Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation





Abstract
1. Introduction
2. Endpoint and qPCR Techniques with Their Applications in Biodegradation Monitoring
2.1. Endpoint PCR
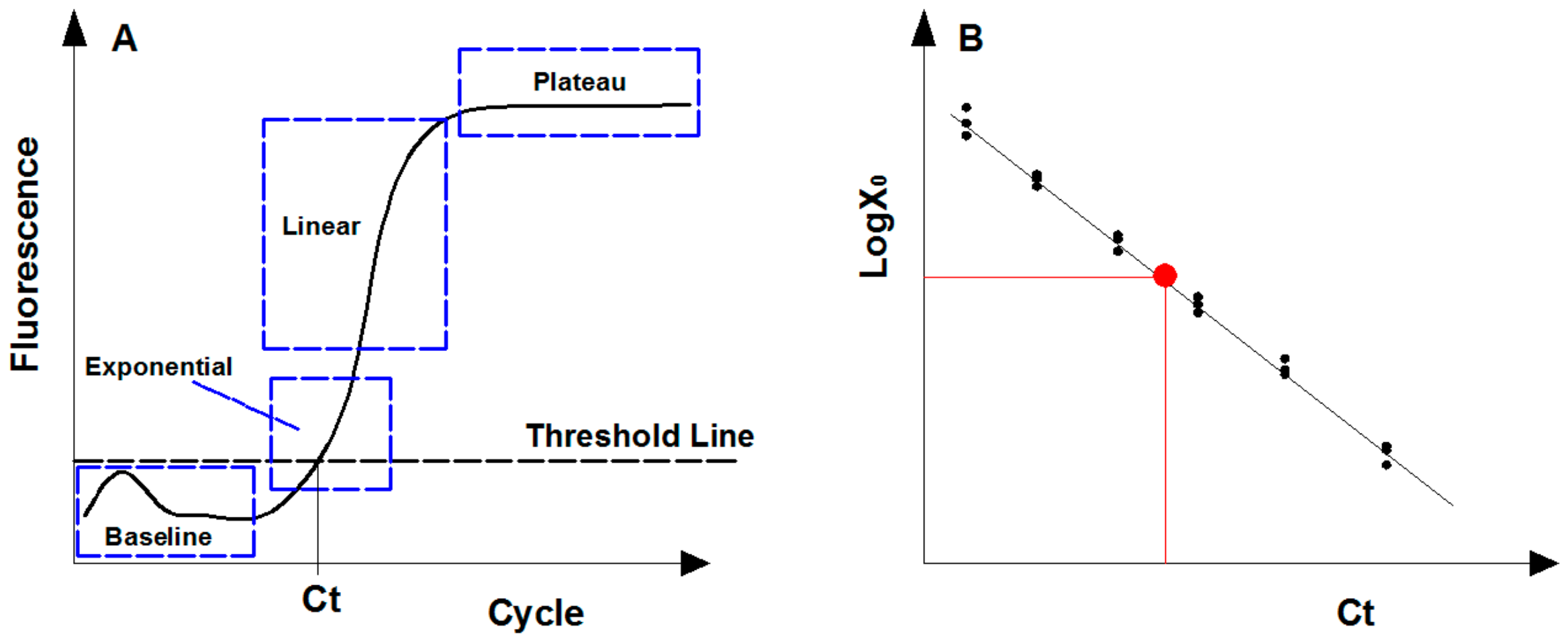
2.2. Real-Time Quantitative PCR
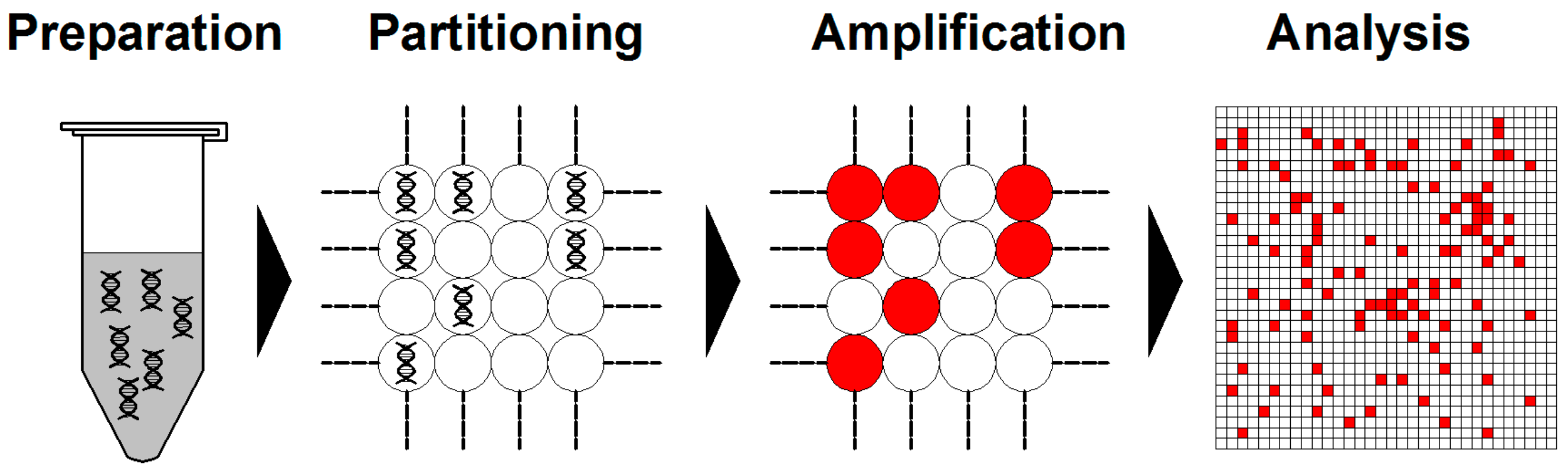
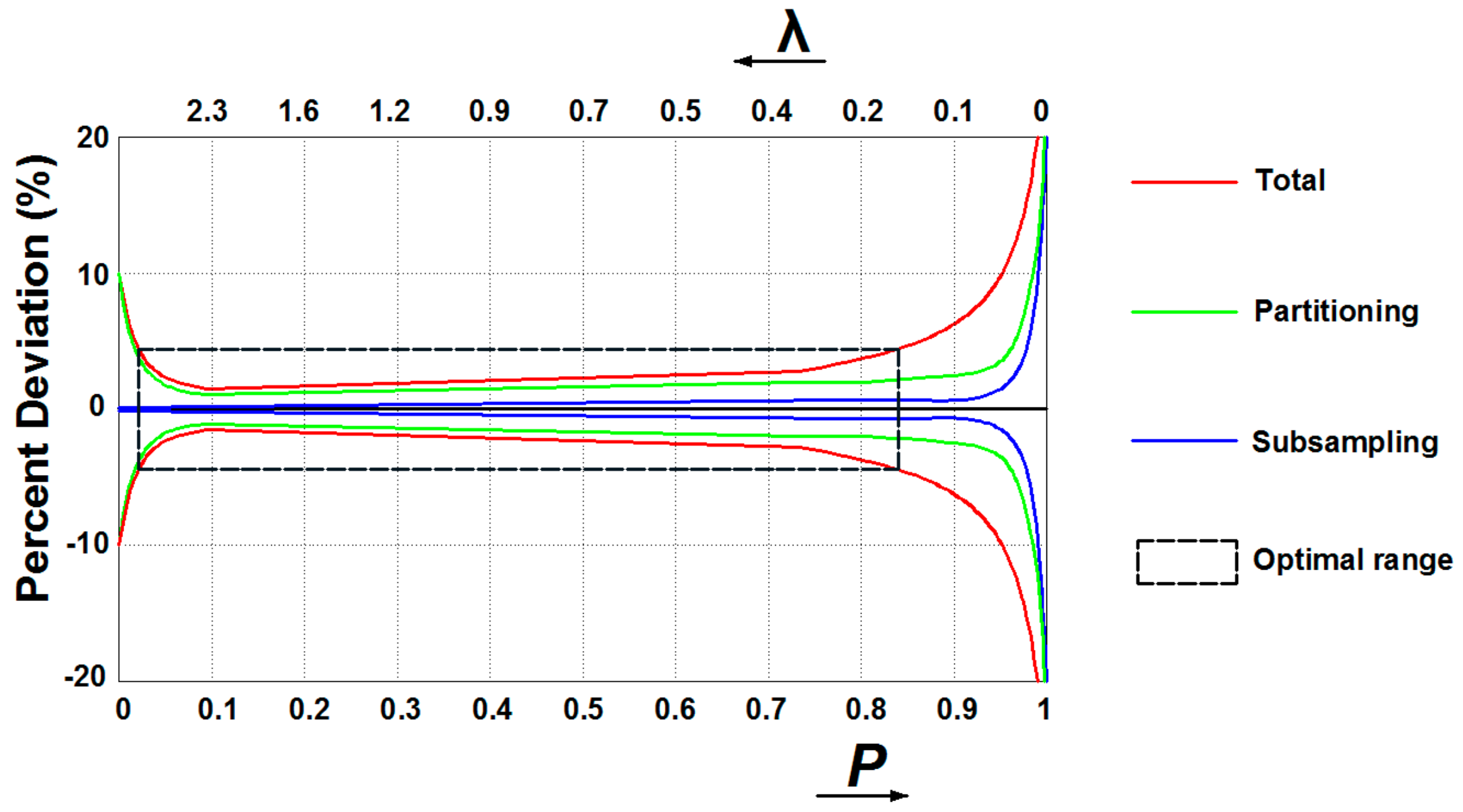
3. Digital PCR and Its Advantages over Previous PCR Techniques
3.1. Digital PCR Systems
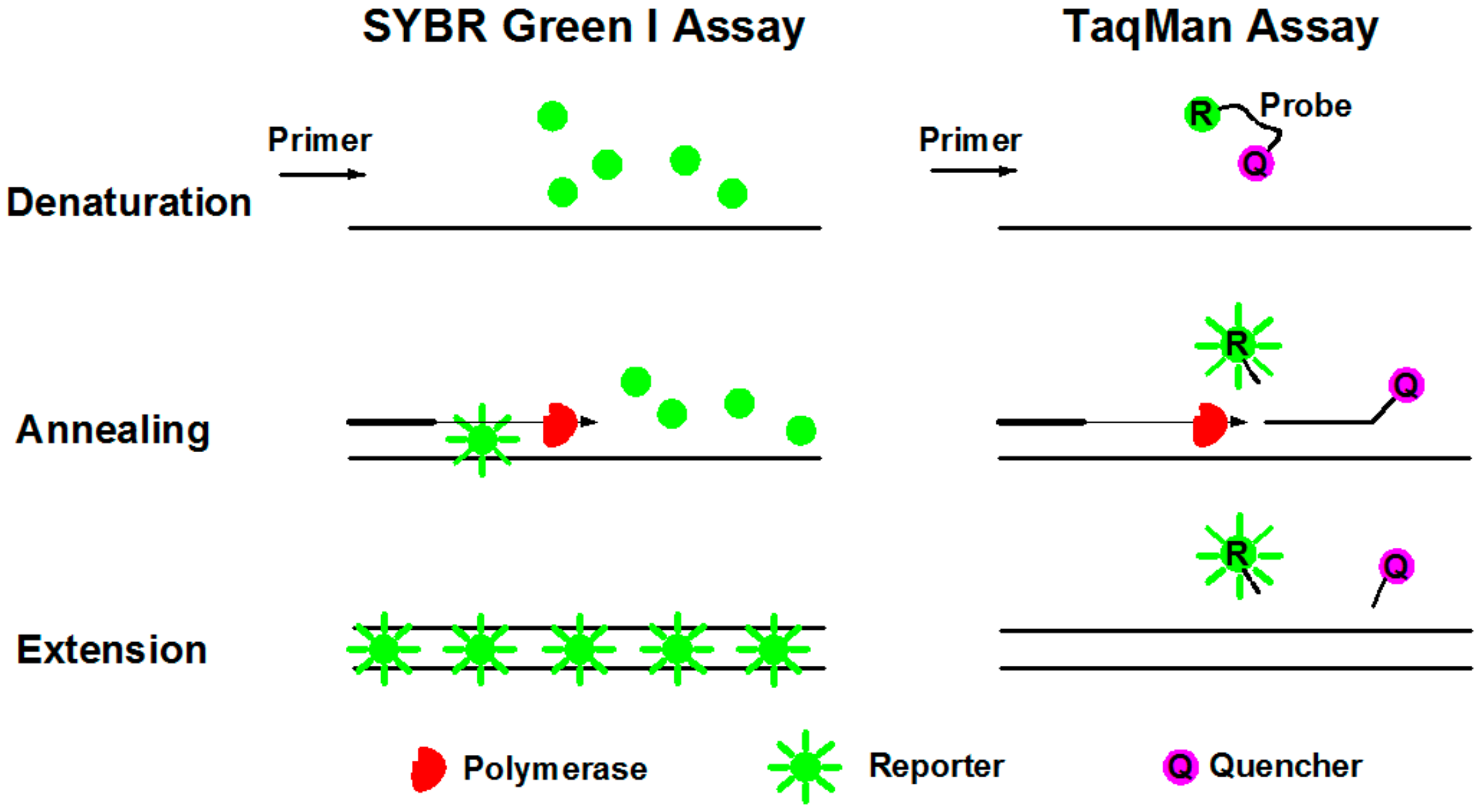
3.2. Fluorescence Reporters in dPCR Systems
4. Applications of dPCR for Monitoring of Biodegradation
4.1. Microbial Enumeration
4.2. Functional Gene Abundance Quantification
4.3. Gene Expressing Determination
5. Limitations of Existing Applications and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Zhang, B.; Matchinski, E.J.; Chen, B.; Ye, X.; Jing, L.; Lee, K. Marine oil spills—Oil pollution, sources and effects. In World Seas: An Environmental Evaluation, 2nd ed.; Sheppard, C., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2019; Volume 3, Chapter 21; pp. 391–406. [Google Scholar]
- Petrie, B.; Barden, R.; Kasprzyk-Hordern, B. A review on emerging contaminants in wastewaters and the environment: Current knowledge, understudied areas and recommendations for future monitoring. Water Res. 2015, 72, 3–27. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.; Boufadel, M.; Chen, B.; Foght, J.; Hodson, P.; Swanson, S.; Venosa, A. Expert Panel Report on the Behaviour and Environmental Impacts of Crude Oil Released into Aqueous Environments; Royal Society of Canada: Ottawa, ON, Canada, 2015; ISBN 978-1-928140-02-3. [Google Scholar]
- Xin, X.; Huang, G.; An, C.; Raina-Fulton, R.; Weger, H. Insights into Long-Term Toxicity of Triclosan to Freshwater Green Algae in Lake Erie. Environ. Sci. Technol. 2019, 53, 2189–2198. [Google Scholar] [CrossRef] [PubMed]
- Xin, X.; Huang, G.; An, C.; Feng, R. Interactive Toxicity of Triclosan and Nano-TiO2 to Green Alga Eremosphaera viridis in Lake Erie: A New Perspective Based on Fourier Transform Infrared Spectromicroscopy and Synchrotron-Based X-ray Fluorescence Imaging. Environ. Sci. Technol. 2019, 53, 9884–9894. [Google Scholar] [CrossRef] [PubMed]
- Lui, M.Y.; Wong, C.Y.Y.; Choi, A.W.-T.; Mui, Y.F.; Qi, L.; Horváth, I.T. Valorization of Carbohydrates of Agricultural Residues and Food Wastes: A Key Strategy for Carbon Conservation. ACS Sustain. Chem. Eng. 2019, 7, 17799–17807. [Google Scholar] [CrossRef]
- Cao, Y.-Q.; Li, Q.; Xia, P.-F.; Wei, L.-J.; Guo, N.; Li, J.-W.; Wang, S.-G. AraBAD based toolkit for gene expression and metabolic robustness improvement in Synechococcus elongatus. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef]
- Chauhan, D.; Agrawal, G.; Deshmukh, S.; Roy, S.S.; Priyadarshini, R. Biofilm formation by Exiguobacterium sp. DR11 and DR14 alter polystyrene surface properties and initiate biodegradation. RSC Adv. 2018, 8, 37590–37599. [Google Scholar] [CrossRef]
- Raddadi, N.; Fava, F. Biodegradation of oil-based plastics in the environment: Existing knowledge and needs of research and innovation. Sci. Total Environ. 2019, 679, 148–158. [Google Scholar] [CrossRef]
- McKew, B.A.; Coulon, F.; Yakimov, M.M.; Denaro, R.; Genovese, M.; Smith, C.J.; Osborn, A.M.; Timmis, K.N.; McGenity, T.J. Efficacy of intervention strategies for bioremediation of crude oil in marine systems and effects on indigenous hydrocarbonoclastic bacteria. Environ. Microbiol. 2007, 9, 1562–1571. [Google Scholar] [CrossRef]
- Chen, Z.; An, C.; Boufadel, M.; Owens, E.; Chen, Z.; Lee, K.; Cao, Y.; Cai, M. Use of Surface-Washing Agents for the Treatment of Oiled Shorelines: Research Advancements, Technical Applications and Future Challenges. Chem. Eng. J. 2019. [Google Scholar] [CrossRef]
- McGenity, T.J.; Folwell, B.D.; McKew, B.A.; Sanni, G.O. Marine crude-oil biodegradation: A central role for interspecies interactions. Aquat. Biosyst. 2012, 8, 10. [Google Scholar] [CrossRef]
- Trueba-Santiso, A.; Fernández-Verdejo, D.; Marco-Rius, I.; Soder-Walz, J.M.; Casabella, O.; Vicent, T.; Marco-Urrea, E. Interspecies interaction and effect of co-contaminants in an anaerobic dichloromethane-degrading culture. Chemosphere 2020, 240, 124877. [Google Scholar] [CrossRef] [PubMed]
