
New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies

 , , and
, , and
Abstract
1. Introduction
2. Experimental
2.1. Synthesis
2.1.1. General Procedure for the Synthesis of Carboxylic Acid Hydrazides (Series A)
2.1.2. General Procedure for the Synthesis of Thiosemicarbazides (Series B)
B13. 4-(2,4-Dichlorophenyl)-1-(pyridin-3-yl)acetylthiosemicarbazide
B14. 4-(3,4-Dichlorophenyl)-1-(pyridin-4-yl)acetylothiosemicarbazide
2.1.3. General Procedure for the Synthesis of 1,2,4-triazole Derivatives (Series C)
C1. 4-(2-Fluorphfenyl)-3-(pyridin-2-yl)-1,2,4-triazoline-5-thione
C2. 4-(2-Chlorophenyl)-3-(pyridin-2-yl)-1,2,4-triazoline-5-thione
C3. 4-(2,4-Dichlorofenylo)-3-(pyridin-2-yl)-1,2,4-triazoline-5-thione
C4. 4-(3,4-Dichlorofenylo)-3-(pyridin-2-yl)-1,2,4-triazoline-5-thione
C5. 4-(2-Chlorofenylo)-3-(pyridin-3-yl)-1,2,4-triazoline-5-thione
C6. 4-(4-Nitrofenylo)-3-(pyridin-3-yl)-1,2,4-triazoline-5-thione
C7. 4-(2,4-Dichlorofenylo)-3-(pyridin-3-yl)-1,2,4-triazoline-5-thione
C8. 4-(3,4-Dichlorofenylo)-3-(pyridin-3-yl)-1,2,4-triazoline-5-htione
C9. 4-(2-Fluorofenylo)-3-(pyridin-4-yl)-1,2,4-triazoline-5-thione
C10. 4-(2,4-Dichlorophenyl)-3-(pyridin-4-yl)-1,2,4-triazoline-5-thione
C11. 4-Phenyl-3-(pyridin-2-yl)-1,2,4-triazoline-5-thione
C12. 4-(4-Methoxyphenyl)-3-(pyridin-3-yl)-1,2,4-triazoline-5-thione
C13. 4-(2,4-Dichlorophenyl)-3-(pyridin-3-ylmethyl)-1,2,4-triazoline-5-thione
C14. 4-(3,4-Dichlorophenyl)-3-(pyridin-4-ylmethyl)-1,2,4-triazoline-5-thione
2.2. X-ray Structure Determination
2.3. Theoretical Calculations
2.4. Antimycobacterial Assay
2.5. Molecular Docking
3. Results and Discussion
3.1. Chemistry
3.2. X-ray Analysis
3.3. Theoretical Calculations
3.4. Antimycobacterial Activity
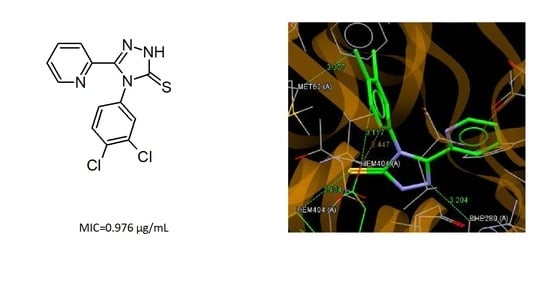
3.5. Molecular Docking
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Beard, C.; Loughman, R.; Smith, A.; Speijers, J. Baseline sensitivity to Tyree triazole fungicides in Pyrenophora tritici-repentis. Aust. Plant Pathol. 2009, 28, 168–172. [Google Scholar] [CrossRef]
- Wu, C.; Sun, J.; Zhang, A.; Liu, W. Dissipation and Enantioselective Degradation of Plant Growth Retardants Paclobutrazol and Uniconazole in Open Field, Greenhouse, and Laboratory Soils. Environ. Sci. Technol. 2013, 47, 843–849. [Google Scholar] [CrossRef] [PubMed]
- Kokalj, A.; Kovacevic, N.; Peljhan, S.; Finšgar, M.; Lesar, A.; Milošev, I. Triazole, Benzotriazole, and Naphthotriazole as Copper Corrosion Inhibitors: I. Molecular Electronic and Adsorption Properties. ChemPhysChem 2011, 12, 3547–3555. [Google Scholar] [CrossRef] [PubMed]
