
Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding


Abstract
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
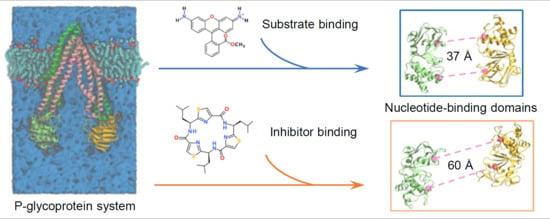
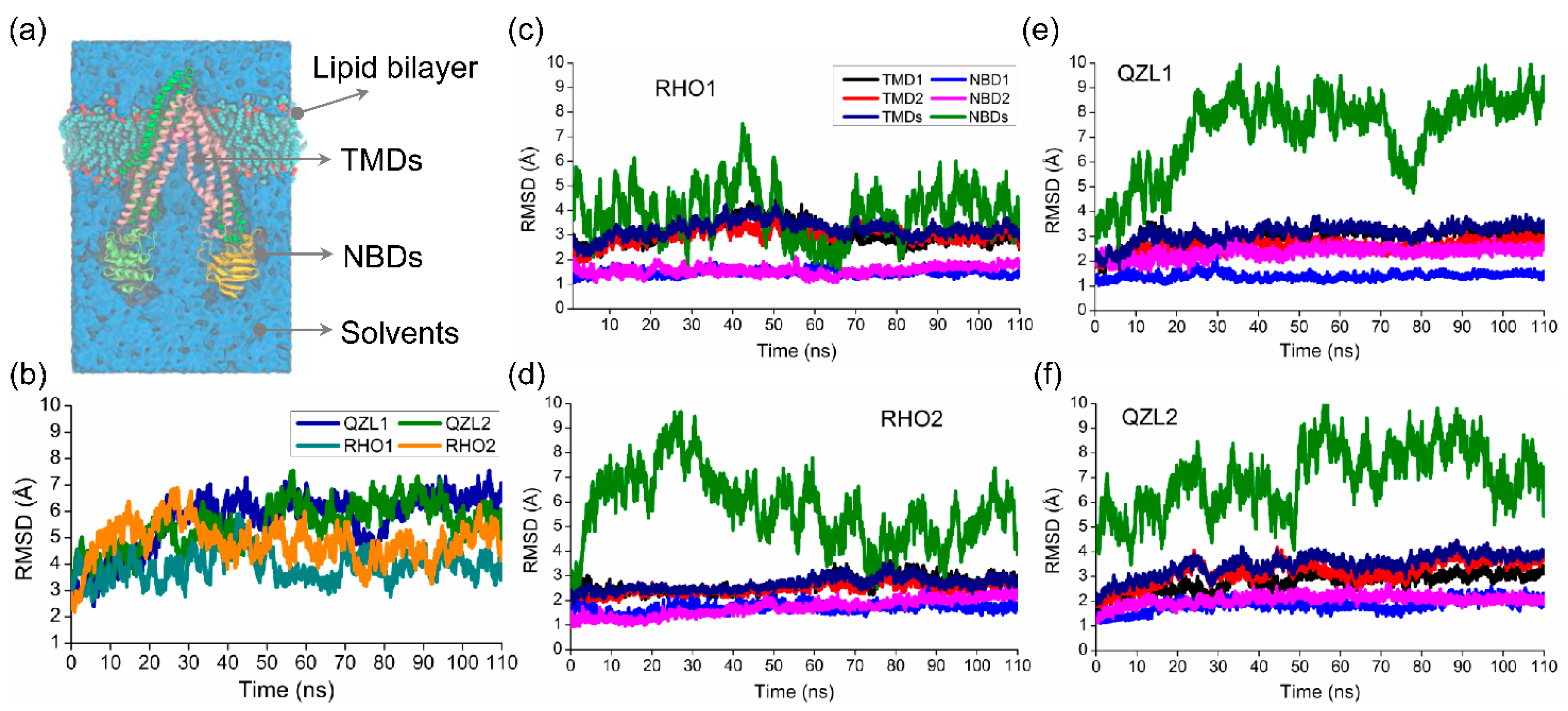
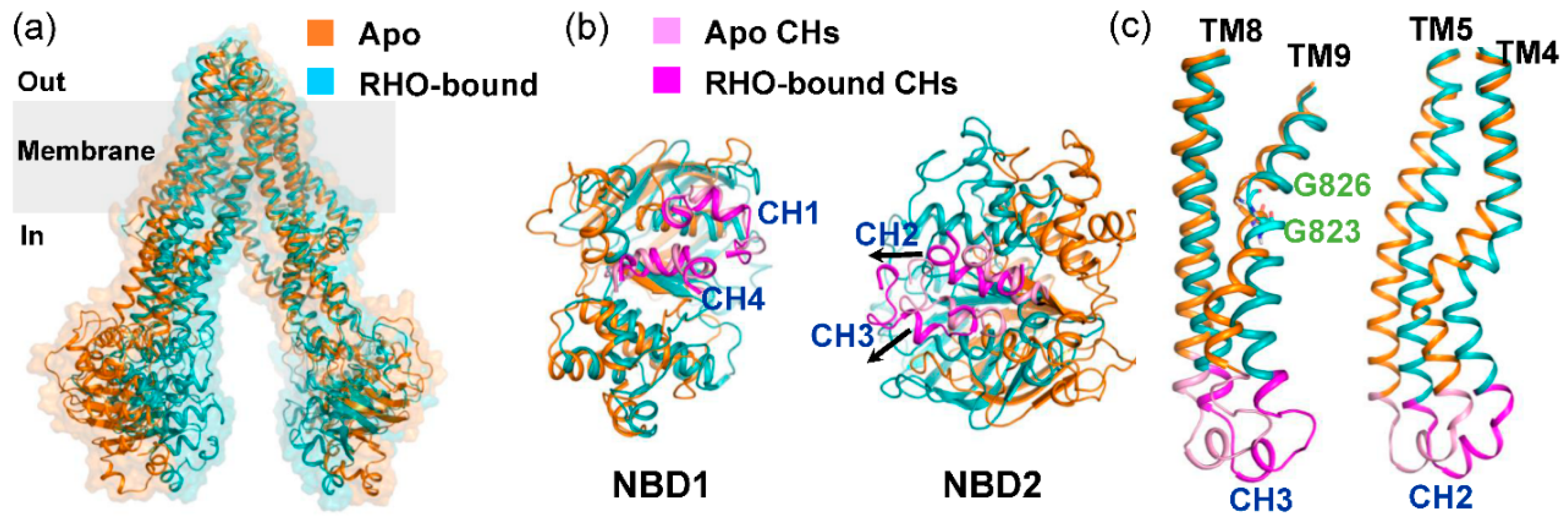
2.1. Different Conformational Changes Mainly Occurred in the NBDs Upon Substrate and Inhibitor Binding to the TMD Cavity
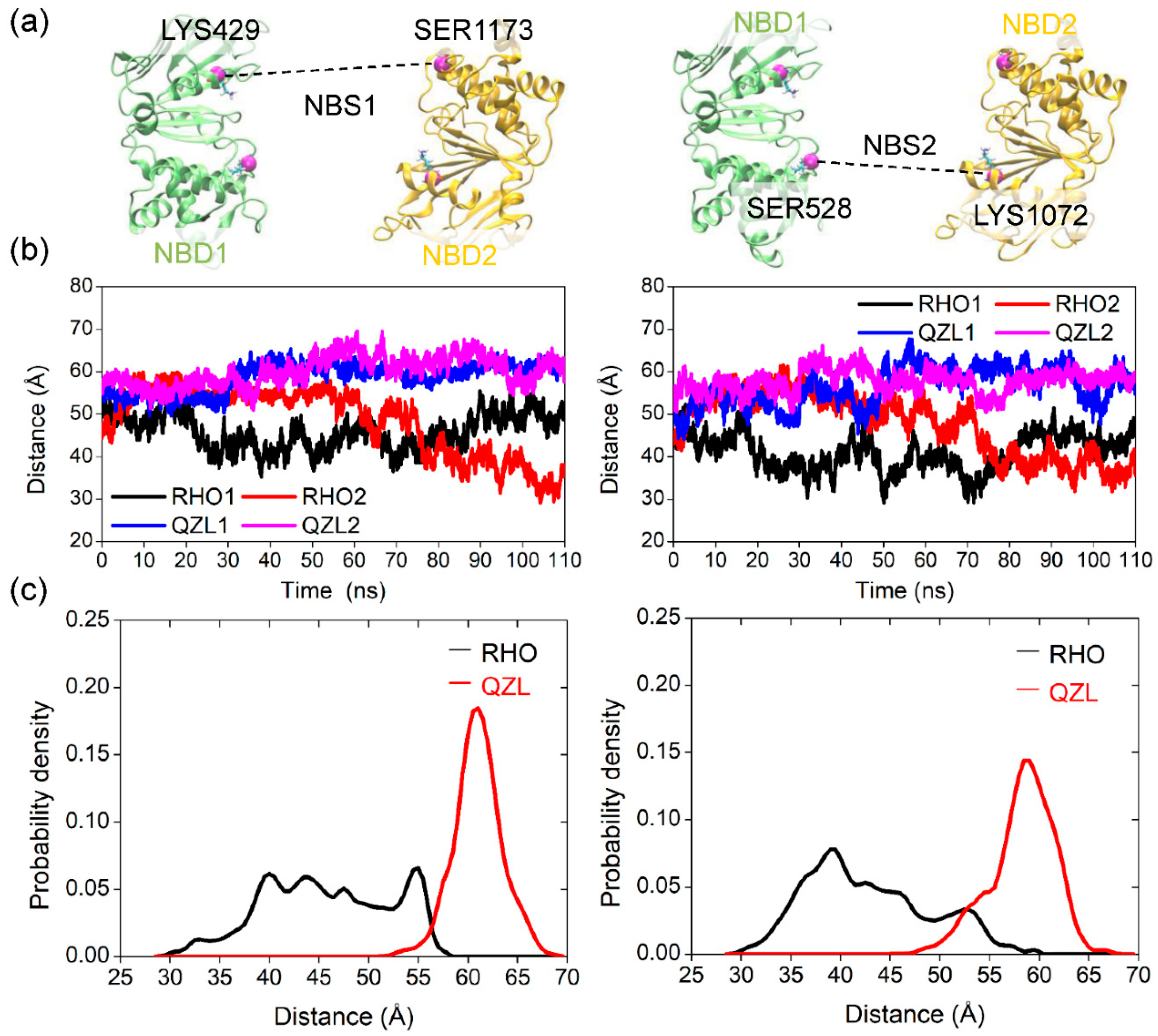
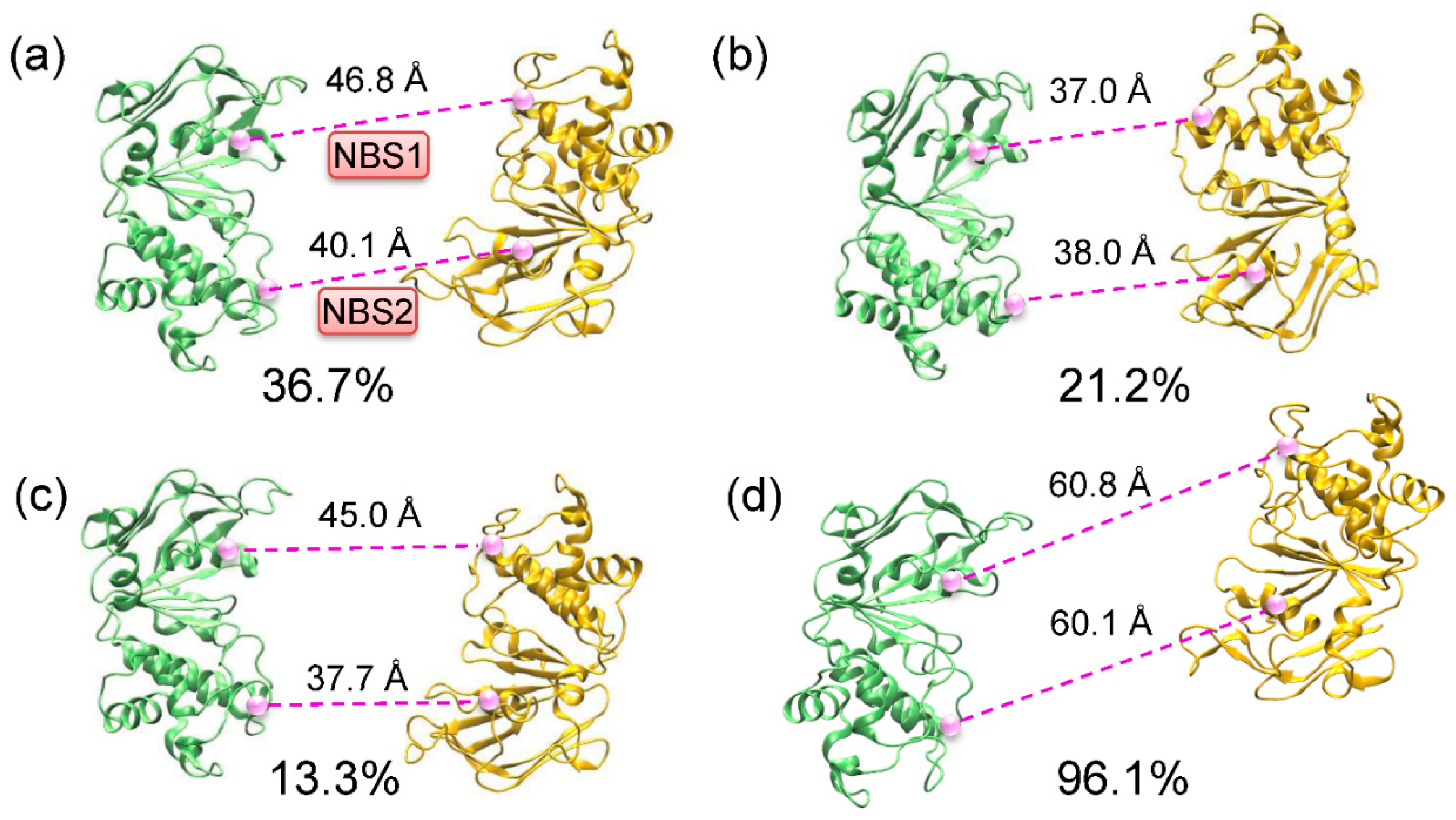
2.2. Conformational Homogeneity in the Substrate- and Inhibitor-Bound States
2.3. Implications of Substrate- and Inhibitor-Coupled Conformational Transitions
2.4. Allosteric Communication between TMDs and NBDs upon Substrate Binding
3. Materials and Methods
3.1. Structural Preparation
3.1.1. Construction of Inhibitor- and Substrate-Bound P-gp Models
3.1.2. Molecular Docking
3.1.3. Preparation of Simulation Systems
3.2. MD Simulations
3.2.1. Simulation Parameters
3.2.2. Simulation Protocol
3.3. Trajectory Analysis
3.3.1. Principal Component Analysis
3.3.2. Cluster Analysis
3.4. Free Energy Calculations
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| P-gp | P-glycoprotein |
| TMDs | transmembrane domains |
| NBDs | nucleotide-binding domains |
| ABC | ATP-binding cassette |
| NBSs | nucleotide-binding sites |
| MD | molecular dynamics |
| RMSDs | root-mean-square deviations |
| PCA | principal component analysis |
| MM/GBSA | molecular mechanics/generalized born surface area |
References
