
Redox-Modulations of Photophysical and Single-molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads

 , , , ,
, , , ,  and
and 
Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
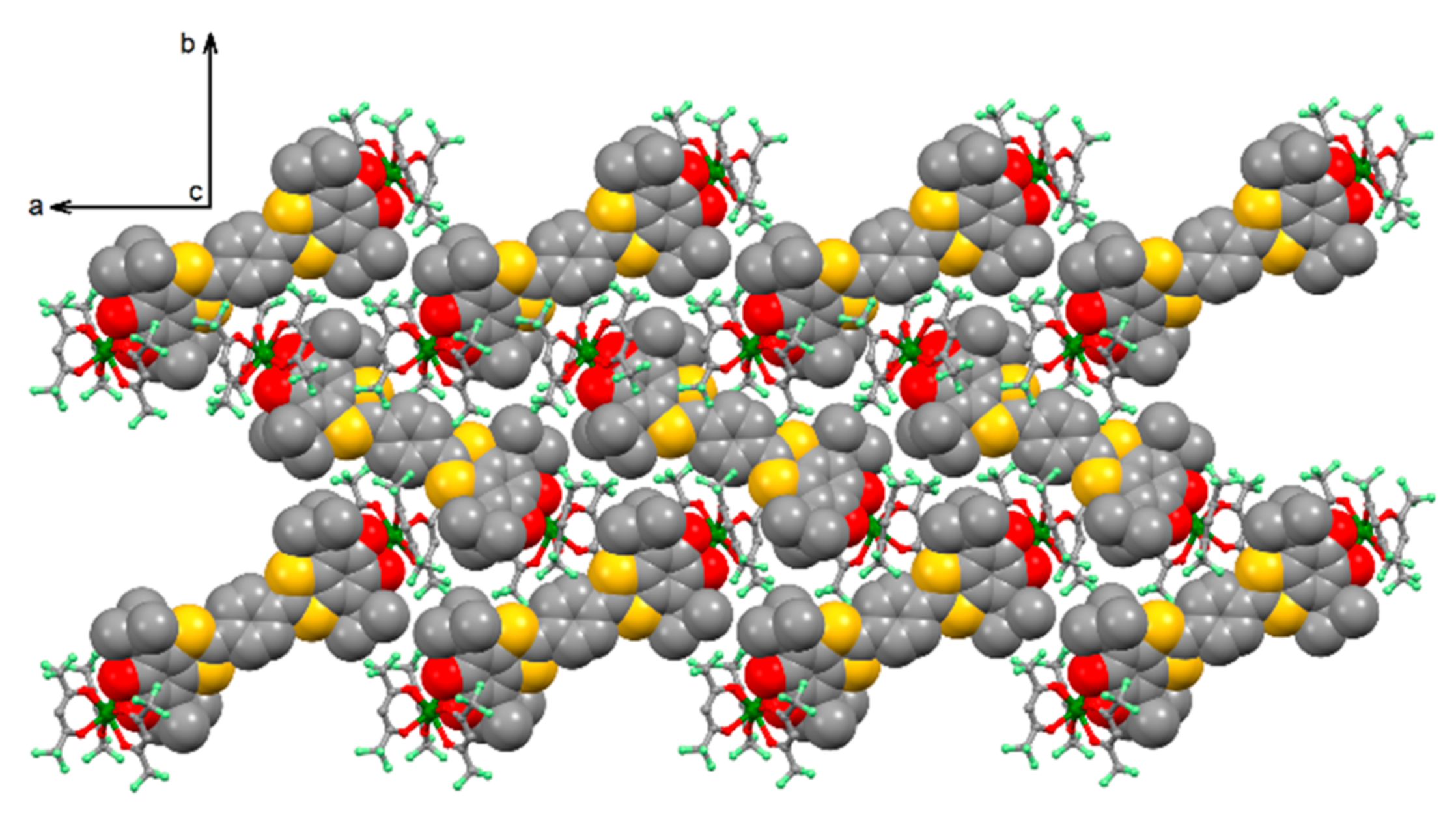
2.1. X-Ray Structures
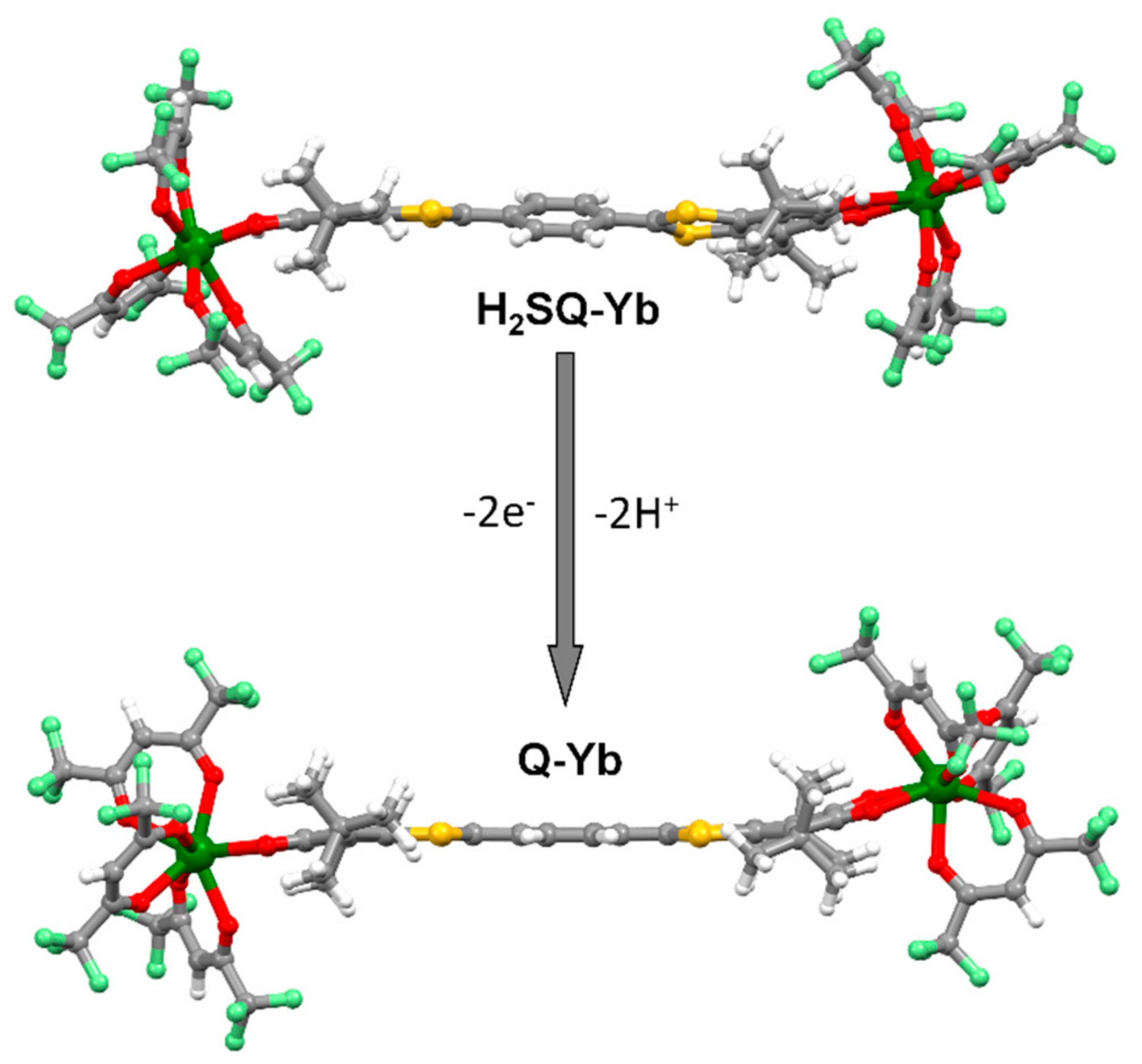
2.1.1. [Yb2(hfac)6(H2SQ)]⋅0.5CH2Cl2 (H2SQ-Yb)
2.1.2. [Yb2(hfac)6(Q)] (Q-Yb)
2.2. Magnetic Properties
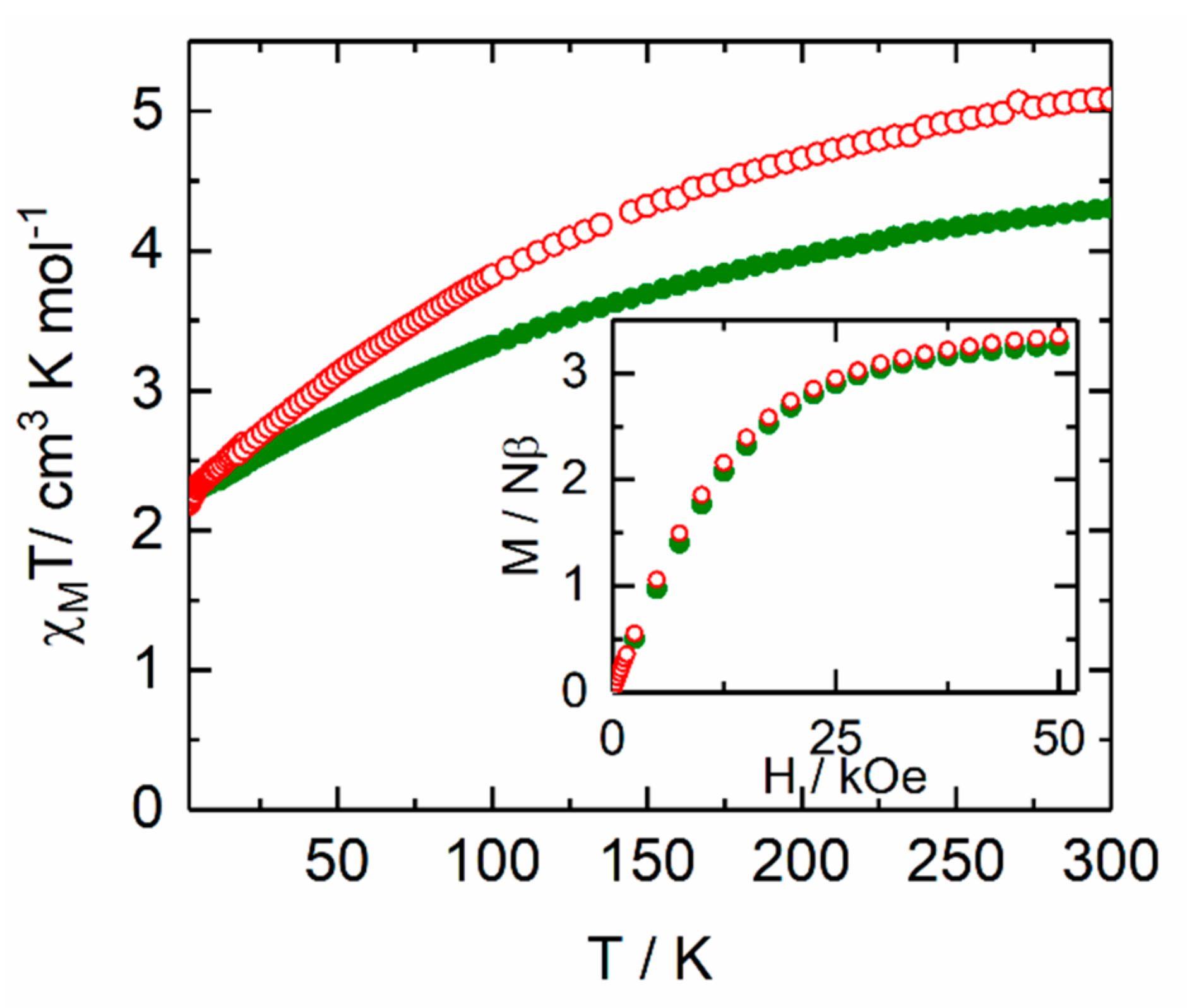
2.2.1. Static Magnetic Measurements
2.2.2. Dynamic Magnetic Measurements
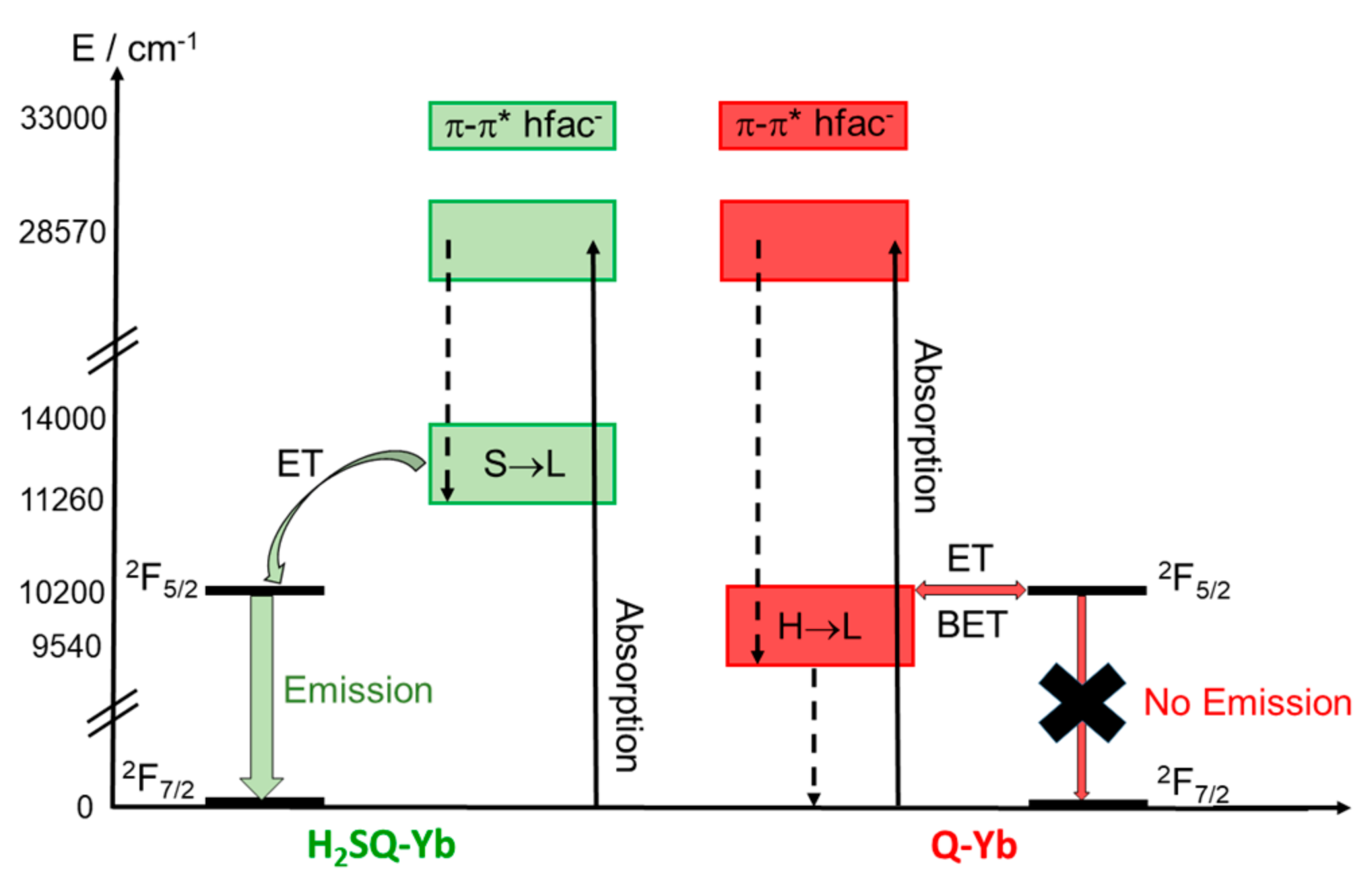
2.2.3. Photophysical Properties
3. Materials and Methods
3.1. Synthesis. General Procedures and Materials
3.2. Synthesis of complexes [Yb2(hfac)6(H2SQ)]⋅0.5CH2Cl2 (H2SQ-Yb) and [Yb2(hfac)6(Q)] (Q-Yb)
3.3. Crystallography
3.4. Physical Measurements
3.5. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sessoli, R.; Gatteschi, D.; Caneschi, A.; Novak, M.A. Magnetic bistability in a metal-ion cluster. Nature 1993, 365, 141–143. [Google Scholar] [CrossRef]
- Ishikawa, N.; Sugita, M.; Ishikawa, T.; Koshihara, S.-Y.; Kaizu, Y. Lanthanide Double-Decker Complexes Functioning as Magnets at the Single-Molecular Level. J. Am. Chem. Soc. 2003, 125, 8694–8695. [Google Scholar] [CrossRef]
- Mannini, M.; Pineider, F.; Sainctavit, P.; Danieli, C.; Otero, E.; Sciancalepore, C.; Talarico, A.M.; Arrio, M.-A.; Cornia, A.; Gatteschi, D.; et al. Magnetic memory of a single-molecule quantum magnet wired to a gold surface. Nat. Mater. 2009, 8, 194–197. [Google Scholar] [CrossRef]
- Affronte, M. Molecular nanomagnets for information technologies. J. Mater. Chem. 2009, 19, 1731–1737. [Google Scholar] [CrossRef]
- Guo, F.-S.; Day, B.-M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. A Dysprosium Metallocene Single-Molecule Magnet Functioning at the Axial Limit. Angew. Chem. Int. Ed. 2017, 56, 11445–11449. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, C.A.P.; Ortu, F.; Reta, D.; Chilton, N.F.; Mills, D.P. Molecular magnetic hysteresis at 60 kelvin in dysprosocenium. Nature 2017, 548, 439–442. [Google Scholar] [CrossRef] [PubMed]
