
Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study




Abstract
1. Introduction
2. Results and Discussion
2.1. Computational Protocol
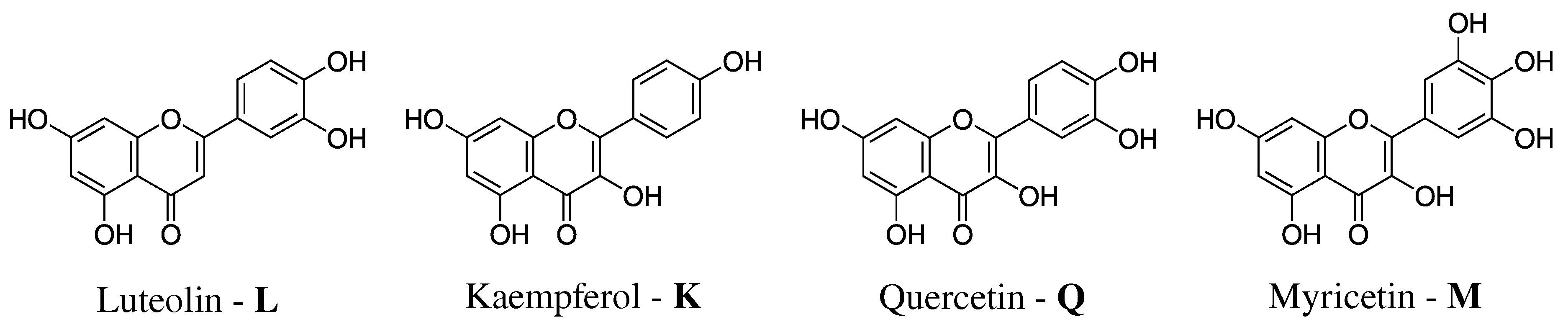
- Definition of the system. The geometry of each of the four flavonoids depicted in Figure 1 was first optimized by resorting to an implicit Polarizable Continuum Model (PCM) [40] description of the solvent, and then surrounded by a number of randomly-placed water molecules large enough to represent the solvation shell.
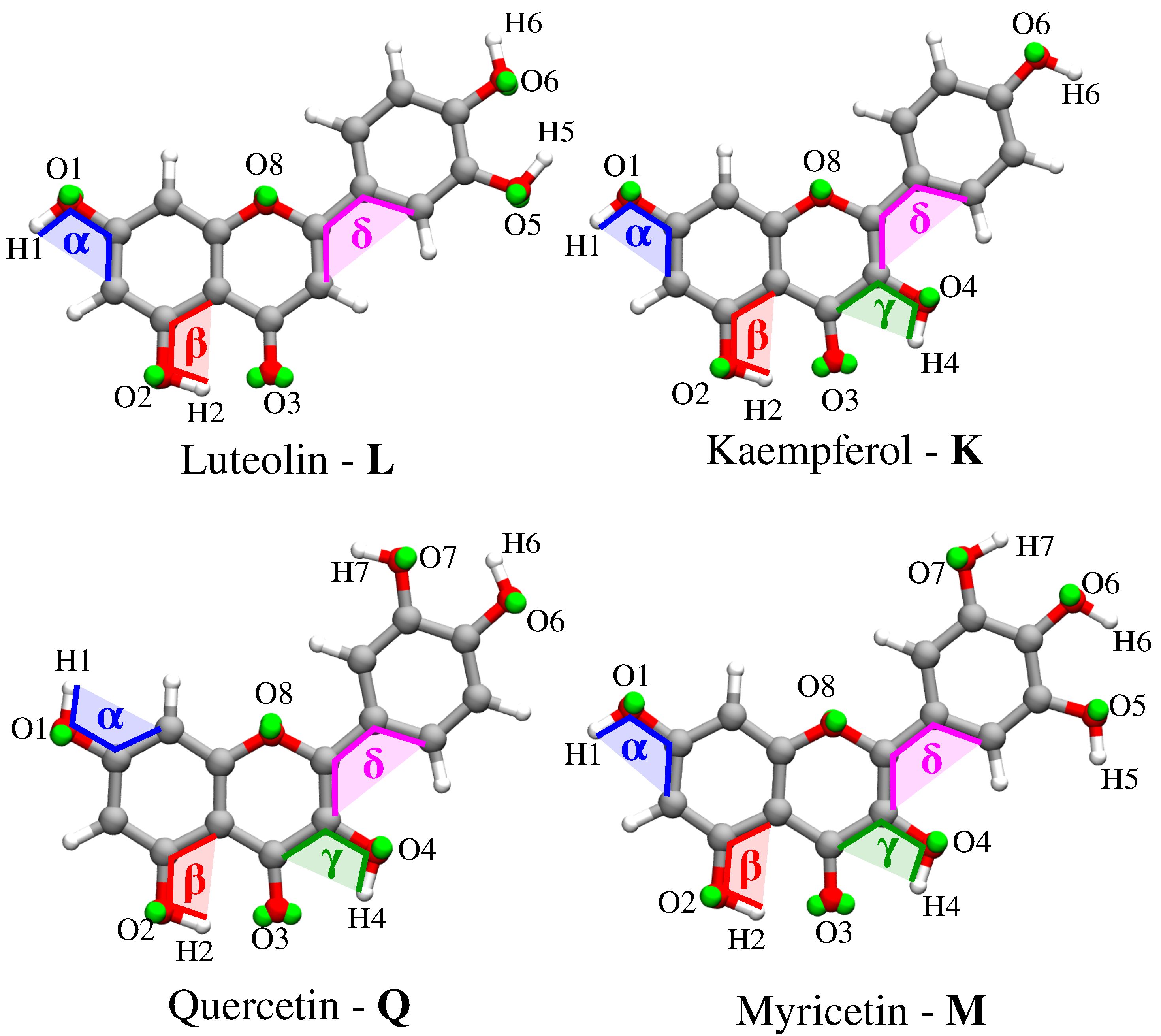
- Classical MD simulations. An equilibration (NPT) and a subsequent production (NVT) runs were performed in order to sample the system under study. In particular, MD production runs were carried out for each of the four molecules for a time long enough to obtain an accurate sampling of the phase space, in order to correctly reproduce all possible system configurations and their relative energy.In order to recover the directionality of hydrogen bonding (HB) interactions, we also placed off-site charges (the so-called virtual sites (VS) or dummy atoms) to