- Wong, K.; Ma, J.; Rothnie, A.; Biggin, P.C.; Kerr, I.D. Towards understanding promiscuity in multidrug efflux pumps. Trends Biochem. Sci. 2014, 39, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Szakacs, G.; Varadi, A.; Ozvegy-Laczka, C.; Sarkadi, B. The role of ABC transporters in drug absorption, distribution, metabolism, excretion and toxicity (ADME-Tox). Drug Discov. Today 2008, 13, 379–393. [Google Scholar] [CrossRef] [PubMed]
- Giacomini, K.M.; Huang, S.M.; Tweedie, D.J.; Benet, L.Z.; Brouwer, K.L.; Chu, X.; Dahlin, A.; Evers, R.; Fischer, V.; Hillgren, K.M.; et al. Membrane transporters in drug development. Nat. Rev. Drug Discov. 2010, 9, 215–236. [Google Scholar] [PubMed]
- Szakacs, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef]
- Chufan, E.E.; Sim, H.M.; Ambudkar, S.V. Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): Recent biochemical and structural studies. Adv. Cancer Res. 2015, 125, 71–96. [Google Scholar]
- Eckford, P.D.; Sharom, F.J. ABC efflux pump-based resistance to chemotherapy drugs. Chem. Rev. 2009, 109, 2989–3011. [Google Scholar] [CrossRef]
- Aller, S.G.; Yu, J.; Ward, A.; Weng, Y.; Chittaboina, S.; Zhuo, R.; Harrell, P.M.; Trinh, Y.T.; Zhang, Q.; Urbatsch, I.L.; et al. Structure of P-glycoprotein reveals a molecular basis for poly-specific drug binding. Science 2009, 323, 1718–1722. [Google Scholar] [CrossRef]
- Ward, A.B.; Szewczyk, P.; Grimard, V.; Lee, C.W.; Martinez, L.; Doshi, R.; Caya, A.; Villaluz, M.; Pardon, E.; Cregger, C.; et al. Structures of P-glycoprotein reveal its conformational flexibility and an epitope on the nucleotide-binding domain. Proc. Natl. Acad. Sci. USA 2013, 110, 13386–13391. [Google Scholar] [CrossRef]