- Asami, T.; Min, Y.K.; Nagata, N.; Yamagishi, K.; Takatsuto, S.; Fujioka, S.; Murofushi, N.; Yamaguchi, I.; Yoshida, S. Characterization of Brassinazole, a Triazole-Type Brassinosteroid Biosynthesis Inhibitor. Plant Physiol. 2000, 123, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, M.M.; Nagarjuna, U.; Padmavathi, V.; Padmaja, A.; Padmaja, A.; Vijaya, T. Synthesis and antimicrobial activity of pyrimidinyl 1,3,4-oxadiazoles, 1,3,4-thiadiazoles and 1,2,4-triazoles. Eur. J. Med. Chem. 2018, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Dubovis, M.; Rudakov, G.; Kulagin, A.; Tsarkova, K.; Popkov, S.; Goloveshkin, A.; Charkaer, G. A new metod of synthesis of substituted 1-(1H-imidazole-4-yl)-1H-1,2,3-triazoles and their fungicidal actvity. Tetrahedron 2018, 78, 672–683. [Google Scholar] [CrossRef]
- Wu, J.; Ni, T.; Chai, X.; Wang, T.; Wang, H.; Chen, J.; Jin, Y.; Zhang, D.-Z.; Yu, S.; Jiang, Y. Molecular docking, design, synthesis and antifungal activity study of novel triazole derivatives. Eur. J. Med. Chem. 2018, 143, 1840–1846. [Google Scholar] [CrossRef]
- Cao, X.; Wang, W.; Wang, S.; Bao, L. Asymmetric synthesis of novel triazole derivatives and their in vitro antiviral activity and mechanism of action. Eur. J. Med. Chem. 2017, 139, 718–725. [Google Scholar] [CrossRef]
- Wu, M.-J.; Wu, D.-M.; Chen, J.-B.; Zhao, J.-F.; Gong, L.; Gong, Y.-X.; Li, Y.; Yang, X.-D.; Zhang, H. Synthesis and anti-proliferative activity of allogibberic acid derivatives containing 1,2,3-triazole pharmacophore. Bioorganic Med. Chem. Lett. 2018, 28, 2543–2549. [Google Scholar] [CrossRef]
- Mustafa, M.; Abdelhamid, D.; Abdelhafez, E.M.N.; Ibrahim, M.A.; Gamal-Eldeen, A.M.; Aly, O.M. Synthesis, antiproliferative, anti-tubulin activity, and docking study of new 1,2,4-triazoles as potential combretastatin analogues. Eur. J. Med. Chem. 2017, 141, 293–305. [Google Scholar] [CrossRef]
- Yamada, M.; Takahashi, T.; Hasegawa, M.; Matsumura, M.; Ono, K.; Fujimoto, R.; Kitamura, Y.; Murata, Y.; Kakusawa, N.; Tanaka, M.; et al. Synthesis, antitumor activity, and cytotoxicity of 4-substituted 1-benzyl-5-diphenylstibano-1H-1,2,3-triazoles. Bioorganic Med. Chem. Lett. 2018, 28, 152–154. [Google Scholar] [CrossRef] [PubMed]
- Saadaoui, I.; Krichen, F.; Ben-Salah, B.; Ben-Mansour, R.; Miled, N.; Bougatef, A.; Kossentini, M. Design, synthesis and biological evaluation of Schiff bases of 4-amino-1,2,4-triazole derivativates as potent angiotensin converting enzyme inhibitors and antioxidant activities. J. Mol. Str. 2019, 1180, 344–354. [Google Scholar] [CrossRef]
- Pawar, S.; Upadhyay, P.; Burade, S.; Kumbhar, N.; Patil, R.; Dhavale, D. Synthesis and anti-leishmanial activity of TRIS-glycine-B-alanine dipeptidic triazole dendron coated with nonameric mannoside glycocluster. Carbohydr. Res. 2019, 485, 1–7. [Google Scholar] [CrossRef]
- Zhang, S.; Xu, Z.; Gao, C.; Ren, Q.-C.; Chang, L.; Lv, Z.-S.; Feng, L.-S. Triazole derivatives and their anti-tubercular activity. Eur. J. Med. Chem. 2017, 138, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Ouellette, W.; Jones, S.; Zubieta, J. Solid state coordination chemistry of metal-1,2,4 triazolates and the related metal-5-(pyrid-4-yl)tetrazolates. CrystEngComm 2011, 13, 4457–4485. [Google Scholar] [CrossRef]