- McClain, K.R.; Gould, C.A.; Chakarawet, K.; Teat, S.J.; Groshens, T.J.; Long, J.R.; Harvey, B.G. High-temperature magnetic blocking and magneto-structural correlations in a series of dysprosium(III) metallocenium single-molecule magnets. Chem. Sci. 2018, 9, 8492–8503. [Google Scholar] [CrossRef] [PubMed]
- Guo, F.-S.; Day, B.-M.; Chen, Y.-C.; Tong, M.-L.; Mansikkamäki, A.; Layfield, R.A. Magnetic hysteresis up to 80 kelvin in a dysprosium metallocene single-molecule magnet. Science 2018, 362, 1400–1403. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Sato, R.; Mizuno, N. Reversible switching of single-molecule magnet behaviors by transformation of dinuclear dysprosium cores in polyoxometalates. Chem. Sci. 2013, 4, 596–600. [Google Scholar] [CrossRef]
- Liu, J.-L.; Chen, Y.-C.; Zheng, Y.-Z.; Lin, W.-Q.; Ungur, L.; Wernsdorfer, W.; Chibotaru, L.F.; Tong, M.-L. Switching the anisotropy barrier of a single-ion magnet by symmetry change from quasi-D5h to quasi-Oh. Chem. Sci. 2013, 4, 3310–3316. [Google Scholar]
- Zhang, X.; Vieru, V.; Feng, X.; Liu, J.-L.; Zhang, Z.; Na, B.; Shi, W.; Wang, B.-W.; Powell, A.K.; Chibotaru, L.F.; et al. Influence of Guest Exchange on the Magnetization Dynamics of Dilanthanide Single-Molecule-Magnet Nodes within a Metal-Organic Framework. Angew. Chem. Int. Ed. 2015, 54, 9861–9865. [Google Scholar] [CrossRef] [PubMed]
- Ge, J.-Y.; Cui, L.; Li, J.; Yu, F.; Song, Y.; Zhang, Y.-Q.; Zuo, J.-L.; Kurmoo, M. Modulation Single-Molecule Magnetic Behavior of a Dinuclear Erbium(III) Complex by Solvent Exchange. Inorg. Chem. 2017, 56, 336–343. [Google Scholar] [CrossRef] [PubMed]
- Pinkowicz, D.; Ren, M.; Zheng, L.-M.; Sato, S.; Hasegawa, M.; Morimoto, M.; Irie, M.; Breedlove, B.K.; Cosquer, G.; Katoh, K.; et al. Control of the Single-Molecule Magnet Behavior of Lanthanide-Diarylethene Photochromic Assemblies by Irradiation with Light. Chem. Eur. J. 2014, 20, 12502–12513. [Google Scholar] [CrossRef] [PubMed]
- Cosquer, G.; Morimoto, M.; Irie, M.; Fetoh, A.; Breedlove, B.K.; Yamashita, M. Photo-control of the magnetic properties of Dy(III) and Ho(III) homometal coordination polymers bridged by a diarylethene ligand. Dalton Trans. 2015, 44, 5996–6002. [Google Scholar] [CrossRef]
