
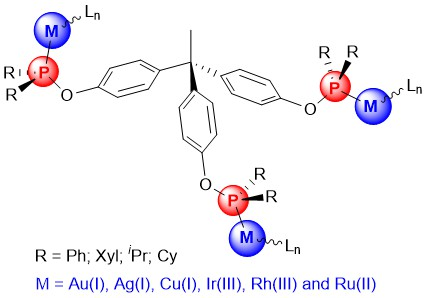
A Versatile Approach to Access Trimetallic Complexes Based on Trisphosphinite Ligands

 ,
,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
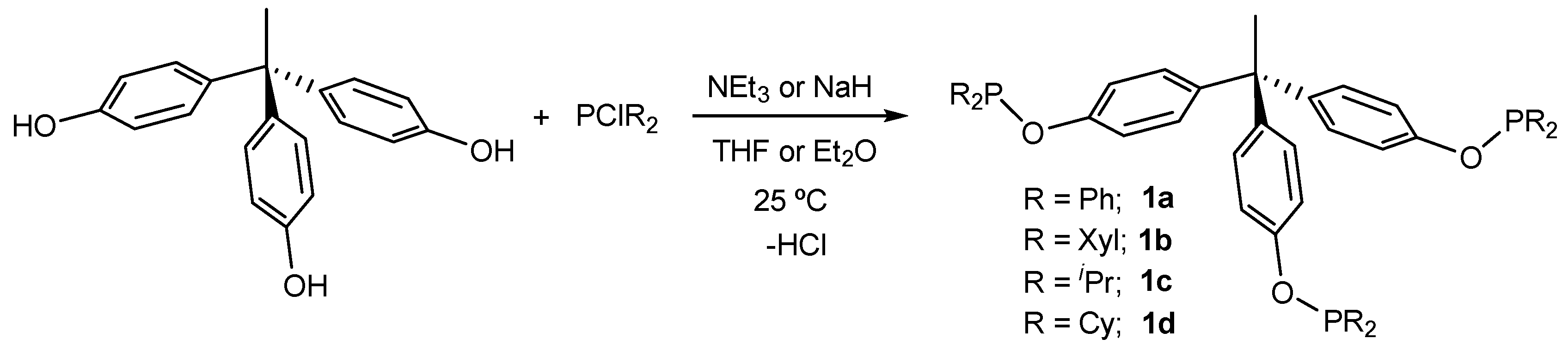{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
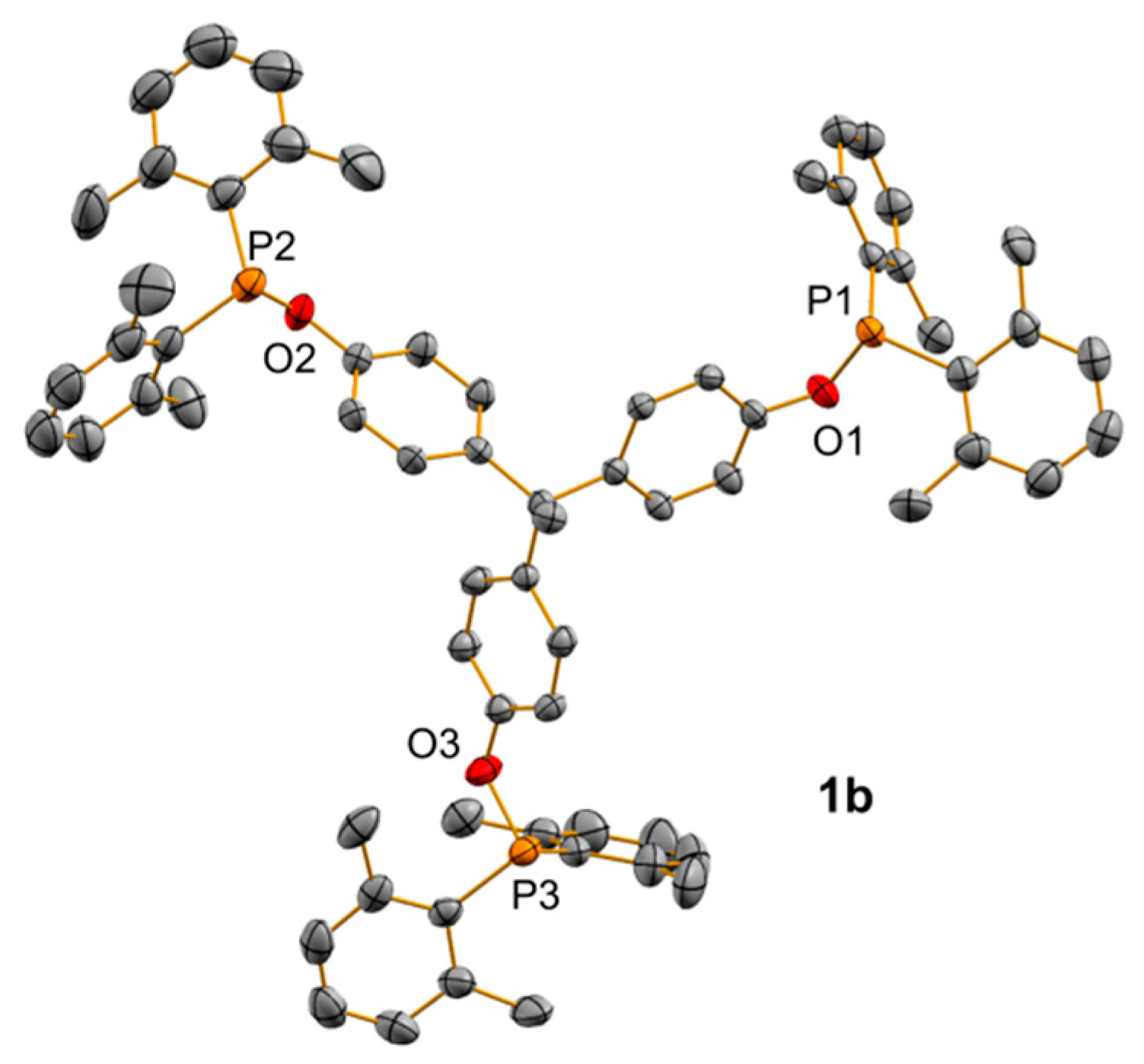
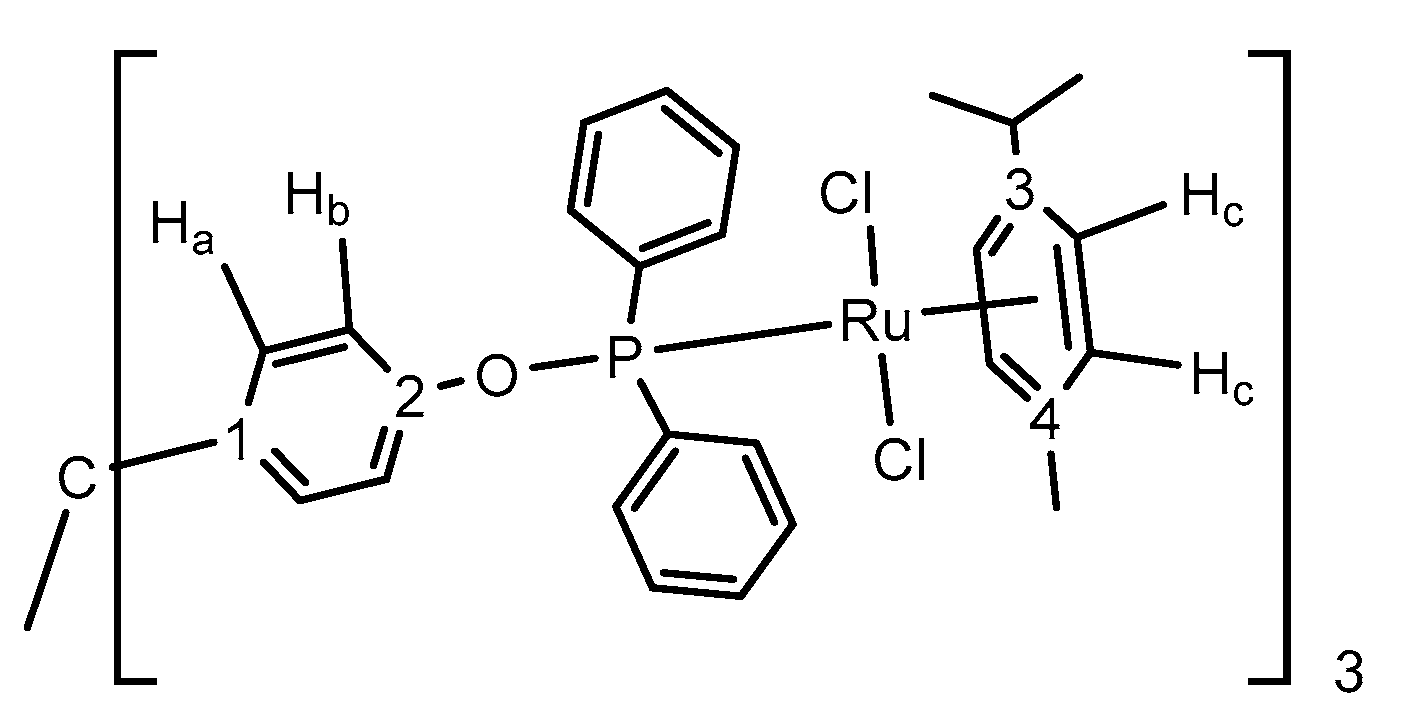
2.1. Synthesis of Tripodal Phosphinites
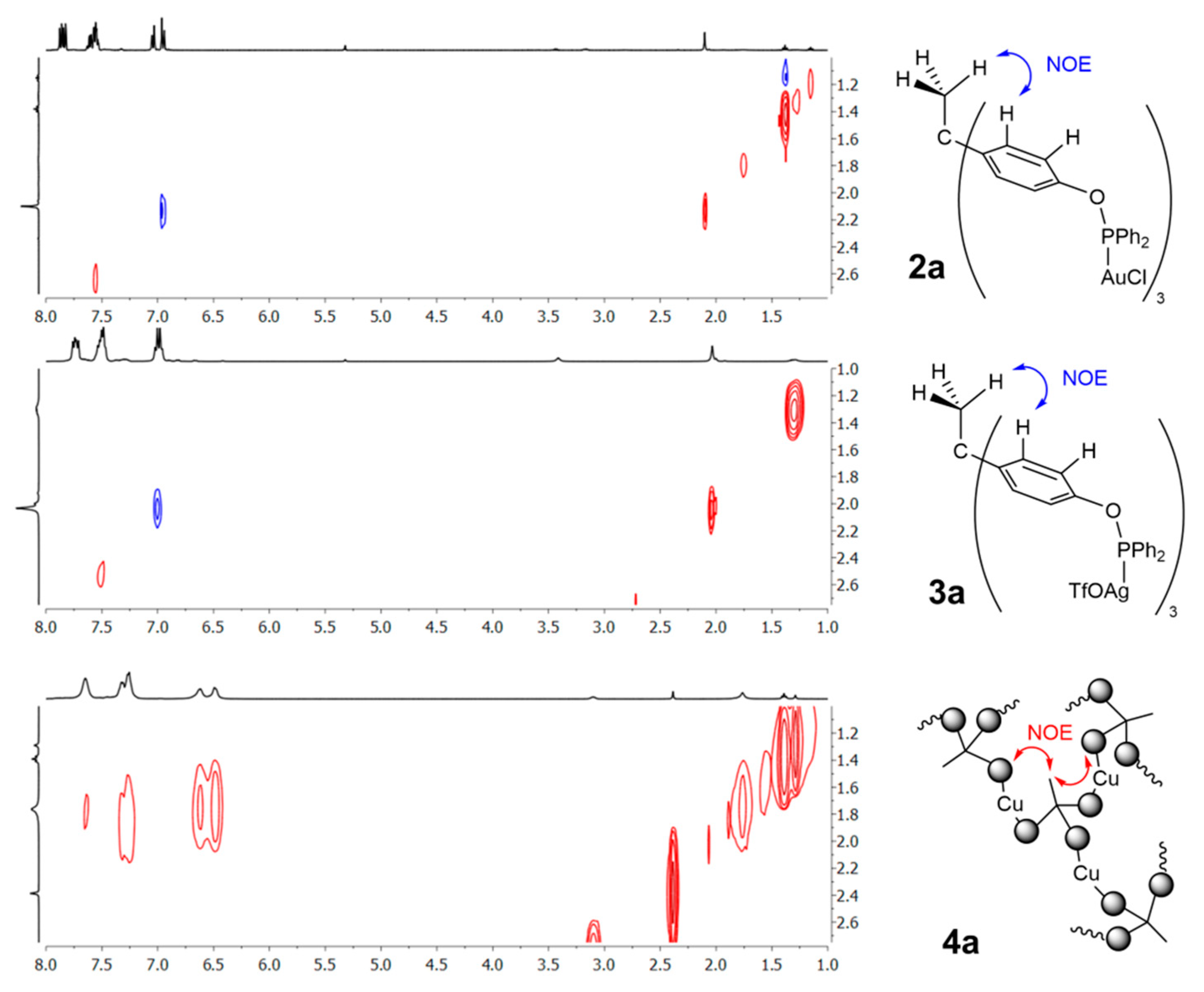
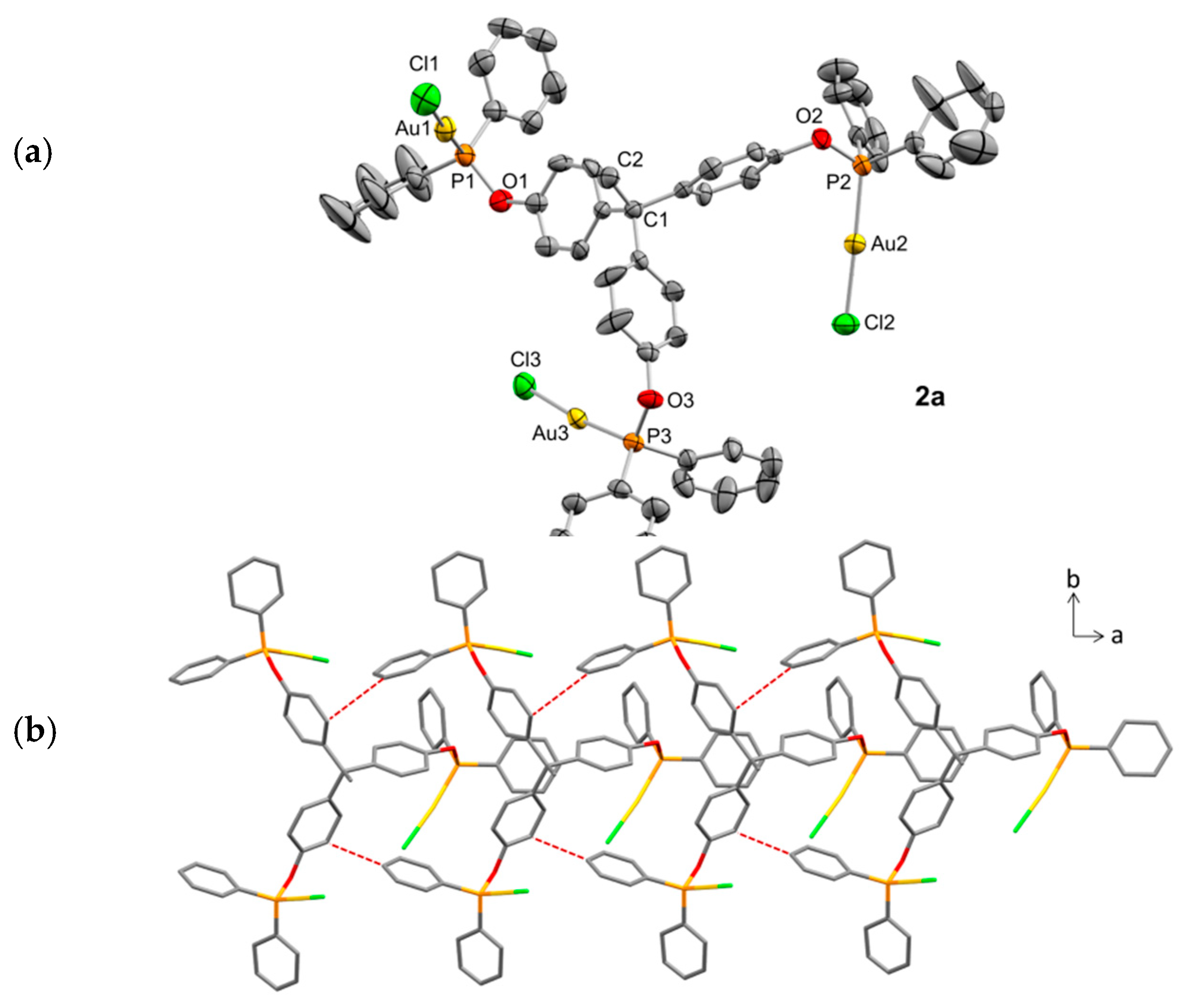
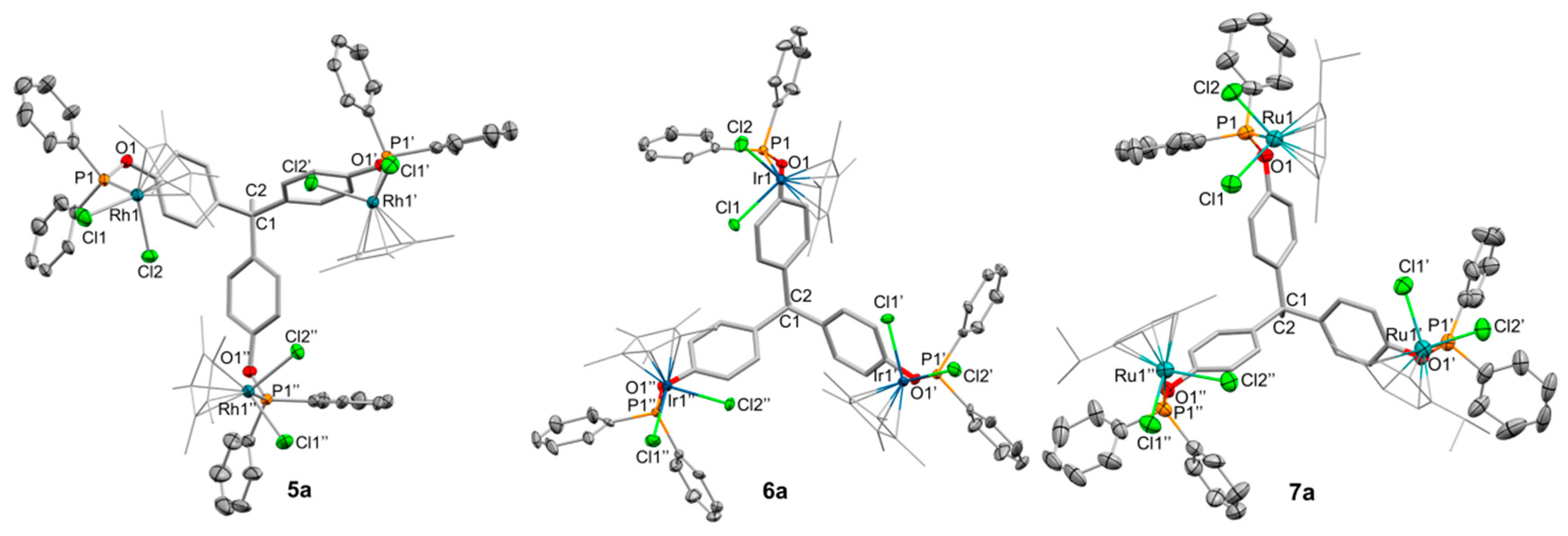
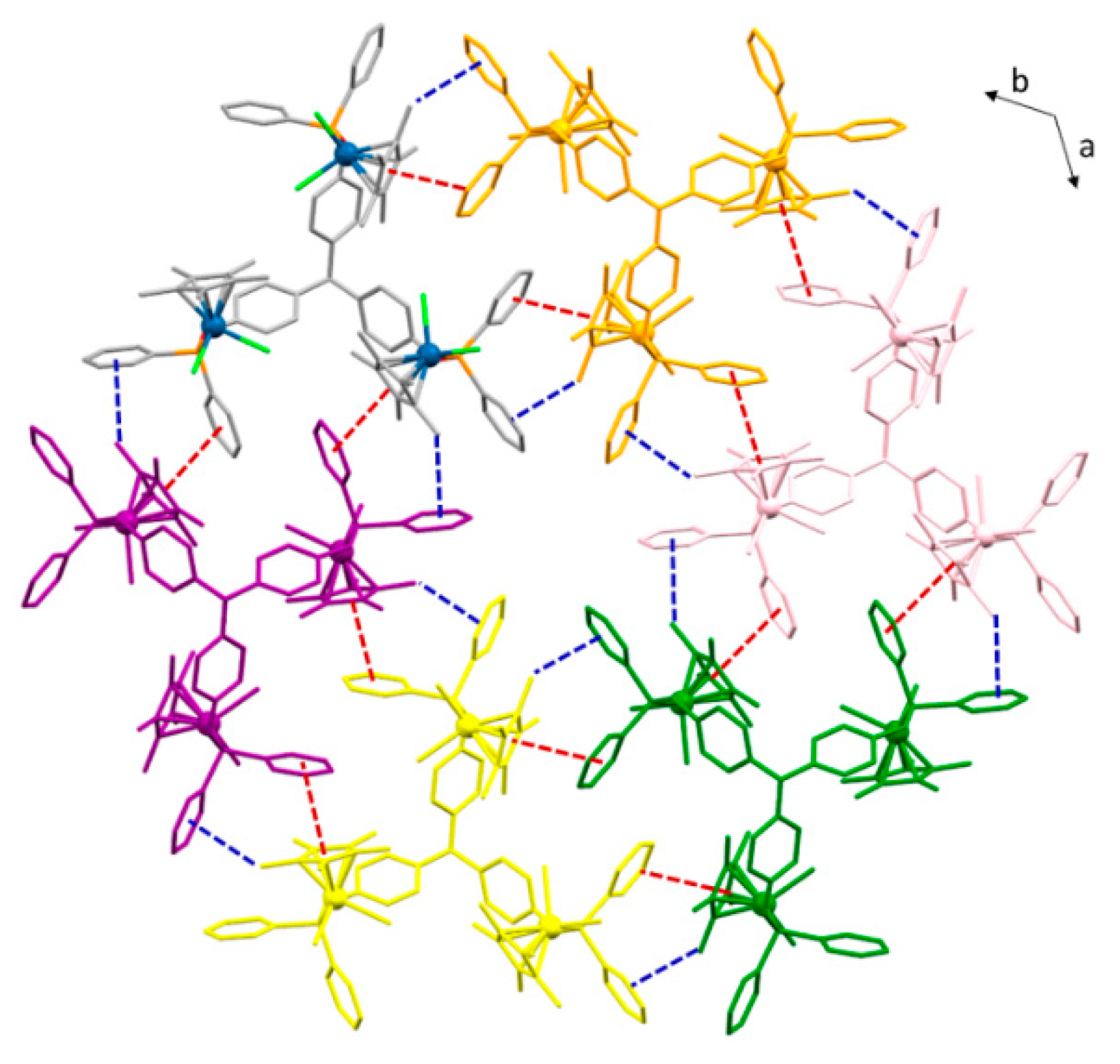
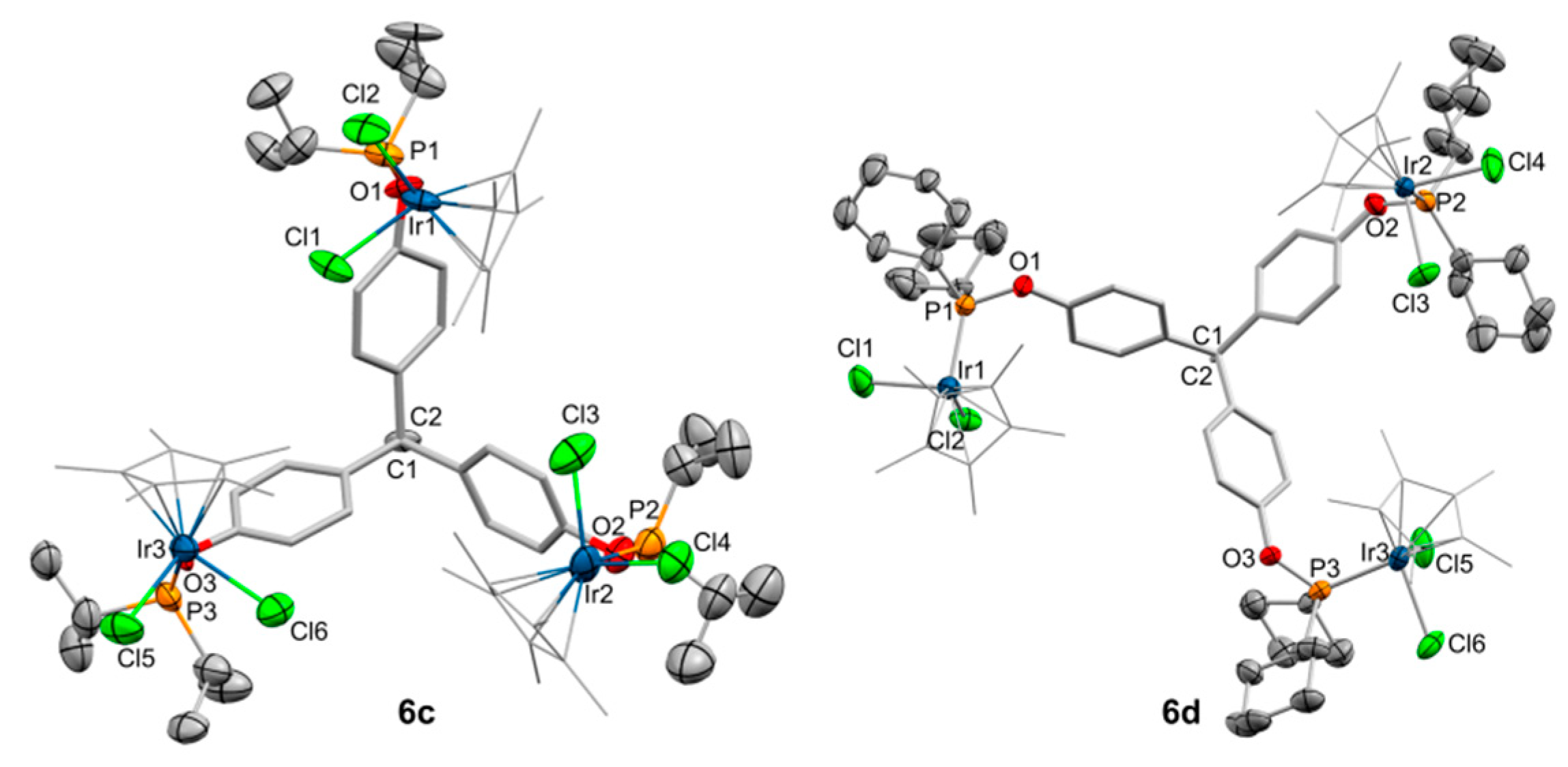
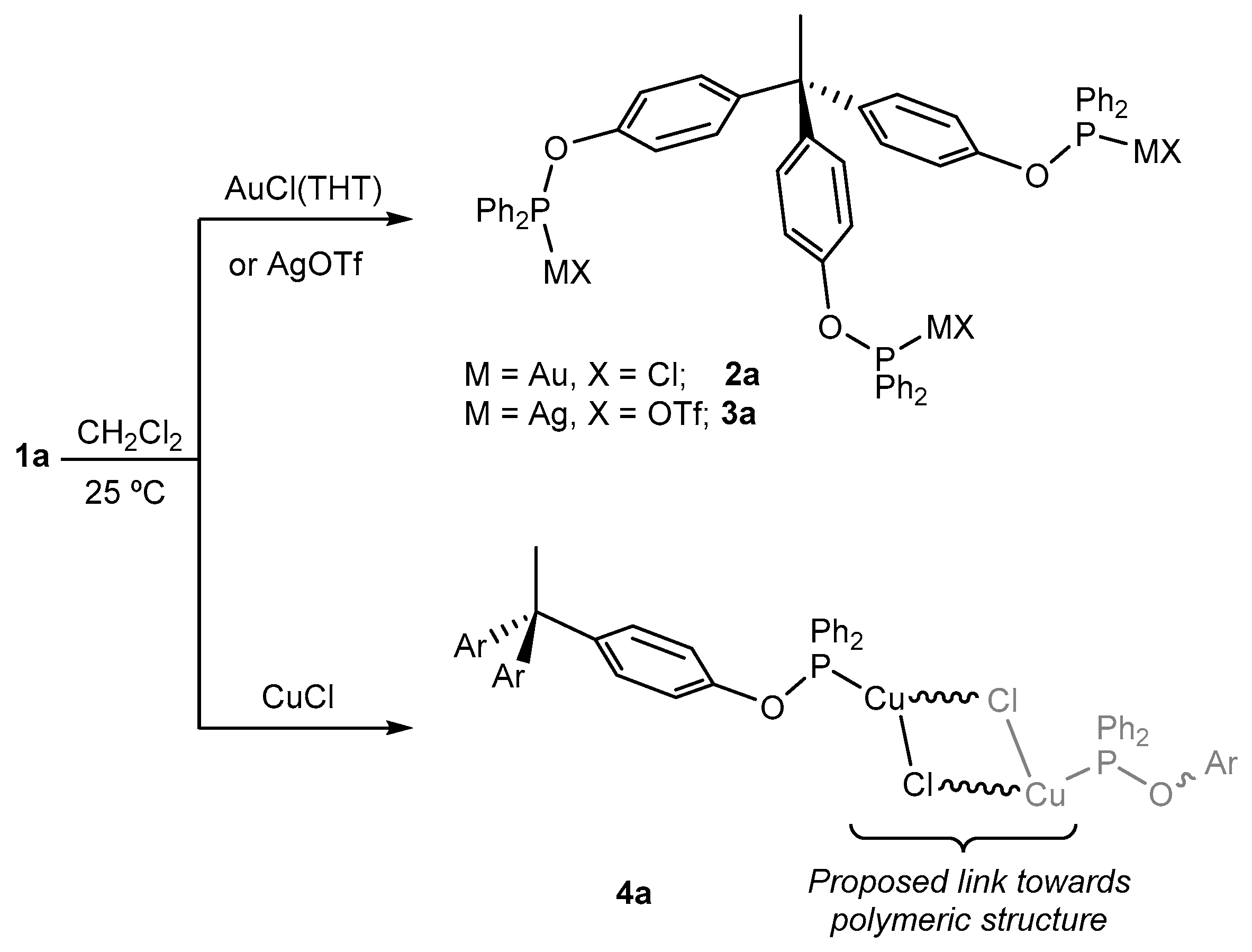
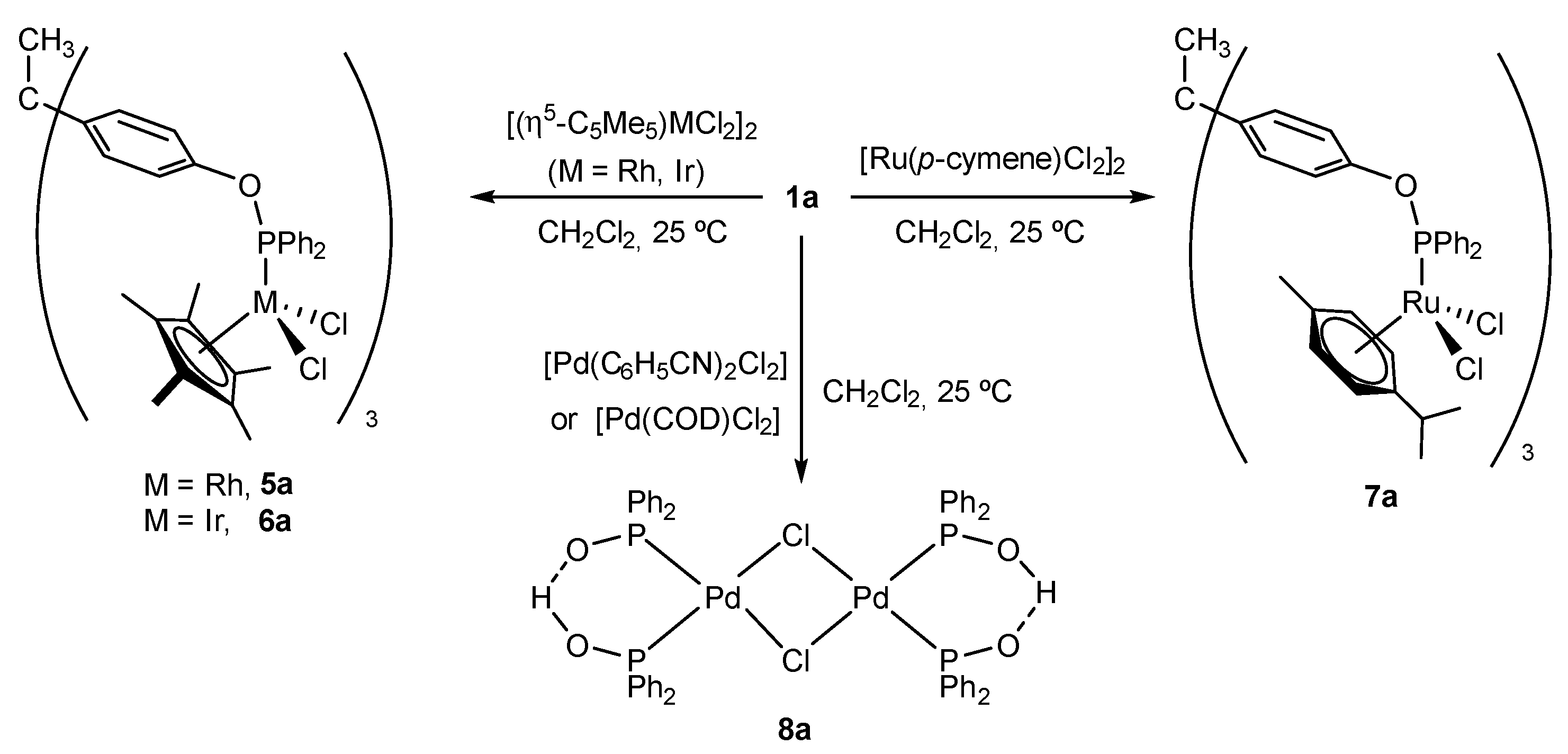
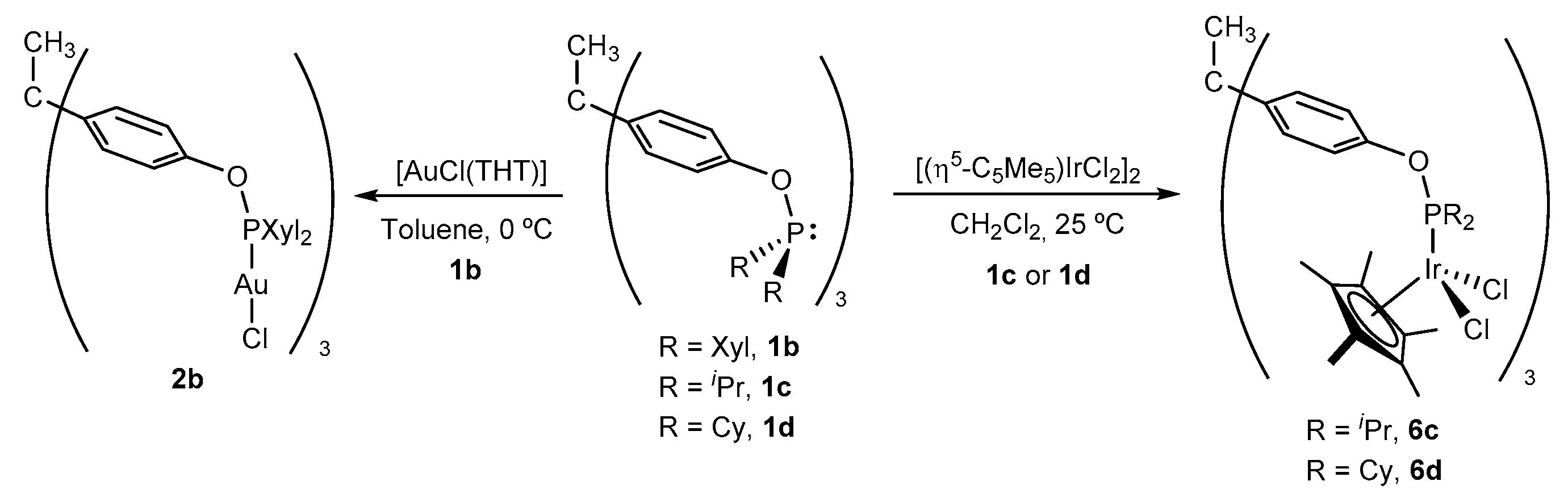
2.2. Synthesis and Characterization of Trimetallic Complexes
3. Materials and Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sweigers, G.F.; Malafetse, T.J. Classification of coordination polygons and polyhedra according to their mode of self-assembly. 2. Review of the literature. Coord. Chem. Rev. 2002, 225, 91–121. [Google Scholar] [CrossRef]
- Caulder, D.L.; Raymond, K.N. Supermolecules by Design. Acc. Chem. Res. 1999, 32, 975–982. [Google Scholar] [CrossRef]
- Stang, P.J. Molecular Architecture: Coordination as the Motif in the Rational Design and Assembly of Discrete Supramolecular Species—Self-Assembly of Metallacyclic Polygons and Polyhedra. Chem. Eur. J. 1998, 4, 19–27. [Google Scholar] [CrossRef]
- Janiak, C. Engineering coordination polymers towards applications. Dalton Trans. 2003, 2781–2804. [Google Scholar] [CrossRef]
- Chakrabarty, R.; Mukherjee, P.S.; Stang, P.J. Supramolecular coordination: Self-assembly of finite two- and three-dimensional ensembles. Chem. Rev. 2011, 111, 6810–6918. [Google Scholar] [CrossRef]
- James, S.L. Phosphines as building blocks in coordination-based self-assembly. Chem. Soc. Rev. 2009, 38, 1744–1758. [Google Scholar] [CrossRef]