better describe the lone pairs of the Oxygen atom (see Figure 2 for a graphical representation) [44,65,66]. Therefore, two different classical MD simulation runs were performed for each molecule, i.e., with or without the inclusion of VS (MD or MD, respectively). From MD runs, a set of snapshots was extracted to be used in QM/FQ calculations.
- Definition of the different regions of the two-layer QM/FQ scheme and their boundaries. For each snapshot extracted from MD runs, a sphere centered on the solute was cut. The radius of the droplet was chosen to retain specific solute–water interactions.
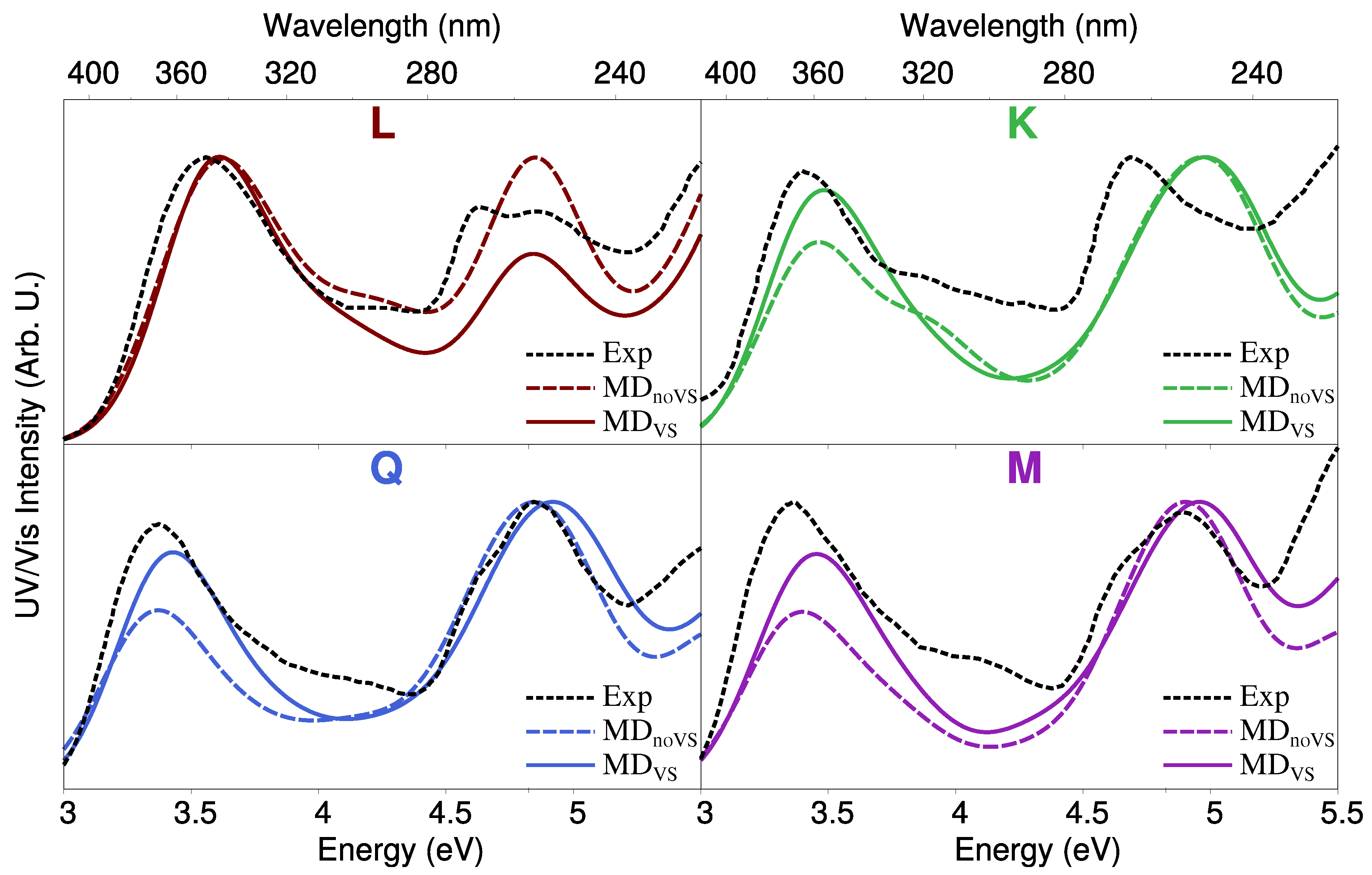
- QM/FQ calculations and comparison with experimental data reported in [28,67]. QM/FQ excitation energies calculations were performed on the set of structures obtained for the four molecules at step 2 of the protocol. The results obtained for each spherical snapshot were then extracted and averaged to produce the final spectrum, which was compared with experiments.
2.2. MD Analysis
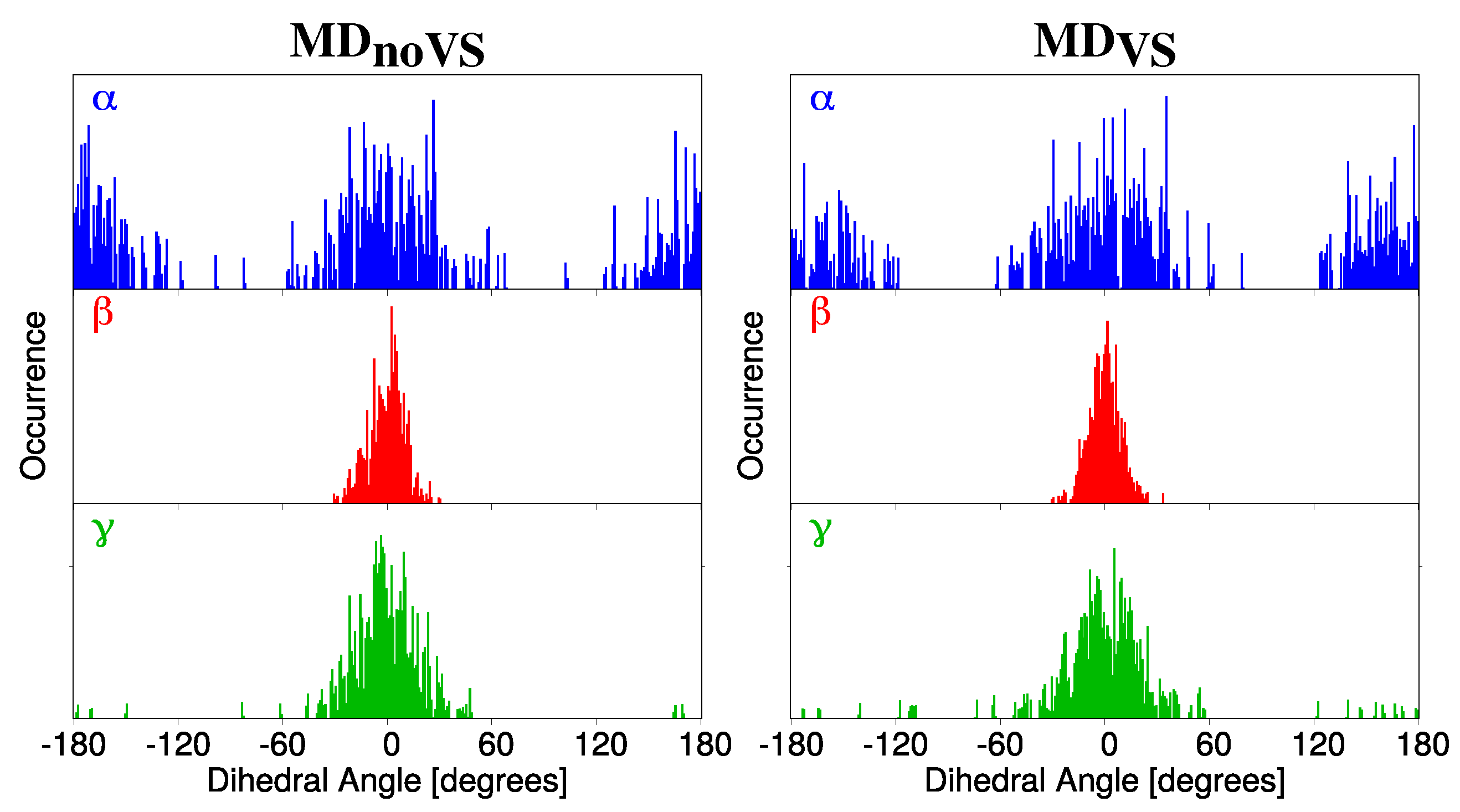
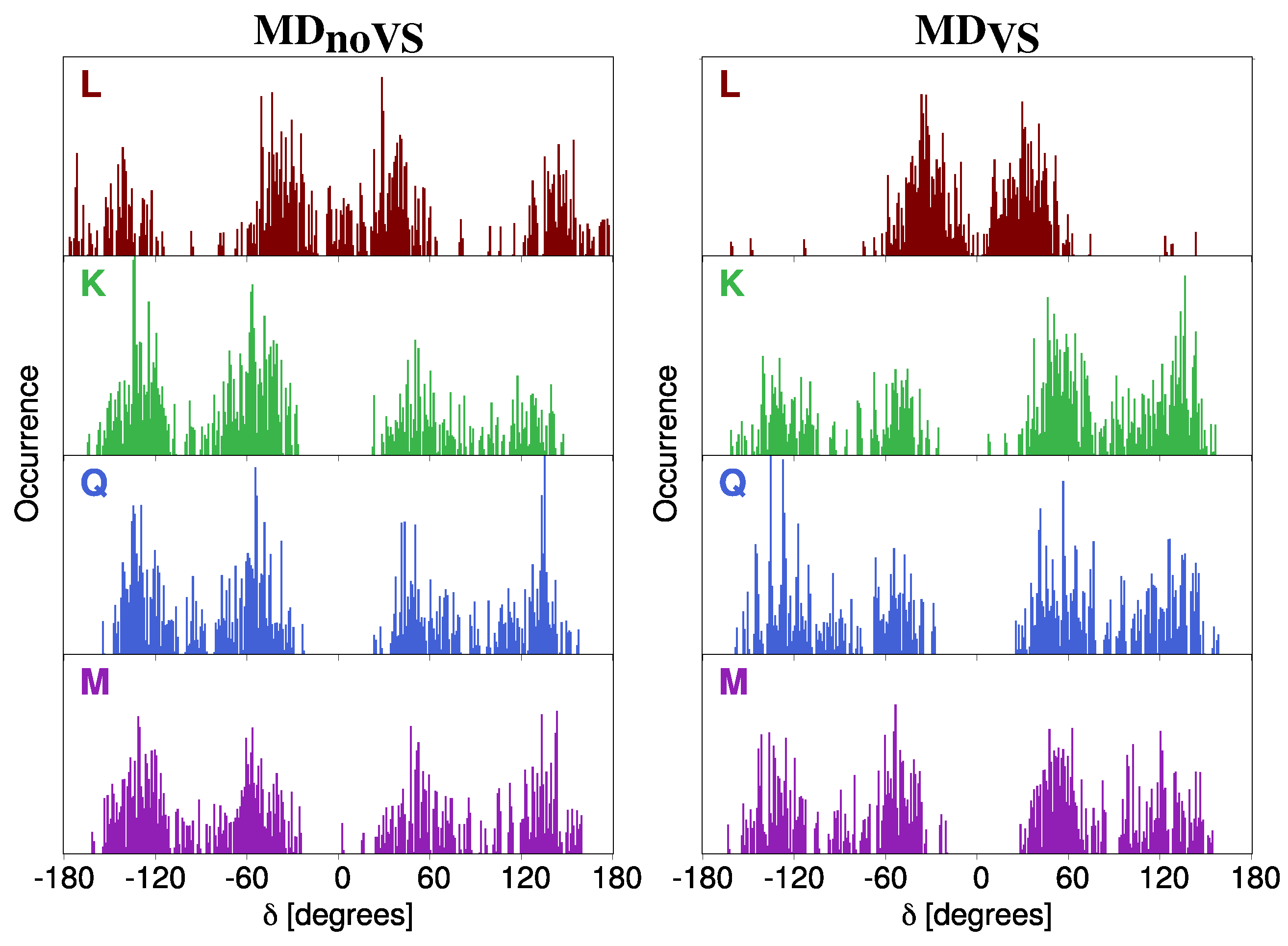