- Wang, L.-F.; Qiu, J.-Z.; Liu, J.-L.; Chen, Y.-C.; Jia, J.-H.; Jover, J.; Ruiz, E.; Tong, M.-L. Modulation of single-molecule magnet behaviour via photochemical [2+2] cycloaddition. Chem. Commun. 2015, 51, 15358–15361. [Google Scholar] [CrossRef]
- Selvanathan, P.; Huang, G.; Guizouarn, T.; Roisnel, T.; Fernandez-Garcia, G.; Totti, F.; Le Guennic, B.; Calvez, G.; Bernot, K.; Norel, L.; et al. Highly Axial Magnetic Anisotropy in a N3O5 Dysprosium(III) Coordination Environment Generated by a Merocyanine Ligand. Chem. Eur. J. 2016, 22, 15222–15226. [Google Scholar] [CrossRef]
- Tian, H.; Su, J.-B.; Bao, S.S.; Kurmoo, M.; Huang, X.-D.; Zhang, Y.-Q.; Zheng, L.-M. Reversible ON-OFF switching of single-molecule-magnetism associated with single-crystal-to-single-crystal structural transformation of a decanuclear dysprosium phosphate. Chem. Sci. 2018, 9, 6424–6433. [Google Scholar] [CrossRef]
- Liang, Z.; Damjanovic, M.; Kamila, M.; Cosquer, G.; Breedlove, B.K.; Enders, M.; Yamashita, M. Proton Control of the Lanthanoid Single-Ion Magnet Behavior of a Double-Desker Complex with an Indolenine-Substituted Annulene Ligand. Inorg. Chem. 2017, 56, 6512–6521. [Google Scholar] [CrossRef]
- Zhang, P.; Perfetti, M.; Kern, M.; Hallmen, P.P.; Ungur, L.; Lenz, S.; Ringenberg, M.R.; Frey, W.; Stoll, H.; Rauhut, G.; et al. Exchange coupling and single molecule magnetism in redox-active tetraoxolene-bridged dilanthanide complexes. Chem. Sci. 2018, 9, 1221–1230. [Google Scholar] [CrossRef]
- Dolinar, B.S.; Gomez-Coca, S.; Alexandropoulos, D.I.; Dunbar, K.R. An air stable radical-bridged dysprosium single molecule magnet and its neutral counterpart: Redox switching of magnetic relaxation dynamics. Chem. Commun. 2017, 53, 2283–2286. [Google Scholar] [CrossRef]
- Takamatsu, S.; Isikawa, T.; Koshihara, S.Y.; Ishikawa, N. Signifant Increase of the Barrier Energy for Magnetization Reversal of a Single-4f-Ionic Single-Molecule Magnet by a Longitudinal Contraction of the Coordination Space. Inorg. Chem. 2007, 46, 7250–7252. [Google Scholar] [CrossRef] [PubMed]