- Nikel, P.I.; Pérez-Pantoja, D.; de Lorenzo, V. Pyridine nucleotide transhydrogenases enable redox balance of Pseudomonas putida during biodegradation of aromatic compounds. Environ. Microbiol. 2016, 18, 3565–3582. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, Y.; Feng, H.; Wang, J.; Yang, X.; Wang, Z. Genome-guided identification and characterization of bacteria for simultaneous degradation of polycyclic aromatic hydrocarbons and resistance to hexavalent chromium. Int. Biodeterior. Biodegrad. 2019, 138, 78–86. [Google Scholar] [CrossRef]
- Varjani, S.J. Microbial degradation of petroleum hydrocarbons. Bioresour. Technol. 2017, 223, 277–286. [Google Scholar] [CrossRef]
- Emadian, S.M.; Onay, T.T.; Demirel, B. Biodegradation of bioplastics in natural environments. Waste Manag. 2017, 59, 526–536. [Google Scholar] [CrossRef]
- Chen, Y.-A.; Liu, P.-W.G.; Whang, L.-M.; Wu, Y.-J.; Cheng, S.-S. Biodegradability and microbial community investigation for soil contaminated with diesel blending with biodiesel. Process Saf. Environ. 2019, 130, 115–125. [Google Scholar] [CrossRef]
- Hansen, S.J.; Morovic, W.; DeMeules, M.; Stahl, B.; Sindelar, C.W. Absolute enumeration of probiotic strains Lactobacillus acidophilus NCFM® and Bifidobacterium animalis subsp. lactis Bl-04® via chip-based digital PCR. Front. Microbiol. 2018, 9, 704. [Google Scholar] [CrossRef]
- Handelsman, J. Metagenomics: Application of genomics to uncultured microorganisms. Microbiol. Mol. Biol. Rev. 2004, 68, 669–685. [Google Scholar] [CrossRef]
- Nesslany, F. The current limitations of in vitro genotoxicity testing and their relevance to the in vivo situation. Food Chem. Toxicol. 2017, 106, 609–615. [Google Scholar] [CrossRef]
- Rocca, J.D.; Hall, E.K.; Lennon, J.T.; Evans, S.E.; Waldrop, M.P.; Cotner, J.B.; Nemergut, D.R.; Graham, E.B.; Wallenstein, M.D. Relationships between protein-encoding gene abundance and corresponding process are commonly assumed yet rarely observed. ISME J. 2015, 9, 1693. [Google Scholar] [CrossRef]
- Meng, L.; Li, W.; Bao, M.; Sun, P. Promoting the treatment of crude oil alkane pollution through the study of enzyme activity. Int. J. Biol. Macromol. 2018, 119, 708–716. [Google Scholar] [CrossRef]
- Scopes, R.K. Enzyme activity and assays. Encycl. Life Sci. 2002, 1–6. [Google Scholar] [CrossRef]
- Szczepaniak, Z.; Cyplik, P.; Juzwa, W.; Czarny, J.; Staninska, J.; Piotrowska-Cyplik, A. Antibacterial effect of the Trichoderma viride fungi on soil microbiome during PAH’s biodegradation. Int. Biodeterior. Biodegrad. 2015, 104, 170–177. [Google Scholar] [CrossRef]
- Jahan-Tigh, R.R.; Ryan, C.; Obermoser, G.; Schwarzenberger, K. Flow cytometry. J. Investig. Dermatol. 2012, 132, e1. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-C.; Duan, G.-L.; Ding, K.; Huang, F.-Y.; Zhu, Y.-G. DNA stable-isotope probing identifies uncultivated members of Pseudonocardia associated with biodegradation of pyrene in agricultural soil. FEMS Microbiol. Ecol. 2018, 94, fiy026. [Google Scholar] [CrossRef]
- Steffan, R.; Atlas, R. Polymerase chain reaction: Applications in environmental microbiology. Annu. Rev. Microbiol. 1991, 45, 137–161. [Google Scholar] [CrossRef]
- Pfaffl, M.W.; Vandesompele, J.; Kubista, M. Real-time PCR: Current technology and applications. In Data Analysis Softw.; Logan, J., Edwards, K., Saunders, N., Eds.; Caister Academic Press: Norfold, UK, 2009; pp. 65–83. [Google Scholar]
- Hindson, C.M.; Chevillet, J.R.; Briggs, H.A.; Gallichotte, E.N.; Ruf, I.K.; Hindson, B.J.; Vessella, R.L.; Tewari, M. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nat. Methods 2013, 10, 1003. [Google Scholar] [CrossRef]
- Hall Sedlak, R.; Jerome, K.R. The potential advantages of digital PCR for clinical virology diagnostics. Expert Rev. Mol. Diagn. 2014, 14, 501–507. [Google Scholar] [CrossRef]