- van Calcar, P.M.; Olmstead, M.M.; Balch, A.L. Self-assembly of metal complexes with cyclophane or polymeric chain structures about a 1,3,5-tris(diphenylphosphino)benzene core. Chem. Commun. 1996, 2597–2598. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Miller, P.W.; Nieuwenhuyzen, M.; James, S.L. Polar Self-Assembly: Steric Effects Leading to Polar Mixed-Ligand Coordination Cages. Chem. Eur. J. 2006, 12, 2448–2453; [Google Scholar] [CrossRef]
- Miller, P.W.; Nieuwenhuyzen, M.; Charmant, J.P.H.; James, S.L. The cyclic “Silver-Diphos” motif [Ag2(μ-diphosphine)2]2+ as a synthon for building up larger structures. Inorg. Chem 2008, 47, 8367–8379. [Google Scholar] [CrossRef]
- Che, C.-M.; Yip, H.-K.; Yam, V.W.-W.; Cheung, P.-Y.; Lai, T.-F.; Shieh, S.-J.; Peng, S.-M. Spectroscopy, photoredox properties and X-ray crystal structures of triangular gold(I) and silver(I) phosphine complexes. J. Chem. Soc. Dalton Trans. 1992, 427–433. [Google Scholar] [CrossRef]
- James, S.L.; Mingos, D.M.P.; White, A.J.P.; Williams, D.J. Anion-templated formation of a unique inorganic ‘super-adamantoid’ cage [Ag6(triphos)4(O3SCF3)4]2+ [triphos = (PPh2CH2)3CMe]. Chem. Commun. 1998, 2323–2324. [Google Scholar] [CrossRef]
- Xu, X.; Nieuwenhuyzen, M.; James, S.L. A Nanoporous Metal–Organic Framework Based on Bulky Phosphane Ligands. Angew. Chem. Int. Ed. 2002, 41, 764–767. [Google Scholar] [CrossRef]
- Lim, S.H.; Cohen, S.M. Self-Assembled Supramolecular Clusters Based on Phosphines and Coinage Metals: Tetrahedra, Helicates, and Mesocates. Inorg. Chem. 2013, 52, 7862–7872. [Google Scholar] [CrossRef] [PubMed]
- Bowyer, P.K.; Cook, V.C.; Gharib-Naseri, N.; Gugger, P.A.; Rae, A.D.; Sweigers, G.F.; Willis, A.C.; Zank, J.; Wild, S.B. Configurationally homogeneous diastereomers of a linear hexa(tertiary phosphine): Enantioselective self-assembly of a double-stranded parallel helicate of the type (P)-[Cu3(hexaphos)2](PF6)3. Proc. Natl. Acad. Sci. USA 2002, 99, 4877–4882. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Nieuwenhuyzen, M.; Chambers, M.; MacLean, E.; Teat, S.J.; James, S.L. Labile coordination dendrimers. Chem. Commun. 2002, 78–79. [Google Scholar] [CrossRef]