- Norel, L.; Feng, M.; Bernot, K.; Roisnel, T.; Guizouarn, T.; Costuas, K.; Rigaut, S. Redox Modulation of Magnetic Slow Relaxation in a 4f-Based Single-Molecule Magnet with a 4d Carbon-Rich Ligand. Inorg. Chem. 2014, 53, 2361–2363. [Google Scholar] [CrossRef] [PubMed]
- Dickie, C.M.; Laughlin, A.L.; Wofford, J.D.; Bhuvanesh, N.S.; Nippe, M. Transition metal redox switches for reversible “on/off” and “slow/fast” single-molecule magnet behaviour in dysprosium and erbium bis-diamidoferrocene complexes. Chem. Sci. 2017, 8, 8039–8049. [Google Scholar] [CrossRef] [PubMed]
- Cador, O.; Le Guennic, B.; Pointillart, F. Electro-Activity and Magnetic Switching in Lanthanide-Based Single-Molecule Magnets. Inorg. Chem. Front. 2019, 6, 3398–3417. [Google Scholar] [CrossRef]
- Sy, M.; Nonat, A.; Hildebrandt, N.; Charbonnière, L.J. Lanthanide-based luminescence biolabelling. Chem. Commun. 2016, 52, 5080–5095. [Google Scholar] [CrossRef]
- Zhang, K.Y.; Yu, Q.; Wei, H.; Liu, S.; Zhao, Q.; Huang, W. Long-Lived Emission Probes for time-Resolved Photoluminescence Bioimaging and Biosensing. Chem. Rev. 2018, 118, 1770–1839. [Google Scholar] [CrossRef]
- D’Aléo, A.; Pointillart, F.; Ouahab, L.; Andraud, C.; Maury, O. Charge transfer excited states sensitization of lanthanide emitting from the visible to the near-infra-red. Coord. Chem. Rev. 2012, 256, 1604–1620. [Google Scholar] [CrossRef]
- Pointillart, F.; Le Guennic, B.; Cador, O.; Maury, O.; Ouahab, L. Lanthanide Ion and Tetrathiafulvalene-Based Ligand as a “Magic” Couple toward Luminescence, Single Molecule Magnets, and Magnetostructural Correlations. Acc. Chem. Res. 2015, 48, 2834–2842. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G.; Piguet, C. Taking advantage of luminescent lanthanide ions. Coord. Chem. Rev. 2005, 34, 1048–1077. [Google Scholar] [CrossRef]
- Eliseeva, S.V.; Bünzli, J.-C.G. Lanthanide luminescence for functional materials and bio-sciences. Chem. Soc. Rev. 2010, 39, 189–227. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. On the design of highly luminescent lanthanide complexes. Coord. Chem. Rev. 2015, 293-294, 19–47. [Google Scholar] [CrossRef]