- Szewczyk, P.; Tao, H.; McGrath, A.P.; Villaluz, M.; Rees, S.D.; Lee, S.C.; Doshi, R.; Urbatsch, I.L.; Zhang, Q.; Chang, G. Snapshots of ligand entry, malleable binding and induced helical movement in P-glycoprotein. Acta Crystallogr. D Biol. Crystallogr. 2015, 71, 732–741. [Google Scholar] [CrossRef]
- Alam, A.; Kung, R.; Kowal, J.; McLeod, R.A.; Tremp, N.; Broude, E.V.; Roninson, I.B.; Stahlberg, H.; Locher, K.P. Structure of a zosuquidar and UIC2-bound human-mouse chimeric ABCB1. Proc. Natl. Acad. Sci. USA 2018, 115, 1973–1982. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Substrate-induced conformational changes in the transmembrane segments of human P-glycoprotein. Direct evidence for the substrate-induced fit mechanism for drug binding. J. Biol. Chem. 2003, 278, 13603–13606. [Google Scholar] [CrossRef]
- Pan, X.; Mei, H.; Qu, S.; Huang, S.; Sun, J.; Yang, L.; Chen, H. Prediction and characterization of P-glycoprotein substrates potentially bound to different sites by emerging chemical pattern and hierarchical cluster analysis. Int. J. Pharm. 2016, 502, 61–69. [Google Scholar] [CrossRef] [PubMed]
- Tan, W.; Mei, H.; Chao, L.; Liu, T.; Pan, X.; Shu, M.; Yang, L. Combined QSAR and molecule docking studies on predicting P-glycoprotein inhibitors. J. Comput. Aided Mol. Des. 2013, 27, 1067–1073. [Google Scholar] [CrossRef]
- Collu, F.; Cascella, M. Multidrug resistance and efflux pumps: Insights from molecular dynamics simulations. Curr. Top Med. Chem. 2013, 13, 3165–3183. [Google Scholar] [CrossRef]