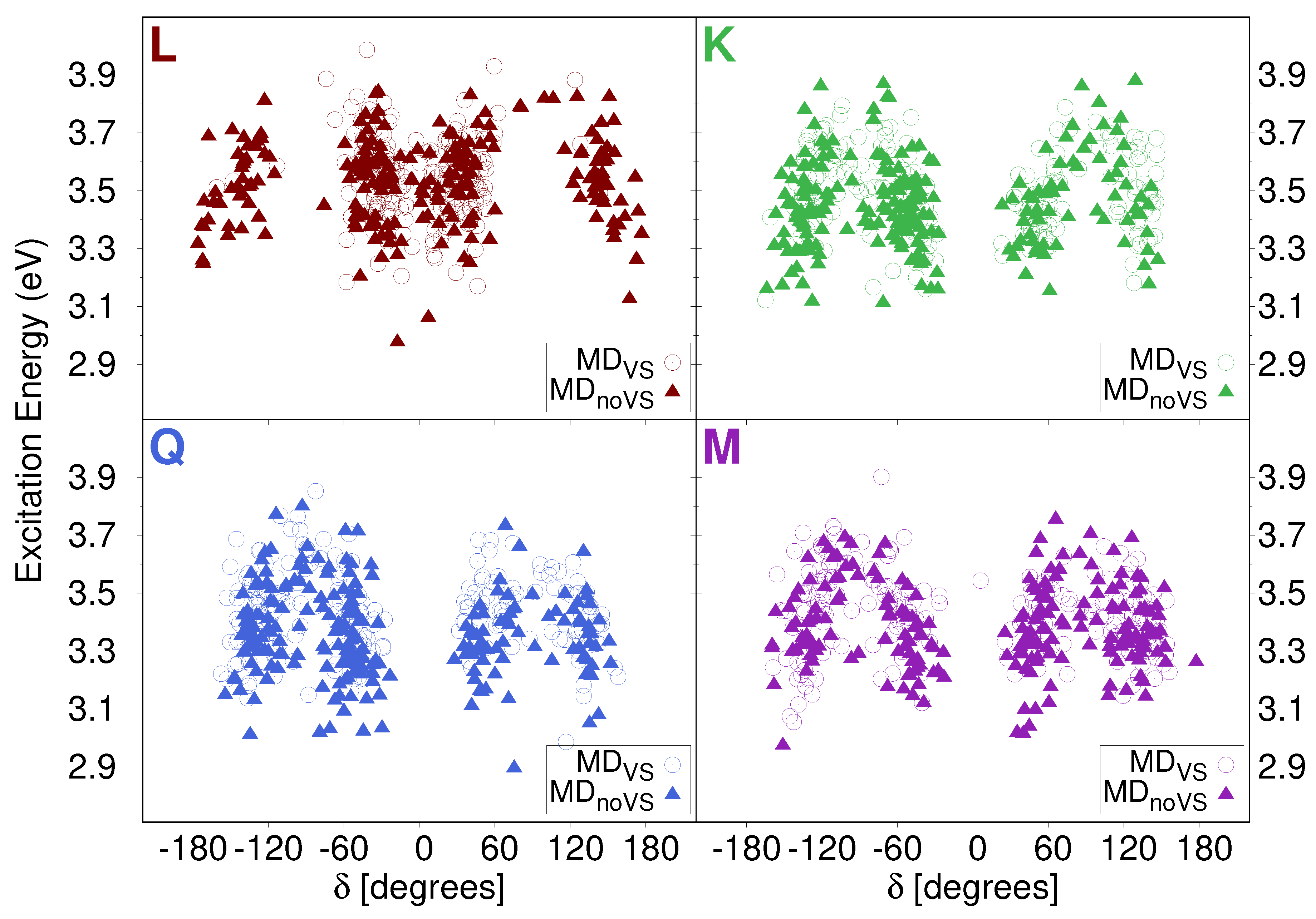
2.2.1. Conformational Analysis Based on MD Simulations
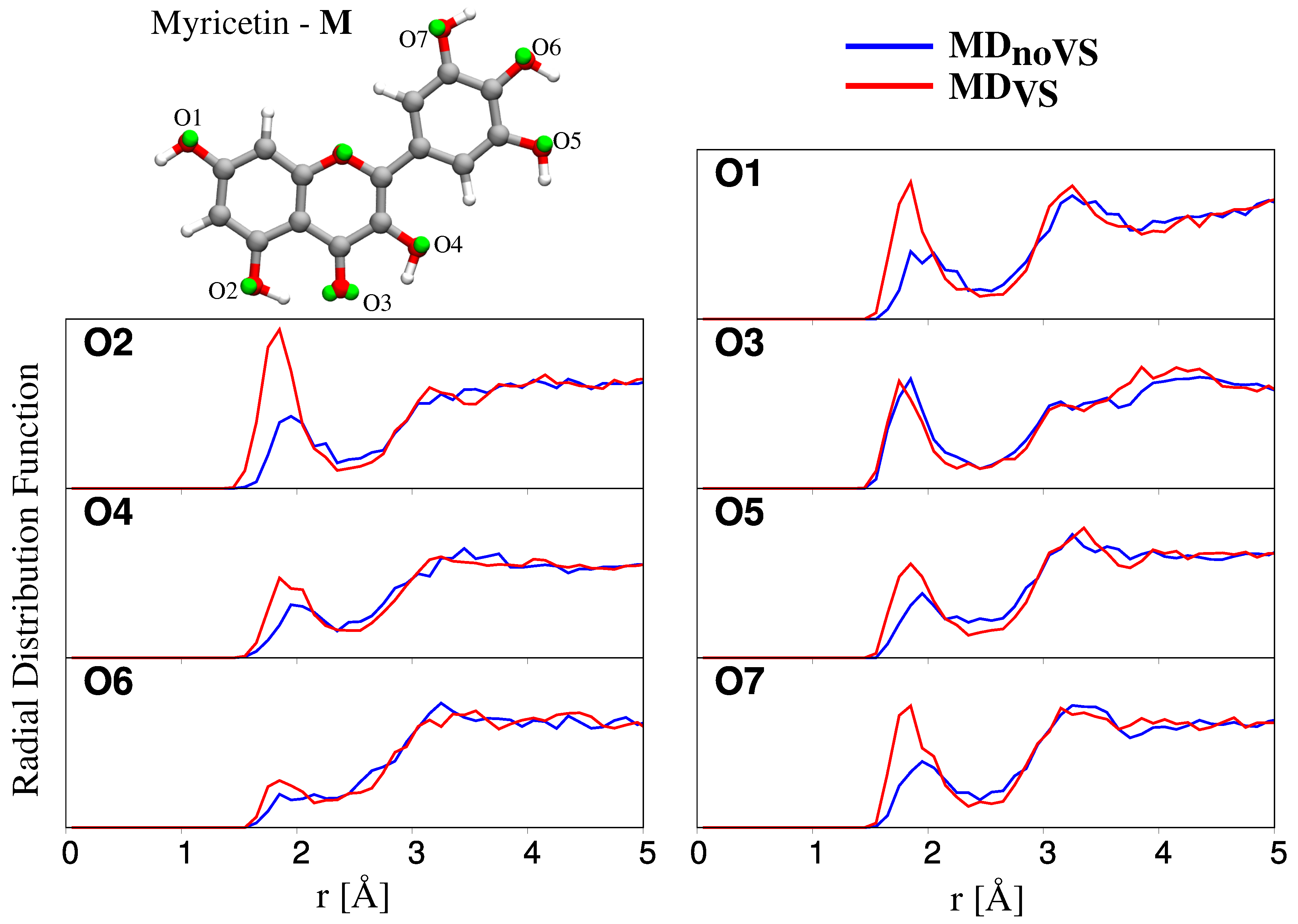
2.2.2. Hydration Pattern
2.3. Excitation Energies
3. Summary and Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Harborne, J.B.; Marby, H.; Marby, T. The Flavonoids; Springer: Boston, MA, USA, 1988. [Google Scholar]
- Kapešová, J.; Petrásková, L.; Markošová, K.; Rebroš, M.; Kotik, M.; Bojarová, P.; Křen, V. Bioproduction of quercetin and rutinose catalyzed by rutinosidase: Novel concept of “solid state biocatalysis”. Int. J. Mol. Sci. 2019, 20, 1112. [Google Scholar] [CrossRef]
- Burak, M.; Imen, Y. Flavonoids and their antioxidant properties. Turkiye Klin. Tip Bil. Derg. 1999, 19, 296–304. [Google Scholar]
- Casta eda Ovando, A.; de Lourdes Pacheco-Hernández, M.; Pez-Hern ndez, M.E.; Gal n Vidal, C.A.S. Chemical studies of anthocyanins: A review. Food Chem. 2009, 113, 859–871. [Google Scholar] [CrossRef]