- Yang, R.; Paparini, A.; Monis, P.; Ryan, U. Comparison of next-generation droplet digital PCR (ddPCR) with quantitative PCR (qPCR) for enumeration of Cryptosporidium oocysts in faecal samples. Int. J. Parasitol. 2014, 44, 1105–1113. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gärtner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by Droplet Digital PCR through application of a novel open access bioinformatic pipeline, definetherain. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef]
- Doi, H.; Takahara, T.; Minamoto, T.; Matsuhashi, S.; Uchii, K.; Yamanaka, H. Droplet digital polymerase chain reaction (PCR) outperforms real-time PCR in the detection of environmental DNA from an invasive fish species. Environ. Sci. Technol. 2015, 49, 5601–5608. [Google Scholar] [CrossRef] [PubMed]
- Castellanos-Rizaldos, E.; Paweletz, C.; Song, C.; Oxnard, G.R.; Mamon, H.; Jänne, P.A.; Makrigiorgos, G.M. Enhanced ratio of signals enables digital mutation scanning for rare allele detection. J. Mol. Diagn. 2015, 17, 284–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Xu, P.; Zeng, G.; Yang, C.; Huang, D.; Zhang, J. Bioremediation of soils contaminated with polycyclic aromatic hydrocarbons, petroleum, pesticides, chlorophenols and heavy metals by composting: Applications, microbes and future research needs. Biotechnol. Adv. 2015, 33, 745–755. [Google Scholar] [CrossRef]
- Wiencke, J.K.; Bracci, P.M.; Hsuang, G.; Zheng, S.; Hansen, H.; Wrensch, M.R.; Rice, T.; Eliot, M.; Kelsey, K.T. A comparison of DNA methylation specific droplet digital PCR (ddPCR) and real time qPCR with flow cytometry in characterizing human T cells in peripheral blood. Epigenetics 2014, 9, 1360–1365. [Google Scholar] [CrossRef] [PubMed]
- Arnheim, N.; Erlich, H. Polymerase chain reaction strategy. Annu. Rev. Biochem. 1992, 61, 131–156. [Google Scholar] [CrossRef] [PubMed]
- Kainz, P. The PCR plateau phase–towards an understanding of its limitations. Biochim. Biophys. Acta Gene Struct. Expr. 2000, 1494, 23–27. [Google Scholar] [CrossRef]
- Deschaght, P.; De Baere, T.; Van Simaey, L.; De Baets, F.; De Vos, D.; Pirnay, J.-P.; Vaneechoutte, M. Comparison of the sensitivity of culture, PCR and quantitative real-time PCR for the detection of Pseudomonas aeruginosa in sputum of cystic fibrosis patients. BMC Microbiol. 2009, 9, 244. [Google Scholar] [CrossRef]
- Antiabong, J.F.; Ngoepe, M.G.; Abechi, A.S. Semi-quantitative digital analysis of polymerase chain reaction-electrophoresis gel: Potential applications in low-income veterinary laboratories. Vet. World 2016, 9, 935. [Google Scholar] [CrossRef]
- Mauffrey, F.; Baccara, P.-Y.; Gruffaz, C.; Vuilleumier, S.; Imfeld, G. Bacterial community composition and genes for herbicide degradation in a stormwater wetland collecting herbicide runoff. Water Air Soil Pollut. 2017, 228, 452. [Google Scholar] [CrossRef]
- Andrade, L.L.; Leite, D.C.; Ferreira, E.M.; Ferreira, L.Q.; Paula, G.R.; Maguire, M.J.; Hubert, C.R.; Peixoto, R.S.; Domingues, R.M.; Rosado, A.S. Microbial diversity and anaerobic hydrocarbon degradation potential in an oil-contaminated mangrove sediment. BMC Microbiol. 2012, 12, 186. [Google Scholar] [CrossRef]
- Oka, A.R.; Phelps, C.D.; Zhu, X.; Saber, D.L.; Young, L. Dual biomarkers of anaerobic hydrocarbon degradation in historically contaminated groundwater. Environ. Sci. Technol. 2011, 45, 3407–3414. [Google Scholar] [CrossRef] [PubMed]
- Muyzer, G. DGGE/TGGE a method for identifying genes from natural ecosystems. Curr. Opin. Microbiol. 1999, 2, 317–322. [Google Scholar] [CrossRef]
- Luo, Y.; Tian, Y.; Huang, X.; Yan, C.; Hong, H.; Lin, G.; Zheng, T. Analysis of community structure of a microbial consortium capable of degrading benzo (a) pyrene by DGGE. Mar. Pollut. Bull. 2009, 58, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Arya, M.; Shergill, I.S.; Williamson, M.; Gommersall, L.; Arya, N.; Patel, H.R. Basic principles of real-time quantitative PCR. Expert Rev. Mol. Diagn. 2005, 5, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Villalba, A.; van Pelt-Verkuil, E.; Gunst, Q.D.; Ruijter, J.M.; van den Hoff, M.J. Amplification of nonspecific products in quantitative polymerase chain reactions (qPCR). Biomol. Detect. Quantif. 2017, 14, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Feretzaki, M.; Lingner, J. A practical qPCR approach to detect TERRA, the elusive telomeric repeat-containing RNA. Methods 2017, 114, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Larionov, A.; Krause, A.; Miller, W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005, 6, 62. [Google Scholar] [CrossRef]