- Kamboj, V.K.; Verma, P.K.; Dhanda, A.; Ranjan, S. 1,2,4-Triazole Derivatives as Potential Scaffold for Anticonvulsant Activity. Central Nerv. Syst. Agents Med. Chem. 2015, 15, 17–22. [Google Scholar] [CrossRef]
- Zhou, C.-H.; Gan, L.; Zhang, Y.; Zhang, F.; Wang, G.; Jin, L.; Geng, R. Review on supermolecules as chemical drugs. Sci. China Ser. B Chem. 2009, 52, 415–458. [Google Scholar] [CrossRef]
- Chu, X.-M.; Wang, C.; Wang, W.-L.; Liang, L.-L.; Liu, W.; Gong, K.-K.; Sun, K.-L. Triazole derivatives and their antiplasmodial and antimalarial activities. Eur. J. Med. Chem. 2019, 166, 206–223. [Google Scholar] [CrossRef]
- Xu, M.; Peng, Y.; Zhu, L.; Wang, S.; Ji, J.; Rakesh, K. Triazole derivatives as inhibitors of Alzheimer’s disease: Current developments and structure-activity relationships. Eur. J. Med. Chem. 2019, 180, 656–672. [Google Scholar] [CrossRef]
- Ceesay, M.M.; Couchman, L.; Smith, M.; Wade, J.; Flanagan, R.J.; Pagliuca, A. Triazole antifungals used for prophylaxis and treatment of invasive fungal disease in adult haematology patients: Trough serum concentrations in relation to outcome. Med. Mycol. 2016, 54, 691–698. [Google Scholar] [CrossRef]
- Srivastava, S.; Bimal, D.; Bohra, K.; Singh, B.; Ponnan, P.; Jain, R.; Varma-Basil, M.; Muty, J.; Thirumal, M.; Prasad, A. Synthesis and antimycobacterial activity of 1-(B-D-Ribofuranosyl)-4-coumarinyloxymethyl-/-coumarinyl-1,2,3-triazole. Eur. J. Med. Chem. 2018, 150, 268–281. [Google Scholar] [CrossRef] [PubMed]
- Sajja, Y.; Vanguru, S.; Vulupala, H.R.; Bantu, R.; Yogeswari, P.; Sriram, D.; Nagarapu, L. Design, synthesis and in vitro anti-tuberculosis activity of benzo[6,7]cyclohepta[1,2-b]pyridine-1,2,3-triazole derivatives. Bioorganic Med. Chem. Lett. 2017, 27, 5119–5121. [Google Scholar] [CrossRef] [PubMed]
- Wilde, F.; Lemmerhirt, H.; Emmrich, T.; Bednarski, P.J.; Link, A. 2-Pyridineacetic acid hydrazide Microwave assisted synthesis and evaluation of acylhydrazones as potential inhibitors of bovine glutathione peroxidise. Mol. Divers. 2014, 18, 307.e322. [Google Scholar] [CrossRef] [PubMed]
- Monjas, L.; Swier, L.J.Y.M.; Setyawati, I.; Slotboom, D.J.; Hirsch, A.K.H. Dynamic Combinatorial Chemistry to Identify Binders of ThiT, an S-Component of the Energy-Coupling Factor Transporter for Thiamine. ChemMedChem 2017, 12, 1693–1696. [Google Scholar] [CrossRef]
- Tran, S.B.; Maxwell, B.D.; Burrell, R.; Bonacorsi, S.J. The syntheses of isotopically labelled CB-1 antagonists for the treatment of obesity. J. Label. Compd. Radiopharm. 2016, 59, 665–672. [Google Scholar] [CrossRef] [PubMed]
- Pitucha, M.; Woś, M.; Miazga-Karska, M.; Klimek, K.; Mirosław, B.; Pachuta-Stec, A.; Gładysz, A.; Ginalska, G. Synthesis, antibacterial and antiproliferative potential of some new 1-pyridinecarbonyl-4-substituted thiosemicarbazide derivatives. Med. Chem. Res. 2016, 25, 1666–1677. [Google Scholar] [CrossRef]