- Lee, Y.K.; Yuk, D.Y.; Lee, J.W.; Lee, S.Y.; Ha, T.Y.; Oh, K.W.; Yun, Y.P.; Hong, J.T. (-)-Epigallocatechin-3-gallate prevents lipopolysaccharide-induced elevation of beta-amyloid generation and memory deficiency. Brain Res. 2009, 1250, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Panche, A.; Diwan, A.; Chandra, S. Flavonoids: An overview. J. Nutr. Sci. 2016, 5, e47. [Google Scholar] [CrossRef]
- Metodiewa, D.; Kochman, A.; Karolczak, S. Evidence for antiradical and antioxidant properties of four biologically active N, N-Diethylaminoethyl ethers of flavaone oximes: A comparison with natural polyphenolic flavonoid rutin action. IUBMB Life 1997, 41, 1067–1075. [Google Scholar] [CrossRef]
- Hayashi, T.; Sawa, K.; Kawasaki, M.; Arisawa, M.; Shimizu, M.; Morita, N. Inhibition of cow’s milk xanthine oxidase by flavonoids. J. Nat. Prod. 1988, 51, 345–348. [Google Scholar] [CrossRef]
- Ngwa, W.; Kumar, R.; Thompson, D.; Lyerly, W.; Moore, R.; Reid, T.E.; Lowe, H.; Toyang, N. Potential of Flavonoid-Inspired Phytomedicines against COVID-19. Molecules 2020, 25, 2707. [Google Scholar] [CrossRef] [PubMed]