- Xing, J.; Mei, H.; Huang, S.; Zhang, D.; Pan, X. An Energetically Favorable Ligand Entrance Gate of a Multidrug Transporter Revealed by Partial Nudged Elastic Band Simulations. Comput. Struct. Biotechnol. J. 2019, 17, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Wen, P.C.; Verhalen, B.; Wilkens, S.; McHaourab, H.S.; Tajkhorshid, E. On the origin of large flexibility of P-glycoprotein in the inward-facing state. J. Biol. Chem. 2013, 288, 19211–19220. [Google Scholar] [CrossRef] [PubMed]
- Moeller, A.; Lee, S.C.; Tao, H.; Speir, J.A.; Chang, G.; Urbatsch, I.L.; Potter, C.S.; Carragher, B.; Zhang, Q. Distinct Conformational Spectrum of Homologous Multidrug ABC Transporters. Structure 2015, 23, 450–460. [Google Scholar] [CrossRef] [PubMed]
- Sim, H.M.; Bhatnagar, J.; Chufan, E.E.; Kapoor, K.; Ambudkar, S.V. Conserved Walker A cysteines 431 and 1074 in human P-glycoprotein are accessible to thiol-specific agents in the apo and ADP-vanadate trapped conformations. Biochemistry 2013, 52, 7327–7338. [Google Scholar] [CrossRef]
- Kimura, Y.; Kioka, N.; Kato, H.; Matsuo, M.; Ueda, K. Modulation of drug-stimulated ATPase activity of human MDR1/P-glycoprotein by cholesterol. Biochem. J. 2007, 401, 597–605. [Google Scholar] [CrossRef]
- Doshi, R.; van Veen, H.W. Substrate binding stabilizes a pre-translocation intermediate in the ATP-binding cassette transport protein MsbA. J. Biol. Chem. 2013, 288, 21638–21647. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. Structural Basis of Substrate Recognition by the Multidrug Resistance Protein MRP1. Cell 2017, 168, 1075–1085. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Chen, J. Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation. Science 2018, 359, 915–919. [Google Scholar] [CrossRef]