- Choi, H.; Yun, W.; Lee, J.; Jang, S.; Won Park, S.; Kim, D.H.; Seon, K.P.; Hyun, J.; Jeong, K.; Ku, J.; et al. Synthesis and anti-endoplasmic reticulum stress activity of n-substituted-2-arylcarbonylhydrazine carbothioamides. Med. Chem. Res. 2019, 28, 2142–2152. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Z.; Zeng, F. Studies on acylthiosemicarbazides and related heterocyclic derivatives. (XII). Synthetic and spectroscopic studies on 1-nicotinoyl-4-aryl thiosemicarbazides and substituted 1,2,4-triazoles. Gaodeng Xuexiao Huaxue Xuebao 1990, 11, 148–153. [Google Scholar]
- Pitucha, M.; Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Wos, M.; Chudzik, K.; Ginalska, G.; Fruziński, A. Synthesis, In Vitro Screening and Docking Studies of New Thiosemicarbazide Derivatives as Antitubercular Agents. Molecules 2019, 24, 251. [Google Scholar] [CrossRef]
- Khalifa, M.E.; Rakha, T.H.; Bekheit, M.M. Ligational Behaviour of 1-Picolinoyl–4-phenyl–3-thiosemicarbazide (H2PTS) Towards Some Transition Metal Ions. Synth. React. Inorg. Met. Chem. 1996, 26, 1149–1161. [Google Scholar] [CrossRef]
- Dobosz, M.; Pitucha, M.; Wujec, M. The reactions of cyclization of thiosemicarbazide derivatives to 1,2,4-triazole or 1,3,4-thiadiazole system. Acta Polon. Pharm. 1996, 53, 31–38. [Google Scholar]
- Pitucha, M.; Janeczko, M.; Klimek, K.; Fornal, E.; Wos, M.; Pachuta-Stec, A.; Ginalska, G.; Kaczor, A.A. 1,2,4-Triazolin-5-thione derivatives with anticancer activity as CK1γ kinase inhibitors. Bioorganic Chem. 2020, 99, 103806. [Google Scholar] [CrossRef] [PubMed]
- Modzelewska-Banachiewicz, B.; Jagiełło-Wójtowicz, E.; Tokarzewska-Wielosz, E. Synthesis and biological activity of bis-1,2,4-triazole and bis-1,3,4-thiadiazole derivatives. Acta Polon. Pharm. 2000, 57, 199–204. [Google Scholar]
- CrysAlisPro, Agilent Technologies, Version 1.171.37.35h, Release 09-02-2015 CrysAlis171.NET. Available online: https://www.selectscience.net/products/crysalispro/?prodID=197116 (accessed on 19 December 2020).
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, 64, 112–122. [Google Scholar] [CrossRef]
- Farrugia, L.J. WinGX and ORTEP for Windows: An update. J. Appl. Cryst. 2012, 45, 849–854. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A.; Vreven, T.; Kudin, K.N.; Burant, J.C.; et al. Gaussian 03, Revision E.01; Gaussian Inc.: Wallingford, CT, USA, 2004. [Google Scholar]
- Dennington, R.; Keith, T.; Millam, J. GaussView 4.1; Semichem, Inc.: Shawnee Mission, KS, USA, 2007. [Google Scholar]
- Ghose, A.K.; Pritchett, A.; Crippen, G.M. Atomic physicochemical parameters for three dimensional structure directed quantitative structure-activity relationships III: Modeling hydrophobic interactions. J. Comput. Chem. 1988, 9, 80–90. [Google Scholar] [CrossRef]
- Hyper-Chem, Release 8.0.10 for Windows Molecular Modeling System; Hypercube Inc.: Gainesville, FL, USA, 2006; p. 32601.