- Hoy, R.; Lönnecke, P.; Hey-Hawkins, E. Selective formation of a two-dimensional coordination polymer based on a tridentate phospholane ligand and gold(I). Dalton Trans. 2018, 47, 14515–14520. [Google Scholar] [CrossRef]
- Xu, X.; Nieuwenhuyzen, M.; Zhang, J.; James, S.L. Effect of Coordinating Solvents on Solution Speciation and the Crystallisation via ROP of a Triphos-Silver Coordination Cage. J. Inorg. Organomet. Polym. Mater. 2005, 15, 431–437. [Google Scholar] [CrossRef]
- Wan, X.-K.; Yuan, S.-F.; Lin, Z.-W.; Wang, Q.-M. A Chiral Gold Nanocluster Au20 Protected by Tetradentate Phosphine Ligand. Angew. Chem. Int. Ed. 2014, 53, 2923–2926. [Google Scholar] [CrossRef]
- Lin, R.; Yip, J.H.K.; Zhang, K.; Koh, L.L.; Wong, K.-Y.; Ho, K.P. Self-Assembly and Molecular Recognition of a Luminescent Gold Rectangle. J. Am. Chem. Soc. 2004, 126, 15852–15869. [Google Scholar] [CrossRef]
- Gianneschi, N.C.; Masar, M.S.; Mirkin, C.A. Development of a Coordination Chemistry-Based Approach for Functional Supramolecular Structures. Acc. Chem. Res. 2005, 38, 825–837. [Google Scholar] [CrossRef]
- Bardají, M.; Laguna, A.; Vicente, J.; Jones, P.G. Synthesis of Luminescent Gold(I) and Gold(III) Complexes with a Triphosphine Ligand. Inorg. Chem. 2001, 40, 2675–2681. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Yang, Q.; Zhu, Y.; Liu, H.; Chi, Z.; Su, C.-Y. Tetraphenylethylene-based phosphine: Tuneable emission and carbon doixide fixation. Dalton Trans. 2014, 43, 15785–15790. [Google Scholar] [CrossRef] [PubMed]
- Feng, J.; Yao, L.; Zhang, J.; Mu, Y.; Chi, Z.; Su, C.-Y. A luminescent silver–phosphine tetragonal cage based on tetraphenylethylene. Dalton Trans. 2016, 45, 1668–1673. [Google Scholar] [CrossRef] [PubMed]