- Dhanasekaran, S.; Doherty, T.M.; Kenneth, J.; Group, T.T.S. Comparison of different standards for real-time PCR-based absolute quantification. J. Immunol. Methods 2010, 354, 34–39. [Google Scholar] [CrossRef]
- Wang, D.; Yamahara, K.M.; Cao, Y.; Boehm, A.B. Absolute quantification of Enterococcal 23S rRNA gene using digital PCR. Environ. Sci. Technol. 2016, 50, 3399–3408. [Google Scholar] [CrossRef]
- Wang, D.; Green, H.C.; Shanks, O.C.; Boehm, A.B. New performance metrics for quantitative polymerase chain reaction-based microbial source tracking methods. Environ. Sci. Technol. Lett. 2013, 1, 20–25. [Google Scholar] [CrossRef]
- Laurie, A.D.; Lloyd-Jones, G. Quantification of phnAc andnahAc in contaminated New Zealand soils by competitive PCR. Appl. Environ. Microbiol. 2000, 66, 1814–1817. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Shimizu, K.; Sakharkar, M.K.; Utsumi, M.; Zhang, Z.; Sugiura, N. Comparative study for the effects of variable nutrient conditions on the biodegradation of microcystin-LR and concurrent dynamics in microcystin-degrading gene abundance. Bioresour. Technol. 2011, 102, 9509–9517. [Google Scholar] [CrossRef] [PubMed]
- Wilson, F.P.; Liu, X.; Mattes, T.E.; Cupples, A.M. Nocardioides, Sediminibacterium, Aquabacterium, Variovorax, and Pseudomonas linked to carbon uptake during aerobic vinyl chloride biodegradation. Environ. Sci. Pollut. Res. 2016, 23, 19062–19070. [Google Scholar] [CrossRef] [PubMed]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef] [PubMed]
- Baker, M. Digital PCR hits its stride. Nat. Methods 2012, 9, 541–544. [Google Scholar] [CrossRef]
- Majumdar, N.; Banerjee, S.; Pallas, M.; Wessel, T.; Hegerich, P. Poisson plus quantification for digital PCR systems. Sci. Rep. 2017, 7, 9617. [Google Scholar] [CrossRef]
- Quan, P.-L.; Sauzade, M.; Brouzes, E. dPCR: A technology review. Sensors 2018, 18, 1271. [Google Scholar] [CrossRef]
- Whale, A.S.; Cowen, S.; Foy, C.A.; Huggett, J.F. Methods for applying accurate digital PCR analysis on low copy DNA samples. PLoS ONE 2013, 8, e58177. [Google Scholar] [CrossRef] [PubMed]
- Morley, A.A. Digital PCR: A brief history. Biomol. Detect. Quantif. 2014, 1, 1–2. [Google Scholar] [CrossRef]
- Groth, S.F.D.S. The evaluation of limiting dilution assays. J. Immunol. Methods 1982, 49, R11–R23. [Google Scholar] [CrossRef]
- Jacobs, B.K.; Goetghebeur, E.; Clement, L. Impact of variance components on reliability of absolute quantification using digital PCR. BMC Bioinform. 2014, 15, 283. [Google Scholar] [CrossRef] [PubMed]
- Bizouarn, F. Introduction to digital PCR. Methods Mol. Biol. 2014, 1160, 27–41. [Google Scholar] [PubMed]
- Dube, S.; Qin, J.; Ramakrishnan, R. Mathematical analysis of copy number variation in a DNA sample using digital PCR on a nanofluidic device. PLoS ONE 2008, 3, e2876. [Google Scholar] [CrossRef] [PubMed]
- Cao, L.; Cui, X.; Hu, J.; Li, Z.; Choi, J.R.; Yang, Q.; Lin, M.; Hui, L.Y.; Xu, F. Advances in digital polymerase chain reaction (dPCR) and its emerging biomedical applications. Biosens. Bioelectron. 2017, 90, 459–474. [Google Scholar] [CrossRef]
- Broeders, S.; Huber, I.; Grohmann, L.; Berben, G.; Taverniers, I.; Mazzara, M.; Roosens, N.; Morisset, D. Guidelines for validation of qualitative real-time PCR methods. Trends Food Sci. Technol. 2014, 37, 115–126. [Google Scholar] [CrossRef]
- Dragan, A.; Pavlovic, R.; McGivney, J.; Casas-Finet, J.; Bishop, E.; Strouse, R.; Schenerman, M.; Geddes, C. SYBR Green I: Fluorescence properties and interaction with DNA. J. Fluoresc. 2012, 22, 1189–1199. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Lu, X.; Su, F.; Wang, L.; Liu, C.; Duan, X.; Li, Z. Real-time fluorescence ligase chain reaction for sensitive detection of single nucleotide polymorphism based on fluorescence resonance energy transfer. Biosens. Bioelectron. 2015, 74, 705–710. [Google Scholar] [CrossRef]