- Turnidge, J.D.; Jorgensen, J.H. Antimicrobial Susceptibility Testing: General Cosiderations. In Manual of Clinical Microbiology, 7th ed.; Murray, P.R., Baron, E.J., Pfaller, M.A., Tenover, F.C., Yolken, R., Eds.; American Society for Microbiology: Washington, DC, USA, 1999; pp. 1469–1473. [Google Scholar]
- Taban, I.M.; Elshihawy, H.E.A.E.; Torun, B.; Zucchini, B.; Williamson, C.J.; Altuwairigi, D.; Ngu, A.S.T.; McLean, K.J.; Levy, C.W.; Sood, S.; et al. Novel Aryl Substituted Pyrazoles as Small Molecule Inhibitors of Cytochrome P450 CYP121A1: Synthesis and Antimycobacterial Evaluation. J. Med. Chem. 2017, 60, 10257–10267. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical Scoring Functions for Advanced Protein−Ligand Docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Allen, F.H.; Kennard, O.; Watson, D.G.; Brammer, L.; Orpen, A.G.; Taylor, R. Tables of bond lengths determined by X-ray and neutron diffraction. Part 1. Bond lengths in organic compounds. J. Chem. Soc. Perkin Trans. 1987, S1–S19. [Google Scholar] [CrossRef]
- Pennington, L.D.; Moustakas, D.T. The Necessary Nitrogen Atom: A Versatile High-Impact Design Element for Multiparameter Optimization. J. Med. Chem. 2017, 60, 3552–3579. [Google Scholar] [CrossRef] [PubMed]
- Rode, N.D.; Sonawane, A.D.; Nawale, L.; Khedkar, V.M.; Joshi, R.A.; Likhite, A.P.; Sarkar, D.; Joshi, R.R. Synthesis, biological evaluation, and molecular docking studies of novel 3-aryl-5-(alkyl-thio)-1H-1,2,4-triazoles derivatives targeting Mycobacterium tuberculosis. Chem. Biol. Drug Des. 2017, 90, 1206–1214. [Google Scholar] [CrossRef] [PubMed]
- Hudson, S.A.; McLean, K.J.; Surade, S.; Yang, Y.-Q.; Leys, D.; Ciulli, A.; Munro, A.W.; Abell, C. Application of Fragment Screening and Merging to the Discovery of Inhibitors of the Mycobacterium tuberculosis Cytochrome P450 CYP121. Angew. Chem. Int. Ed. 2012, 51, 9311–9316. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| D–H…A | D–H | H…A | D…A | D–H…A |
|---|---|---|---|---|
| C1 | ||||
| N1–H1…S5i | 0.89(3) | 2.37(4) | 3.261(2) | 175(3) |
| i = −x, 2 − y, −z | ||||
| C12 | ||||
| N1–H1…N63i | 0.92(3) | 1.88(3) | 2.789(3) | 171(2) |
| i = x, 3/2 − y, −1/2 + z | ||||
| C13 | ||||
| N1–H1…N63i | 0.99(3) | 1.83(3) | 2.815(4) | 170(3) |
| C43–H43…N2ii | 0.93 | 2.53 | 3.435(4) | 164 |
| C46–H46…S5iii | 0.93 | 2.84 | 3.675(4) | 150 |
| C65–H65…S5iv | 0.93 | 2.87 | 3.714(4) | 152 |
| C31–H312…Sv | 0.97 | 2.87 | 3.552(4) | 129 |
| i = −1 + x, 1 + y, z; ii = x, −1+y, z; iii = −x, 1 − y, 1 − z; iv = −x, 1 − y, −z; v = 1 + x, y, z | ||||