- Fernández, E.J.; Laguna, A.; López-de-Luzuriaga, J.M.; Monge, M.; Sánchez-Forcada, E. Different phosphorescent excited states of tetra- and octanuclear dendritic-like phosphine gold(I) thiolate complexes: Photophysical and theoretical studies. Dalton Trans. 2011, 40, 3287–3294. [Google Scholar] [CrossRef] [PubMed]
- Ananthnag, G.S.; Mague, J.T.; Balakrishna, M.S. Self-Assembled Cyclophane-Type Copper(I) Complexes of 2,4,6-Tris(diphenylphosphino)-1,3,5-triazine and Their Catalytic Application. Inorg. Chem. 2015, 54, 10985–10992. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Pérez, H.; Etayo, P.; Panossian, A.; Vidal-Ferran, A. Phosphine–Phosphinite and Phosphine–Phosphite Ligands: Preparation and Applications in Asymmetric Catalysis. Chem. Rev. 2011, 111, 2119–2176. [Google Scholar] [CrossRef] [PubMed]
- Agbossou-Niedercorn, F.; Suisse, I. Chiral aminophosphine phosphinite ligands and related auxiliaries: Recent advances in their design, coordination chemistry, and use in enantioselective catalysis. Coord. Chem. Rev. 2003, 242, 145–158. [Google Scholar] [CrossRef]
- Vicente, B.C.; Huang, Z.; Brookhart, M.; Goldman, A.S.; Scott, S.L. Reactions of phosphinites with oxide surfaces: A new method for anchoring organic and organometallic complexes. Dalton Trans. 2011, 40, 4268–4274. [Google Scholar] [CrossRef]
- Huang, Z.; Brookhart, M.; Goldman, A.S.; Kundu, S.; Ray, A.; Scott, S.L.; Vicente, B.C. Highly Active and Recyclable Heterogeneous Iridium Pincer Catalysts for Transfer Dehydrogenation of Alkanes. Adv. Synth. Catal. 2009, 351, 188–206. [Google Scholar] [CrossRef]
- Dulière, E.; Marchand-Brynaert, J. A Convenient Synthesis of p-Aminobenzyl-tris(hydroxymethyl)methane as Precursor of Solid-Supported Tripodal Ligands. Synthesis 2002, 1, 39–42. [Google Scholar] [CrossRef]
- Hollatz, C.; Schier, A.; Schmidbaur, H. Self-assembly of tripodal tris(diphenylphosphinito)fluoroborate ligands in trinuclear gold(I) complexes Inorg. Chem. Commun. 1998, 1, 115–117. [Google Scholar] [CrossRef]