- Das, P.; Majumder, R.; Mandal, M.; Basak, P. In-Silico approach for identification of effective and stable inhibitors for COVID-19 main protease (Mpro) from flavonoid based phytochemical constituents of Calendula officinalis. J. Biomol. Struct. Dyn. 2020, 1–16. [Google Scholar] [CrossRef]
- Solnier, J.; Fladerer, J.P. Flavonoids: A complementary approach to conventional therapy of COVID-19? Phytochem. Rev. 2020, 1–23. [Google Scholar] [CrossRef]
- Robak, J.; Gryglewski, R. Bioactivity of flavonoids. Pol. J. Pharmacol. 1996, 48, 555–564. [Google Scholar] [PubMed]
- Rice-Evans, C.; Miller, N. Antioxidant activities of flavonoids as bioactive components of food. Biochem. Soc. Trans. 1996, 24, 790–795. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.P.; Abd El Mohsen, M.M.; Rice-Evans, C. Cellular uptake and metabolism of flavonoids and their metabolites: Implications for their bioactivity. Arch. Biochem. Biophys. 2004, 423, 148–161. [Google Scholar] [CrossRef] [PubMed]
- Pinent, M.; Castell, A.; Baiges, I.; Montagut, G.; Arola, L.; Ardévol, A. Bioactivity of flavonoids on insulin-secreting cells. Compr. Rev. Food Sci. 2008, 7, 299–308. [Google Scholar] [CrossRef]