- Johnson, Z.L.; Chen, J. ATP Binding Enables Substrate Release from Multidrug Resistance Protein 1. Cell 2018, 172, 81–89. [Google Scholar] [CrossRef]
- Ward, A.; Reyes, C.L.; Yu, J.; Roth, C.B.; Chang, G. Flexibility in the ABC transporter MsbA: Alternating access with a twist. Proc. Natl. Acad. Sci. USA 2007, 104, 19005–19010. [Google Scholar] [CrossRef]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Drug binding in human P-glycoprotein causes conformational changes in both nucleotide-binding domains. J. Biol. Chem. 2003, 278, 1575–1578. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Location of the rhodamine-binding site in the human multidrug resistance P-glycoprotein. J. Biol. Chem. 2002, 277, 44332–44338. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Bartlett, M.C.; Clarke, D.M. Identification of residues in the drug translocation pathway of the human multidrug resistance P-glycoprotein by arginine mutagenesis. J. Biol. Chem. 2009, 284, 24074–24087. [Google Scholar] [CrossRef] [PubMed]
- Donmez Cakil, Y.; Khunweeraphong, N.; Parveen, Z.; Schmid, D.; Artaker, M.; Ecker, G.F.; Sitte, H.H.; Pusch, O.; Stockner, T.; Chiba, P. Pore-exposed tyrosine residues of P-glycoprotein are important hydrogen-bonding partners for drugs. Mol. Pharmacol. 2014, 85, 420–428. [Google Scholar] [CrossRef]
- Ma, J.; Biggin, P.C. Substrate versus inhibitor dynamics of P-glycoprotein. Proteins 2013, 81, 1653–1668. [Google Scholar] [CrossRef]
- Kodan, A.; Yamaguchi, T.; Nakatsu, T.; Sakiyama, K.; Hipolito, C.J.; Fujioka, A.; Hirokane, R.; Ikeguchi, K.; Watanabe, B.; Hiratake, J.; et al. Structural basis for gating mechanisms of a eukaryotic P-glycoprotein homolog. Proc. Natl. Acad. Sci. USA 2014, 111, 4049–4054. [Google Scholar] [CrossRef]
