciardi, W. Mark Saltzman, and Peter M. Glazer. 2020. "Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair" Molecules 25, no. 3: 735. https://doi.org/10.3390/molecules25030735
APA StyleEconomos, N. G., Oyaghire, S., Quijano, E., Ricciardi, A. S., Saltzman, W. M., & Glazer, P. M. (2020). Peptide Nucleic Acids and Gene Editing: Perspectives on Structure and Repair. Molecules, 25(3), 735. https://doi.org/10.3390/molecules25030735