- Erlund, I. Review of the flavonoids quercetin, hesperetin, and naringenin. Dietary sources, bioactivities, bioavailability, and epidemiology. Nutr. Res. 2004, 24, 851–874. [Google Scholar] [CrossRef]
- Rohan, T.; Connell, M. The precursors of chocolate aroma: A study of the flavonoids and phenolic acids. J. Food Sci. 1964, 29, 460–463. [Google Scholar] [CrossRef]
- Samanta, A.; Das, G.; Das, S.K. Roles of flavonoids in plants. Carbon 2011, 100, 12–35. [Google Scholar]
- Sadighara, P.; Gharibi, S.; Jafari, A.M.; Khaniki, G.J.; Salari, S. The antioxidant and Flavonoids contents of Althaea officinalis L. flowers based on their color. Avicenna J. Phytomed. 2012, 2, 113. [Google Scholar]
- Yoshida, K.; Oyama, K.; Kondo, T. Chemistry of flavonoids in color development. Rec. Adv. Polyphen. Res. 2012, 3, 99–129. [Google Scholar]
- Kennedy, J.A.; Matthews, M.A.; Waterhouse, A.L. Effect of maturity and vine water status on grape skin and wine flavonoids. Am. J. Enol. Viticult. 2002, 53, 268–274. [Google Scholar]
- Waterhouse, A.L. Wine phenolics. Ann. N. Y. Acad. Sci. 2002, 957, 21–36. [Google Scholar] [CrossRef] [PubMed]