- Di Piazza, E.; Norel, L.; Costuas, K.; Bourdolle, A.; Maury, O.; Rigaut, S. D-f Heterobimetallic Association between Ytterbium and Ruthenium Carbon-Rich Complexes: Redox Commutation of Near-IR Luminescence. J. Am. Chem. Soc. 2011, 133, 6174–6176. [Google Scholar] [CrossRef] [PubMed]
- Al Sabea, H.; Norel, L.; Galangau, O.; Hijazi, H.; Métivier, R.; Roisnel, T.; Maury, O.; Bucher, C.; Riobé, F.; Rigaut, S. Dual Light and Redox Control of NIR Luminescence with Complementary Photochromic and Organometallic Antennae. J. Am. Chem. Soc. 2019. [Google Scholar] [CrossRef] [PubMed]
- Tropiano, M.; Kilah, N.L.; Morten, M.; Rahman, H.; Davis, J.J.; Beer, P.D.; Faulkner, S. Reversible Luminescence Switching of a Redox-Active Ferrocene-Europium Dyad. J. Am. Chem. Soc. 2011, 133, 11847–11849. [Google Scholar] [CrossRef] [PubMed]
- Molloy, J.K.; Jarjayes, O.; Philouze, C.; Fedele, L.; Imbert, D.; Thomas, F. A redox active switch for lanthanide luminescence in phenolate complexes. Chem. Commun. 2017, 53, 605–608. [Google Scholar] [CrossRef]
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Romanenko, G.V.; Ketkov, S.Y.; Smolyaninov, I.V.; Starikov, A.G.; Kuropatov, V.A. Compactly Fused o-Quinone-Extended Tetrathiafulvalene-o-Quinone Triad—A Redox-Amphoteric Ligand. Eur. J. Org. Chem. 2014, 4571–4576. [Google Scholar] [CrossRef]
- Flores Gonzalez, J.; Cador, O.; Ouahab, L.; Norkov, S.; Kuropatov, V.; Pointillart, F. Field-Induced Dysprosium Single-Molecule Magnet Involving a Fused o-Semiquinone-Extended-Tetrathiafulvalene-o-Semiquinone Bridging Triad. Inorganics 2018, 6, 45. [Google Scholar] [CrossRef]
- Jones, A.E.; Christensen, C.A.; Perepichka, D.F.; Batsanov, A.S.; Beeby, A.; Low, P.J.; Bryce, M.R.; Parker, A.W. Photochemistry of the π-Extended 9,10-Bis(1,3-dithiol-2-ylidene)-9,10-dihydroanthracene System: Generation and Characterisation of the Radical Cation, Dication, and Derived Products. Chem. Eur. J. 2001, 7, 973–978. [Google Scholar] [CrossRef]