- Martin-Sanchez, P.M.; Gorbushina, A.A.; Toepel, J. Quantification of microbial load in diesel storage tanks using culture-and qPCR-based approaches. Int. Biodeterior. Biodegrad. 2018, 126, 216–223. [Google Scholar] [CrossRef]
- Brown, M.V.; Fuhrman, J.A. Marine bacterial microdiversity as revealed by internal transcribed spacer analysis. Aquat. Microb. Ecol. 2005, 41, 15–23. [Google Scholar] [CrossRef]
- Pornwongthong, P.; Mulchandani, A.; Gedalanga, P.B.; Mahendra, S. Transition metals and organic ligands influence biodegradation of 1, 4-dioxane. Appl. Biochem. Biotechnol. 2014, 173, 291–306. [Google Scholar] [CrossRef]
- Bücker, F.; de Moura, T.M.; da Cunha, M.E.; de Quadros, P.D.; Beker, S.A.; Cazarolli, J.C.; Caramão, E.B.; Frazzon, A.P.G.; Bento, F.M. Evaluation of the deteriogenic microbial community using qPCR, n-alkanes and FAMEs biodegradation in diesel, biodiesel and blends (B5, B10, and B50) during storage. Fuel 2018, 233, 911–917. [Google Scholar] [CrossRef]
- Fayeulle, A.; Veignie, E.; Schroll, R.; Munch, J.C.; Rafin, C. PAH biodegradation by telluric saprotrophic fungi isolated from aged PAH-contaminated soils in mineral medium and historically contaminated soil microcosms. J. Soils Sediments 2019, 19, 3056–3067. [Google Scholar] [CrossRef]
- Richardson, E.L.; King, C.K.; Powell, S.M. The use of microbial gene abundance in the development of fuel remediation guidelines in polar soils. Integr. Environ. Assess. 2015, 11, 235–241. [Google Scholar] [CrossRef] [PubMed]
- Krolicka, A.; Boccadoro, C.; Nilsen, M.M.; Demir-Hilton, E.; Birch, J.; Preston, C.; Scholin, C.; Baussant, T. Identification of microbial key-indicators of oil contamination at sea through tracking of oil biotransformation: An Arctic field and laboratory study. Sci. Total Environ. 2019, 696, 133715. [Google Scholar] [CrossRef]
- Ren, L.; Jia, Y.; Ruth, N.; Qiao, C.; Wang, J.; Zhao, B.; Yan, Y. Biodegradation of phthalic acid esters by a newly isolated Mycobacterium sp. YC-RL4 and the bioprocess with environmental samples. Environ. Sci. Pollut. Res. 2016, 23, 16609–16619. [Google Scholar] [CrossRef]
- Chang, H.-W.; Sung, Y.; Kim, K.-H.; Nam, Y.-D.; Roh, S.W.; Kim, M.-S.; Jeon, C.O.; Bae, J.-W. Development of microbial genome-probing microarrays using digital multiple displacement amplification of uncultivated microbial single cells. Environ. Sci. Technol. 2008, 42, 6058–6064. [Google Scholar] [CrossRef]
- Hoshino, T.; Inagaki, F. Molecular quantification of environmental DNA using microfluidics and digital PCR. Syst. Appl. Microbiol. 2012, 35, 390–395. [Google Scholar] [CrossRef]
- Kim, T.G.; Jeong, S.-Y.; Cho, K.-S. Comparison of droplet digital PCR and quantitative real-time PCR for examining population dynamics of bacteria in soil. Appl. Biochem. Biotechnol. 2014, 98, 6105–6113. [Google Scholar] [CrossRef]
- US EPA. Method 1611: Enterococci in Water by TaqMan® Quantitative Polymerase Chain Reaction (qPCR) Assay; US EPA: Washington, DC, USA, 2012.
- Bian, X.; Jing, F.; Li, G.; Fan, X.; Jia, C.; Zhou, H.; Jin, Q.; Zhao, J. A microfluidic droplet digital PCR for simultaneous detection of pathogenic Escherichia coli O157 and Listeria monocytogenes. Biosens. Bioelectron. 2015, 74, 770–777. [Google Scholar] [CrossRef]
- Cao, Y.; Raith, M.R.; Griffith, J.F. Droplet digital PCR for simultaneous quantification of general and human-associated fecal indicators for water quality assessment. Water Res. 2015, 70, 337–349. [Google Scholar] [CrossRef]
- Te, S.H.; Chen, E.Y.; Gin, K.Y.-H. Comparison of quantitative PCR and droplet digital PCR multiplex assays for two genera of bloom-forming cyanobacteria, Cylindrospermopsis and Microcystis. Appl. Environ. Microbiol. 2015, 81, 5203–5211. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lee, A.H.; Chang, W. Enhanced bioremediation of nutrient-amended, petroleum hydrocarbon-contaminated soils over a cold-climate winter: The rate and extent of hydrocarbon biodegradation and microbial response in a pilot-scale biopile subjected to natural seasonal freeze-thaw temperatures. Sci. Total Environ. 2018, 612, 903–913. [Google Scholar] [PubMed]