- Puddephatt, R.J. Montreal Medal Award Lecture - Coordination chemistry of molecular bowls: Ligands and their complexes derived from resorcinarenes. Can. J. Chem. 2006, 84, 1505–1514. [Google Scholar] [CrossRef]
- Eisler, D.J.; Puddephatt, R.J. Structures and Conformational Dynamics of Gold(I) Halide Complexes of Resorcinarene Tetraphosphinite Ligands. Inorg. Chem. 2003, 42, 6352–6365. [Google Scholar] [CrossRef] [PubMed]
- Eisler, D.J.; Kirby, C.W.; Puddephatt, R.J. Tetraphosphinitoresorcinarene Complexes: Dynamic Clusters with Silver(I) and Copper(I) Halides. Inorg. Chem. 2003, 42, 7626–7634. [Google Scholar] [CrossRef]
- Eisler, D.J.; Puddephatt, R.J. Tetraphosphinitoresorcinarene Complexes: Cationic Silver(I) and Copper(I) Halide Complexes as Mercurate(II) Anion Receptors. Inorg. Chem. 2003, 42, 8192–8202. [Google Scholar] [CrossRef]
- Karakhanov, E.A.; Kardasheva, Y.S.; Runova, E.A.; Terenina, M.V.; Shadrova, A.Y. Hydroformylation of olefins catalyzed by rhodium complexes with phosphinitecalix[4]arenes. Pet. Chem. 2007, 47, 340–344. [Google Scholar] [CrossRef]
- Meriça, N.; Durapa, F.; Aydemira, M.; Baysalb, A. The application of tunable tridendate P-based ligands for the Ru(II)-catalysed transfer hydrogenation of various ketones. Appl. Organometal. Chem. 2014, 28, 803–808. [Google Scholar] [CrossRef]
- Christina, H.; McFarlane, E.; McFarlane, W. Polyphosphorus ligands—V. The synthesis, phosphorus-31 NMR spectra and conformations of the polykis(diphenylphosphino) benzens (Ph2P)nC6H6–n (n = 1–4). Polyhedron 1988, 7, 1875–1879. [Google Scholar] [CrossRef]
- Rosa, P.; Debay, A.; Capes, L.; Chastanet, G.; Bousseksou, A.; Le Floch, P.; Létard, J.-F. Heat- and Light-Induced Spin Transition of an Iron(II) Polymer Containing the 1,2,4,5-Tetrakis(diphenylphosphanyl)benzene Ligand. Eur. J. Inorg. Chem. 2004, 3017–3019. [Google Scholar] [CrossRef]