- Cooper, W.F.; Edmonds, J.W.; Wudl, F.; Coppens, P. The 2-2’-bi-1,3-dithiole. Cryst. Struct. Commun. 1974, 3, 23–26. [Google Scholar]
- Ellern, A.; Bernstein, J.; Becker, J.Y.; Zamir, S.; Shahal, L.; Cohen, S. New Polymorphic Modification of tetrathiafulvalene. Crystal Structure, Lattice Energy and Intermolecular Interactions. Chem. Mater. 1994, 6, 1378–1385. [Google Scholar] [CrossRef]
- Llunell, M.; Casanova, D.; Cirera, J.; Bofill, J.M.; Alemany, P.; Alvarez, S. SHAPE, Version 2.1.; Universitat de Barcelona: Barcelona, Spain, 2013. [Google Scholar]
- Pointillart, F.; Kuropatov, V.; Mitin, A.; Maury, O.; Le Gal, Y.; Golhen, S.; Cador, O.; Cherkasov, V.; Ouahab, L. Lanthanide-Based Dinuclear Complexes Involving an o-Quinone-Tetrathiafulvalene-o-Quinone Bridging Ligand: X-ray Structures, Magnetic and Photophysical Properties. Eur. J. Inorg. Chem. 2012, 4708–4718. [Google Scholar] [CrossRef]
- Pointillart, F.; Cador, O.; Le Guennic, B.; Ouahab, L. Uncommon lanthanide ions in purely 4f Single Molecule Magnets. Coord. Chem. Rev. 2017, 346, 150–175. [Google Scholar] [CrossRef]
- Kahn, O. Molecular Magnetism; VCH: Weinhem, Germany, 1993. [Google Scholar]
- Poneti, G.; Bernot, K.; Bogani, L.; Caneschi, A.; Sessoli, R.; Wernsdorfer, W.; Gatteschi, D. A rational approach to the modulation of the dynamics of the magnetisation in a dysprosium-nitronyl-nitroxide radical complex. Chem. Commun. 2007, 1807–1809. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.S.; Cole, R.H. Dispersion and absorption in dielectrics I. Alternating current characteristics. J. Chem. Phys. 1941, 9, 341–351. [Google Scholar] [CrossRef]
- Orbach, R. Spin-lattice relaxation in rare-earth salts. Proc. R. Soc. Lond. A Math. Phys. Eng. Sci. 1961, 264, 458–484. [Google Scholar]
- Orbach, R. On the theory of spin-lattice relaxation in paramagnetic salts. Proc. Phys. Soc. 1961, 77, 821–826. [Google Scholar] [CrossRef]
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Starikov, A.G.; Kuropatov, V.A. Protonated paramagnetic redox forms of di-o-quinone bridged with p-phenylene-extended TTF: A EPR spectroscopy study. Beilstein J. Org. Chem. 2016, 12, 2450–2456. [Google Scholar] [CrossRef]