- Dong, L.; Meng, Y.; Wang, J.; Liu, Y. Evaluation of droplet digital PCR for characterizing plasmid reference material used for quantifying ammonia oxidizers and denitrifiers. Anal. Bioanal. Chem. 2014, 406, 1701–1712. [Google Scholar] [CrossRef]
- Ren, S.; Boo, C.; Guo, N.; Wang, S.; Elimelech, M.; Wang, Y. Photocatalytic reactive ultrafiltration membrane for removal of antibiotic resistant bacteria and antibiotic resistance genes from wastewater effluent. Environ. Sci. Technol. 2018, 52, 8666–8673. [Google Scholar] [CrossRef]
- Cavé, L.; Brothier, E.; Abrouk, D.; Bouda, P.S.; Hien, E.; Nazaret, S. Efficiency and sensitivity of the digital droplet PCR for the quantification of antibiotic resistance genes in soils and organic residues. Appl. Microbiol. Biotechnol. 2016, 100, 10597–10608. [Google Scholar] [CrossRef]
- Gao, M.; Qiu, T.; Sun, Y.; Wang, X. The abundance and diversity of antibiotic resistance genes in the atmospheric environment of composting plants. Environ. Int. 2018, 116, 229–238. [Google Scholar] [CrossRef]
- Liu, Z.; He, Z.; Huang, H.; Ran, X.; Oluwafunmilayo, A.O.; Lu, Z. pH stress-induced cooperation between Rhodococcus ruber YYL and Bacillus cereus MLY1 in biodegradation of tetrahydrofuran. Front. Microbiol. 2017, 8, 2297. [Google Scholar] [CrossRef]
- Shahi, A.; Aydin, S.; Ince, B.; Ince, O. Evaluation of microbial population and functional genes during the bioremediation of petroleum-contaminated soil as an effective monitoring approach. Ecotoxicol. Environ. Saf. 2016, 125, 153–160. [Google Scholar] [CrossRef] [PubMed]
- Ren, C.; Wang, Y.; Tian, L.; Chen, M.; Sun, J.; Li, L. Genetic bioaugmentation of activated sludge with dioxin-catabolic plasmids harbored by Rhodococcus sp. strain p52. Environ. Sci. Technol. 2018, 52, 5339–5348. [Google Scholar] [CrossRef] [PubMed]
- Ottesen, E.A.; Hong, J.W.; Quake, S.R.; Leadbetter, J.R. Microfluidic digital PCR enables multigene analysis of individual environmental bacteria. Science 2006, 314, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Collier, R.; Dasgupta, K.; Xing, Y.P.; Hernandez, B.T.; Shao, M.; Rohozinski, D.; Kovak, E.; Lin, J.; de Oliveira, M.L.P.; Stover, E. Accurate measurement of transgene copy number in crop plants using droplet digital PCR. Plant J. 2017, 90, 1014–1025. [Google Scholar] [CrossRef] [PubMed]
- Gedalanga, P.B.; Pornwongthong, P.; Mora, R.; Chiang, S.-Y.D.; Baldwin, B.; Ogles, D.; Mahendra, S. Identification of biomarker genes to predict biodegradation of 1, 4-dioxane. Appl. Environ. Microbiol. 2014, 80, 3209–3218. [Google Scholar] [CrossRef] [PubMed]
- Bælum, J.; Nicolaisen, M.H.; Holben, W.E.; Strobel, B.W.; Sørensen, J.; Jacobsen, C.S. Direct analysis of tfdA gene expression by indigenous bacteria in phenoxy acid amended agricultural soil. ISME J. 2008, 2, 677. [Google Scholar] [CrossRef] [PubMed]
- Wagner, A.; Adrian, L.; Kleinsteuber, S.; Andreesen, J.R.; Lechner, U. Transcription analysis of genes encoding homologues of reductive dehalogenases in Dehalococcoides sp. strain CBDB1 by using terminal restriction fragment length polymorphism and quantitative PCR. Appl. Environ. Microbiol. 2009, 75, 1876–1884. [Google Scholar] [CrossRef][Green Version]
- Von Netzer, F.; Kuntze, K.; Vogt, C.; Richnow, H.H.; Boll, M.; Lueders, T. Functional gene markers for fumarate-adding and dearomatizing key enzymes in anaerobic aromatic hydrocarbon degradation in terrestrial environments. J. Mol. Microbiol. Biotechnol. 2016, 26, 180–194. [Google Scholar] [CrossRef]
- Huggett, J.F.; Foy, C.A.; Benes, V.; Emslie, K.; Garson, J.A.; Haynes, R.; Hellemans, J.; Kubista, M.; Mueller, R.D.; Nolan, T. The digital MIQE guidelines: Minimum information for publication of quantitative digital PCR experiments. Clin. Chem. 2013, 59, 892–902. [Google Scholar] [CrossRef]
- Taylor, S.C.; Laperriere, G.; Germain, H. Droplet Digital PCR versus qPCR for gene expression analysis with low abundant targets: From variable nonsense to publication quality data. Sci. Rep. 2017, 7, 2409. [Google Scholar] [CrossRef]
- Kaitu’u-Lino, T.; Hastie, R.; Cannon, P.; Lee, S.; Stock, O.; Hannan, N.J.; Hiscock, R.; Tong, S. Stability of absolute copy number of housekeeping genes in preeclamptic and normal placentas, as measured by digital PCR. Placenta 2014, 35, 1106–1109. [Google Scholar] [CrossRef]