- Lim, S.H.; Su, Y.; Cohen, S.M. Supramolecular Tetrahedra of Phosphines and Coinage Metals. Angew. Chem. Int. Ed. 2012, 51, 5106–5109. [Google Scholar] [CrossRef]
- Naik, S.; Kumaravel, M.; Mague, J.T.; Balakrishna, M.S. Novel Trisphosphine Ligand Containing 1,3,5-Triazine Core, [2,4,6-C3N3{C6H4PPh2-p}3]: Synthesis and Transition Metal Chemistry. Inorg. Chem. 2014, 53, 1370–1381. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Giménez, L.; Gutiérrez, A.; Lima, J.C.; Martínez, M.; Rodríguez, L.; Martín, A.; Puttreddye, R.; Rissanen, K. Polypyridyl-functionalizated alkynyl gold(i) metallaligands supported by tri- and tetradentate phosphanes. Dalton Trans. 2017, 46, 13920–13934. [Google Scholar] [CrossRef] [PubMed]
- Rajanbabu, T.V. Phosphinite and Phosphonite Ligands. In Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis; Kamer, P.C.J., van Leeuwen, P.W.N.M., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2012. [Google Scholar] [CrossRef]
- Cotton, F.A.; Murillo, C.A.; Yu, R.M. Deliberate synthesis of the preselected enantiomer of an enantiorigid molecule with pure rotational symmetry T. Dalton Trans. 2005, 3161–3165. [Google Scholar] [CrossRef] [PubMed]
- Tabatabaei, R.; Dehghanpour, S.; Simpson, J.; Lipkowski, J. Self-assembly of chelating, bridging N, N, N donor ligands and Cu(I), Cu(II), Cd(II), Hg(II): Molecular box versus coordination polymer. Polyhedron 2015, 89, 9–19. [Google Scholar] [CrossRef]
- Mishra, R.; Ülker, E.; Karadas, F. One-Dimensional Copper(II) Coordination Polymeras an Electrocatalyst for Water Oxidation. Chem. Electro. Chem. 2017, 4, 75–80. [Google Scholar] [CrossRef]
- Mishra, R.; Patil, B.; Karadas, F.; Yılmaz, E. Bioinspired Copper Coordination Polymer Catalysts for Oxygen Reduction Reaction. Chem. Select 2017, 2, 8296–8300. [Google Scholar] [CrossRef]
- Di Nicola, C.; Koutsantonis, G.A.; Pettinari, C.; Skelton, B.W.; Somers, N.; White, A.H. The structural definition of some novel adducts of stoichiometry CuX:dpex:MeCN (2:1:1)(n), X = (pseudo-) halogen, dppx = Ph2E(CH2)xEPh2, E = P, As, Sb. Inorg. Chim. Acta 2006, 359, 2159–2169. [Google Scholar] [CrossRef]