- Chalkov, N.O.; Cherkasov, V.K.; Abakumov, G.A.; Starikov, A.G.; Kuropatov, V.A. EPR spectroscopy study of di-o-quinone bridged by π-extended TTF: Redox behavior and binding modes as a ligand. New J. Chem. 2016, 40, 1244–1249. [Google Scholar] [CrossRef]
- Ziessel, R.; Ulrich, G.; Charbonnière, L.; Imbert, D.; Scopelliti, R.; Bünzli, J.-C.G. NIR Lanthanide Luminescence by Energy Transfer from Appended Terpyridine-Boradiazaindacene Dyes. Chem. Eur. J. 2006, 12, 5060–5067. [Google Scholar] [CrossRef]
- Pointillart, F.; Cauchy, T.; Maury, O.; Le Gal, Y.; Golhen, S.; Cador, O.; Ouahab, L. Tetrathiafulvalene-amido-2-pyridine-N-oxide as Efficient Charge-Transfer Antenna Ligand for the Sensitization of YbIII Luminescence in a Series of Lanthanide Paramagnetic Coordination Complexes. Eur. J. Chem. 2010, 16, 11926–11941. [Google Scholar] [CrossRef]
- Bünzli, J.-C.G. Benefiting from the Unique Properties of Lanthanide Ions. Acc. Chem. Res. 2006, 39, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Eliseeva, S.V.; Bünzli, J.-C.G. Rare earths: Jewels for functional materials of the future. New. J. Chem. 2011, 35, 1165–1176. [Google Scholar] [CrossRef]
- Rinehart, J.D.; Long, J.R. Exploiting single-ion anisotropy in the design of f-element single-molecule magnets. Chem. Sci. 2011, 2, 2078–2085. [Google Scholar] [CrossRef]
- Richardson, M.F.; Wagner, W.F.; Sands, D.E. Rare-earth trishexafluoroacetylacetonates and related compounds. J. Inorg. Nucl. Chem. 1968, 30, 1275–1289. [Google Scholar] [CrossRef]
- Sheldrick, G.L. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. Sect. A 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, 71, 3–8. [Google Scholar] [CrossRef]
- Te Velde, G.; Bickelhaupt, F.M.; Baerends, E.J.; van Gisbergen, S.J.A.; Fonseca Guerra, C.; Snijders, J.G.; Ziegler, T. Chemistry with ADF. J. Comput. Chem. 2001, 22, 931–967. [Google Scholar] [CrossRef]
- Fonseca Guerra, C.; Snijders, J.G.; te Velde, G.; Baerends, E.J. Towards an order-N DFT method. Theor. Chem. Acc. 1998, 99, 391–403. [Google Scholar] [CrossRef]