- Zhang, S.; Sun, X.; Wang, X.; Qiu, T.; Gao, M.; Sun, Y.; Cheng, S.; Zhang, Q. Bioaugmentation with Diaphorobacter polyhydroxybutyrativorans to enhance nitrate removal in a poly (3-hydroxybutyrate-co-3-hydroxyvalerate)-supported denitrification reactor. Bioresour. Technol. 2018, 263, 499–507. [Google Scholar] [CrossRef]
- Segawa, T.; Ishii, S.; Ohte, N.; Akiyoshi, A.; Yamada, A.; Maruyama, F.; Li, Z.; Hongoh, Y.; Takeuchi, N. The nitrogen cycle in cryoconites: Naturally occurring nitrification-denitrification granules on a glacier. Environ. Microbiol. 2014, 16, 3250–3262. [Google Scholar] [CrossRef]
- Basu, A.S. Digital assays part I: Partitioning statistics and digital PCR. SLAS Technol. 2017, 22, 369–386. [Google Scholar] [CrossRef]
- Sedlak, R.H.; Nguyen, T.; Palileo, I.; Jerome, K.R.; Kuypers, J. Superiority of digital reverse transcription-PCR (RT-PCR) over real-time RT-PCR for quantitation of highly divergent human rhinoviruses. J. Clin. Microbiol. 2017, 55, 442–449. [Google Scholar] [CrossRef]
- Suslov, O.; Steindler, D.A. PCR inhibition by reverse transcriptase leads to an overestimation of amplification efficiency. Nucleic Acids Res. 2005, 33, e181. [Google Scholar] [CrossRef]
- Zmienko, A.; Samelak-Czajka, A.; Goralski, M.; Sobieszczuk-Nowicka, E.; Kozlowski, P.; Figlerowicz, M. Selection of reference genes for qPCR-and ddPCR-based analyses of gene expression in senescing barley leaves. PLoS ONE 2015, 10, e0118226. [Google Scholar] [CrossRef]
- Vasina, D.V.; Moiseenko, K.V.; Fedorova, T.V.; Tyazhelova, T.V. Lignin-degrading peroxidases in white-rot fungus Trametes hirsuta 072. Absolute expression quantification of full multigene family. PLoS ONE 2017, 12, e0173813. [Google Scholar] [CrossRef] [PubMed]
- Green, H.C.; Field, K.G. Sensitive detection of sample interference in environmental qPCR. Water Res. 2012, 46, 3251–3260. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.; Zhang, H.; Miranda, L.; Lin, S. Serious overestimation in quantitative PCR by circular (supercoiled) plasmid standard: Microalgal pcna as the model gene. PLoS ONE 2010, 5, e9545. [Google Scholar] [CrossRef] [PubMed]
- Svec, D.; Tichopad, A.; Novosadova, V.; Pfaffl, M.W.; Kubista, M. How good is a PCR efficiency estimate: Recommendations for precise and robust qPCR efficiency assessments. Biomol. Detect. Quantif. 2015, 3, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, L.B.; Coleman, V.A.; Hindson, C.M.; Herrmann, J.; Hindson, B.J.; Bhat, S.; Emslie, K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2011, 84, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Case, R.J.; Boucher, Y.; Dahllöf, I.; Holmström, C.; Doolittle, W.F.; Kjelleberg, S. Use of 16S rRNA and rpoB genes as molecular markers for microbial ecology studies. Appl. Environ. Microbiol. 2007, 73, 278–288. [Google Scholar] [CrossRef] [PubMed]
- Weaver, S.; Dube, S.; Mir, A.; Qin, J.; Sun, G.; Ramakrishnan, R.; Jones, R.C.; Livak, K.J. Taking qPCR to a higher level: Analysis of CNV reveals the power of high throughput qPCR to enhance quantitative resolution. Methods 2010, 50, 271–276. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Chung, W.K. Quantitative analysis of copy number variants based on real-time LightCycler PCR. Curr. Protoc. Hum. Genet. 2014, 80, 7–21. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Description | Advantages | Disadvantages | Platform |
|---|---|---|---|
| Microbial enumeration | |||
| Quantifying the 16S rRNA and GyrB markers to assess temporal variability of oleophilic bacteria in seawater when facing oil contaminants | Employment of ddPCR to determine single copy number markers like GryB gene, which is sometimes undetectable through qPCR | qPCR; ddPCR | |
| Quantifying the V3-V4 region of 16S rRNA to explore the dynamic change of inoculated Mycobacterium sp. YC-RL4 in the soil for phthalic acid esters degradation | Absolute quantification of specific microbes in complex environments with known copy number of 16S rRNA | ddPCR | |