- Loo, T.W.; Clarke, D.M. Tariquidar inhibits P-glycoprotein drug efflux but activates ATPase activity by blocking transition to an open conformation. Biochem. Pharmacol. 2014, 92, 558–566. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Mapping the Binding Site of the Inhibitor Tariquidar That Stabilizes the First Transmembrane Domain of P-glycoprotein. J. Biol. Chem. 2015, 290, 29389–29401. [Google Scholar] [CrossRef] [PubMed]
- Loo, T.W.; Clarke, D.M. Locking intracellular helices 2 and 3 together inactivates human P-glycoprotein. J. Biol. Chem. 2014, 289, 229–236. [Google Scholar] [CrossRef]
- Alam, A.; Kowal, J.; Broude, E.; Roninson, I.; Locher, K.P. Structural insight into substrate and inhibitor discrimination by human P-glycoprotein. Science 2019, 363, 753–756. [Google Scholar] [CrossRef] [PubMed]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucl. Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex: Fully automatic flexible molecular docking using a molecular similarity-based search engine. J. Med. Chem. 2003, 46, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI Membrane Builder Toward Realistic Biological Membrane Simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef]
- Price, D.J.; Brooks, C.L., 3rd. A modified TIP3P water potential for simulation with Ewald summation. J. Chem. Phys. 2004, 121, 10096–10103. [Google Scholar] [CrossRef]