- Baerends, E.J.; Ziegler, T.; Atkins, A.J.; Autschbach, J.; Bashford, D.; Baseggio, O.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerritger, P.M.; et al. ADF2017, SCM, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Van Lenthe, E.; Baerends, E.J.; Snijders, J.G. Relativistic Regular two-component Hamiltonians. J. Chem. Phys. 1993, 99, 4597–4610. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Van Lenthe, E.; Baerends, E.J. Optimized Slater-type basis sets for the elements 1–118. J. Comput. Chem. 2003, 24, 1142–1156. [Google Scholar] [CrossRef] [PubMed]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lefeuvre, B.; Flores Gonzalez, J.; Gendron, F.; Dorcet, V.; Riobé, F.; Cherkasov, V.; Maury, O.; Le Guennic, B.; Cador, O.; Kuropatov, V.; et al. Redox-Modulations of Photophysical and Single-molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads. Molecules 2020, 25, 492. https://doi.org/10.3390/molecules25030492
Lefeuvre B, Flores Gonzalez J, Gendron F, Dorcet V, Riobé F, Cherkasov V, Maury O, Le Guennic B, Cador O, Kuropatov V, et al. Redox-Modulations of Photophysical and Single-molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads. Molecules. 2020; 25(3):492. https://doi.org/10.3390/molecules25030492
Chicago/Turabian StyleLefeuvre, Bertrand, Jessica Flores Gonzalez, Frédéric Gendron, Vincent Dorcet, François Riobé, Vladimir Cherkasov, Olivier Maury, Boris Le Guennic, Olivier Cador, Viacheslav Kuropatov, and et al. 2020. "Redox-Modulations of Photophysical and Single-molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads" Molecules 25, no. 3: 492. https://doi.org/10.3390/molecules25030492
APA StyleLefeuvre, B., Flores Gonzalez, J., Gendron, F., Dorcet, V., Riobé, F., Cherkasov, V., Maury, O., Le Guennic, B., Cador, O., Kuropatov, V., & Pointillart, F. (2020). Redox-Modulations of Photophysical and Single-molecule Magnet Properties in Ytterbium Complexes Involving Extended-TTF Triads. Molecules, 25(3), 492. https://doi.org/10.3390/molecules25030492