| Compound | Dm | NBO Charge | EHOMO | ELUMO | ΔE | logP | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| N1 | N2 | N4 | Npyridil | S | ||||||
| C1 | 5.631 | −0.381 | −0.274 | −0.455 | −0.455 | −0.207 | −134.17 | −45.45 | 88.68 | 4.95 |
| C2 | 5.736 | −0.381 | −0.276 | −0.462 | −0.451 | −0.205 | −133.80 | −45.42 | 88.38 | 5.33 |
| C3 | 6.001 | −0.389 | −0.273 | −0.460 | −0.451 | −0.200 | −136.58 | −48.19 | 88.39 | 5.85 |
| C4 | 6.262 | −0.382 | −0.274 | −0.462 | −0.451 | −0.202 | −137.66 | −49.22 | 88.44 | 5.85 |
| C5 | 3.265 | −0.384 | −0.277 | −0.471 | −0.444 | −0.206 | −136.88 | −43.70 | 93.18 | 5.40 |
| C6 | 6.515 | −0.382 | −0.270 | −0.471 | −0.440 | −0.193 | −144.86 | −73.52 | 77.33 | 0.97 |
| C7 | 3.502 | −0.384 | −0.276 | −0.474 | −0.443 | −0.202 | −139.61 | −46.19 | 93.42 | 5.81 |
| C8 | 3.901 | −0.384 | −0.275 | −0.465 | −0.441 | −0.206 | −140.92 | −47.72 | 93.20 | 5.91 |
| C9 | 2.870 | −0.382 | −0.258 | −0.455 | −0.442 | −0.196 | −138.88 | −49.32 | 89.55 | 5.02 |
| C10 | 1.587 | −0.381 | −0.265 | −0.472 | −0.439 | −0.206 | −141.92 | −51.26 | 90.65 | 5.91 |
| C11 | 4.538 | −0.394 | −0.308 | −0.476 | −0.469 | −0.222 | −132.84 | −32.45 | 100.39 | 4.75 |
| C12 | 3.208 | −0.384 | −0.281 | −0.461 | −0.444 | −0.217 | −133.86 | −42.17 | 91.68 | 4.63 |
| C13 | 3.694 | −0.393 | −0.294 | −0.482 | −0.444 | −0.205 | −138.47 | −38.39 | 99.94 | 5.85 |
| C14 | 4.688 | −0.392 | −0.291 | −0.480 | −0.448 | −0.205 | −140.64 | −39.24 | 101.40 | 5.85 |
| Inhibition Zone (mm) 250 μg of Compound PerWell | ||||||
|---|---|---|---|---|---|---|
| Ar | R | Mycobacterium H37Ra | Mycobacterium Phlei | Mycobacterium Smegmatis | Mycobacterium Timereck | |
| C1 | pyridin-2-yl | 2-FC6H4 | 15.1 ± 0.61 | 15.1 ± 0.53 | 16.0 ± 0.81 | 15.1 ± 0.67 |
| C2 | pyridin-2-yl | 2-ClC6H4 | 19.6 ± 0.43 | 21.5 ± 1.12 | 17.2 ± 0.98 | 15.3 ± 0.84 |
| C3 | pyridin-2-yl | 2,4-Cl2C6H3 | 10.2 ± 0.8 | 17.0 ± 0.78 | 17.9 ± 0.77 | 20.1 ± 1.15 |
| C4 | pyridin-2-yl | 3,4-Cl2C6H3 | 26.9 ± 0.78 | 22.7 ± 0.91 | 22.3 ± 1.05 | 21.5 ± 0.9 |
| C5 | pyridin-3-yl | 2-ClC6H4 | 12.3 ± 0.78 | 0.0 | 10.2 ± 0.86 | 16.5 ± 0.41 |
| C6 | pyridin-3-yl | 4-NO2C6H4 | 0.0 | 0.0 | 0.0 | 10.6 ± 0.53 |
| C7 | pyridin-3-yl | 2,4-Cl2C6H3 | 0.0 | 8.9 ± 0.66 | 0.0 | 0.0 |
| C8 | pyridin-3-yl | 3,4-Cl2C6H3 | 22.2 ± 0.72 | 21.9 ± 0.29 | 23.1 ± 0.6 | 24.7 ± 0.87 |
| C9 | pyridin-4-yl | 2-FC6H4 | 10.6 ± 0.53 | 10.9 ± 0.66 | 12.3 ± 0.92 | 19.6 ± 0.53 |
| C10 | pyridin-4-yl | 2,4-Cl2C6H3 | 0.0 | 0.0 | 0.0 | 0.0 |
| C11 | pyridin-2-yl | C6H5 | 35.9 ± 0.49 | 34.0 ± 0.93 | 35.3 ± 0.7 | 35.1 ± 0.58 |
| C12 | pyridin-3-yl | 4-CH3OC6H4 | 10.6 ± 0.85 | 0.0 | 0.0 | 0.0 |
| C13 | pyridin-3-ylmethyl | 2,4-Cl2C6H3 | 12.0 ± 0.67 | 0.0 | 0.0 | 0.0 |