- O’Mara, M.L.; Mark, A.E. The Effect of Environment on the Structure of a Membrane Protein: P-Glycoprotein under Physiological Conditions. J. Chem. Theory Comput. 2012, 8, 3964–3976. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. WIREs Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Maier, J.A.; Martinez, C.; Kasavajhala, K.; Wickstrom, L.; Hauser, K.E.; Simmerling, C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. [Google Scholar] [CrossRef]
- Skjevik, A.A.; Madej, B.D.; Walker, R.C.; Teigen, K. LIPID11: A modular framework for lipid simulations using amber. J. Phys. Chem. B 2012, 116, 11124–11136. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle Mesh Ewald—An N.Log(N) Method for Ewald Sums in Large Systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.-P.; Ciccotti, G.; Berendsen, H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]






Sample Availability: Samples of the compounds rhodamine-123 and QZ-Leu are available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xing, J.; Huang, S.; Heng, Y.; Mei, H.; Pan, X. Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules 2020, 25, 6006. https://doi.org/10.3390/molecules25246006
Xing J, Huang S, Heng Y, Mei H, Pan X. Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules. 2020; 25(24):6006. https://doi.org/10.3390/molecules25246006
Chicago/Turabian StyleXing, Juan, Shuheng Huang, Yu Heng, Hu Mei, and Xianchao Pan. 2020. "Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding" Molecules 25, no. 24: 6006. https://doi.org/10.3390/molecules25246006
APA StyleXing, J., Huang, S., Heng, Y., Mei, H., & Pan, X. (2020). Computational Insights into Allosteric Conformational Modulation of P-Glycoprotein by Substrate and Inhibitor Binding. Molecules, 25(24), 6006. https://doi.org/10.3390/molecules25246006