- Balakrishna, M.S.; Suresha, D.; Kumara, P.; Mague, J.T. Synthesis and transition metal chemistry of a bridging diphosphinite, 1,4 bis(diphenylphosphinoxy)benzene. J. Organomet. Chem. 2011, 696, 3616–3622. [Google Scholar] [CrossRef]
- Eisler, D.J.; Puddephatt, R.J. Tetraphosphinite Resorcinarene Complexes: Silver(I) Capsule Complexes. Inorg. Chem. 2005, 44, 4666–4678. [Google Scholar] [CrossRef]
- Gunther, H. NMR Spectroscopy: Basic Principles, Concepts and Applications in Chemistry; Wiley-VCH: Hoboken, NJ, USA, 2013. [Google Scholar]
- Janiak, C. A critical account on π–π stacking in metal complexes with aromatic nitrogen-containing ligands. J. Chem. Soc. Dalton Trans 2000, 3885–3896. [Google Scholar] [CrossRef]
- Suezawa, H.; Yoshida, T.; Umezawa, Y.; Tsuboyama, S.; Nishio, M. CH/π Interactions Implicated in the Crystal Structure of Transition Metal Compounds—A Database Study. Eur. J. Inorg. Chem. 2002, 2002, 3148–3155. [Google Scholar] [CrossRef]
- Braga, D.; Grepioni, F.; Tedesco, E. X–H---π (X = O, N, C) Hydrogen Bonds in Organometallic Crystals. Organometallics 1998, 17, 2669–2672. [Google Scholar] [CrossRef]
- Campos, J.; Esqueda, A.C.; Carmona, E. Cyclometallation and Hydrogen/Deuterium Exchange Reactions of an Arylphosphine Ligand upon Coordination to {Ir(η5-C5Me5)}. Chem. Eur. J. 2010, 16, 419–422. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; López-Serrano, J.; Álvarez, E.; Carmona, E. Cationic Ir(III) Alkylidenes Are Key Intermediates in C–H Bond Activation and C–C Bond-Forming Reactions. J. Am. Chem. Soc. 2012, 134, 7165–7175. [Google Scholar] [CrossRef]
- Campos, J.; Espada, M.F.; López-Serrano, J.; Carmona, E. Cyclometalated Iridium Complexes of Bis(Aryl) Phosphine Ligands: Catalytic C–H/C–D Exchanges and C–C Coupling Reactions. Inorg. Chem. 2013, 52, 6694–6704. [Google Scholar] [CrossRef] [PubMed]