| C14 | pyridin-4-ylmethyl | 3,4-Cl2C6H3 | 20.7 ± 0.29 | 0.0 | 18.2 ± 0.53 | 18.6 ± 0.98 |
| MIC (µg/mL) | ||||||
|---|---|---|---|---|---|---|
| Ar | R | Mycobacterium H37Ra | Mycobacterium Phlei | Mycobacterium Smegmatis | Mycobacterium Timereck | |
| C1 | pyridin-2-yl | 2-FC6H4 | 62.5 | 500 | >500 | >500 |
| C4 | pyridin-2-yl | 3,4-Cl2C6H3 | 0.976 | 7.81 | 125 | 62.5 |
| C8 | pyridin-3-yl | 3,4-Cl2C6H3 | 62.5 | 31.25 | 125 | 62.5 |
| C11 | pyridin-2-yl | C6H5 | 62.5 | 31.25 | 250 | 62.5 |
| C14 | pyridin-2-ylmethyl | 3,4-Cl2C6H3 | 62.5 | 125 | >500 | 125 |
| Compound | fCHEMPLP | Ligand—Amino Acids Interactions |
|---|---|---|
| C1 | 56.67 | F42…C(HEME); S5…C(MET62); N1…O(HEME); N2…C(HEME) |
| C2 | 57.79 | N1…O(HEME); N2…C(HEME); S5…C(MET62) |
| C3 | 54.59 | S5…C(HEME); N1…O(VAL82) |
| C4 | 57.68 | N2…C(PHE280); S5…C(HEME); C45…O(HEME); C46…O(HEME); Cl44…(MET62) |
| C5 | 57.04 | S5…C(MET62); N2…C(PHE280) |
| C6 | 67.83 | S5…C(MET62) × 2; N63…N(ARG386); O…O(SER237) |
| C7 | 55.14 | S5…C(MET62); N63…C(PHE280); Cl44…(C(PHE280); Cl42…O(HEME) |
| C8 | 53.67 | N63…N(ARG386); Cl43…C(VAL78) |
| C9 | 58.71 | S5…C((MET62); N64…O(SER237) |
| C10 | 60.20 | N2…C(HEME); N64…N(ARG386); C63…O(SER237); C43…O(HEME) |
| C11 | 65.77 | S5…C(MET62) × 2; S5…C(HEME) |
| C12 | 55.89 | N1…O(VAL82) |
| C13 | 66.94 | S5…N(ASN85); S5…C(ASN85); N64…C(MET62); N1…O(VAL82); N2…C(MET62) |
| C14 | 68.29 | N1…O(VAL82); S5…N(ASN85); S5…C(ASN85); Cl43…C(HEME) |
Sample Availability: Samples of the compounds B13, B14, C1–C14 are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Karczmarzyk, Z.; Swatko-Ossor, M.; Wysocki, W.; Drozd, M.; Ginalska, G.; Pachuta-Stec, A.; Pitucha, M. New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies. Molecules 2020, 25, 6033. https://doi.org/10.3390/molecules25246033
Karczmarzyk Z, Swatko-Ossor M, Wysocki W, Drozd M, Ginalska G, Pachuta-Stec A, Pitucha M. New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies. Molecules. 2020; 25(24):6033. https://doi.org/10.3390/molecules25246033
Chicago/Turabian StyleKarczmarzyk, Zbigniew, Marta Swatko-Ossor, Waldemar Wysocki, Monika Drozd, Grazyna Ginalska, Anna Pachuta-Stec, and Monika Pitucha. 2020. "New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies" Molecules 25, no. 24: 6033. https://doi.org/10.3390/molecules25246033
APA StyleKarczmarzyk, Z., Swatko-Ossor, M., Wysocki, W., Drozd, M., Ginalska, G., Pachuta-Stec, A., & Pitucha, M. (2020). New Application of 1,2,4-Triazole Derivatives as Antitubercular Agents. Structure, In Vitro Screening and Docking Studies. Molecules, 25(24), 6033. https://doi.org/10.3390/molecules25246033