- Lairon, D.; Amiot, M.J. Flavonoids in food and natural antioxidants in wine. Curr. Opin. Lipidol. 1999, 10, 23–28. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, I.; Pérez-Gregorio, R.; Soares, S.; Mateus, N.; De Freitas, V. Wine flavonoids in health and disease prevention. Molecules 2017, 22, 292. [Google Scholar] [CrossRef] [PubMed]
- Miyagi, Y.; Miwa, K.; Inoue, H. Inhibition of human low-density lipoprotein oxidation by flavonoids in red wine and grape juice. Am. J. Cardiol. 1997, 80, 1627–1631. [Google Scholar] [CrossRef]
- Tapas, A.R.; Sakarkar, D.; Kakde, R. Flavonoids as nutraceuticals: A review. Trop. J. Pharm. Res. 2008, 7, 1089–1099. [Google Scholar] [CrossRef]
- Takahashi, A.; Ohnishi, T. The significance of the study about the biological effects of solar ultraviolet radiation using the Exposed Facility on the International Space Station. Biol. Sci. Space 2004, 18, 255–260. [Google Scholar] [CrossRef]
- Biler, M.; Biedermann, D.; Valentová, K.; Křen, V.; Kubala, M. Quercetin and its analogues: Optical and acido–basic properties. Phys. Chem. Chem. Phys. 2017, 19, 26870–26879. [Google Scholar] [CrossRef]