- Campos, J.; Carmona, E. Rhodium and Iridium Complexes of Bulky Tertiary Phosphine Ligands. Searching for Isolable Cationic MIII Alkylidenes. Organometallics 2015, 34, 2212–2221. [Google Scholar] [CrossRef]
- Uson, R.; Laguna, A.; Laguna, M.; Briggs, D.A.; Murray, H.H.; Fackler, J.P. (Tetrahydrothiophene)Gold(I) or Gold(III) Complexes. Inorg. Synth. 1989, 26, 85–91. [Google Scholar] [CrossRef]
- White, C.; Yates, A.; Maitlis, P.M.; Heinekey, D.M. (η5-Pentamethylcyclopentadienyl)Rhodium and -Iridium Compounds. Inorg. Synth. 1992, 29, 228–234. [Google Scholar] [CrossRef]
- Bennett, M.A.; Huang, T.N.; Matheson, T.W.; Smith, A.K.; Nickerson, W. (η6-Hexamethylbenzene) ruthenium Complexes. Inorg. Synth. 1982, 21, 74–78. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds 1a–7a are available from the authors. |











© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miranda-Pizarro, J.; Alférez, M.G.; Fernández-Martínez, M.D.; Álvarez, E.; Maya, C.; Campos, J. A Versatile Approach to Access Trimetallic Complexes Based on Trisphosphinite Ligands. Molecules 2020, 25, 593. https://doi.org/10.3390/molecules25030593
Miranda-Pizarro J, Alférez MG, Fernández-Martínez MD, Álvarez E, Maya C, Campos J. A Versatile Approach to Access Trimetallic Complexes Based on Trisphosphinite Ligands. Molecules. 2020; 25(3):593. https://doi.org/10.3390/molecules25030593
Chicago/Turabian StyleMiranda-Pizarro, Juan, Macarena G. Alférez, M. Dolores Fernández-Martínez, Eleuterio Álvarez, Celia Maya, and Jesús Campos. 2020. "A Versatile Approach to Access Trimetallic Complexes Based on Trisphosphinite Ligands" Molecules 25, no. 3: 593. https://doi.org/10.3390/molecules25030593
APA StyleMiranda-Pizarro, J., Alférez, M. G., Fernández-Martínez, M. D., Álvarez, E., Maya, C., & Campos, J. (2020). A Versatile Approach to Access Trimetallic Complexes Based on Trisphosphinite Ligands. Molecules, 25(3), 593. https://doi.org/10.3390/molecules25030593