| Quantifying the 16S rRNA gene of 15 key degraders to uncover the microbial population dynamics in a given culture for dichloromethane dichlorination | Directly monitoring of single uncultivated bacterial cells and their diversity | cdPCR | |
| Quantifying the 16S rRNA genes to compare archaea abundance and evaluating the PCR-inhibitory effects of substances from soil and marine subsurface sediments | Accurate and absolute quantification with little inhibitory effects | qPCR; cdPCR | |
| Quantifying the 16S rRNA using isolated Cupriavidus sp. MBT14 and Sphingopyxis sp. MD2 as model strains to identify population dynamics in soil | Standard curve unrequired; high sensitivity and efficiency for multi targets measurement; less variability among labs | Time consumption for droplets generation; expensive reaction regents; more steps required than qPCR | qPCR; ddPCR |
| Quantifying the 23S rRNA to enumerate Enterococci to assess water quality | Standard curve unrequired; accurate quantification; less affected by inhibitors comparing with qPCR and inhibition could be relieved by dilution | qPCR; cdPCR | |
| Quantifying rfbE and prfA genes simultaneously to detect pathogenic bacterial contamination (i.e., E. coli O157: H7 and L. monocytogenes) in water | Simultaneous genes detection via two-color fluorescence probes without cross-assay interference; high accuracy and sensitivity; low detection limit | qPCR; ddPCR | |
| Functional gene abundance quantification | |||
| Quantifying the copy number variation of alkB1 gene to assess the biodegradation potential of nutrient-amended petroleum hydrocarbon-contaminated soil | Absolute quantification without standard curve | cdPCR | |
| Quantifying the copy number variation of nosZ, nirS and amoA genes in plasmid DNA to assess nitrification and denitrification | Independent of DNA standards | Two measurement bias: (1) plasmid DNA and (2) droplet volume. Linearizing plasmid DNA through restriction and correcting droplet volume could improve reliability and accuracy | ddPCR |
| Quantifying low copy number variation of antibiotic resistance genes Sul1 and qnrB in soil | High sensitivity; lower detection limit; less affected by environmental DNA templates | Lower range of quantification than qPCR | qPCR; ddPCR |
| Quantifying 22 antibiotic resistance genes in composting plants’ atmosphere to assess ecological risk of composts | Absolute and accurate quantification without standard curve | ddPCR | |
| Quantifying transgene behavior of hptII, nptII, bar, ZmUBI1p genes between crop plants | Accurate and efficient determination of transgene copy number; high reliability | ddPCR | |
| Gene expression determination | |||
| Quantifying expressions of narG, nirK and nirS genes in biofilm samples to assess nitrate degradation in denitrification bioreactor with bioaugmented Diaphorobacter | High precision and tolerance to inhibitors and better for complex environmental samples | No reference genes applied may cause inaccuracy | RT-ddPCR |
| Quantifying expression of amoA, narG, nirK and nosZ genes to assess nitrogen cycle in cryoconites | Absolute quantification without standard curve | RT-cdPCR | |
| Quantifying expression of Lip, mnp, vp genes refer to tubulin gene to assess lignin degradation in soil | Absolute quantification; accurate quantification using reference gene; reliable and reproducible measurements of small changes for low abundant cDNA | RT-ddPCR | |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cao, Y.; Yu, M.; Dong, G.; Chen, B.; Zhang, B. Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation. Molecules 2020, 25, 706. https://doi.org/10.3390/molecules25030706
Cao Y, Yu M, Dong G, Chen B, Zhang B. Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation. Molecules. 2020; 25(3):706. https://doi.org/10.3390/molecules25030706
Chicago/Turabian StyleCao, Yiqi, Miao Yu, Guihua Dong, Bing Chen, and Baiyu Zhang. 2020. "Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation" Molecules 25, no. 3: 706. https://doi.org/10.3390/molecules25030706
APA StyleCao, Y., Yu, M., Dong, G., Chen, B., & Zhang, B. (2020). Digital PCR as an Emerging Tool for Monitoring of Microbial Biodegradation. Molecules, 25(3), 706. https://doi.org/10.3390/molecules25030706