- Duan, Y. Ultraviolet-visible spectrum characterizations of quercetin in aqueous ethanol solution with different pH values. J. Chem. Pharm. Res. 2014, 6, 236–240. [Google Scholar]
- Dall’Acqua, S.; Miolo, G.; Innocenti, G.; Caffieri, S. The photodegradation of quercetin: Relation to oxidation. Molecules 2012, 17, 8898–8907. [Google Scholar] [CrossRef]
- Cerrato, A.; De Santis, D.; Moresi, M. Production of luteolin extracts from Reseda luteola and assessment of their dyeing properties. J. Sci. Food Agric. 2002, 82, 1189–1199. [Google Scholar] [CrossRef]
- M Calderon-Montano, J.; Burgos-Morón, E.; Pérez-Guerrero, C.; López-Lázaro, M. A review on the dietary flavonoid kaempferol. Mini-Rev. Med. Chem. 2011, 11, 298–344. [Google Scholar] [CrossRef] [PubMed]
- McDonald, M.S.; Hughes, M.; Burns, J.; Lean, M.E.; Matthews, D.; Crozier, A. Survey of the free and conjugated myricetin and quercetin content of red wines of different geographical origins. J. Agric. Food Chem. 1998, 46, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Fang, F.; Li, J.M.; Pan, Q.H.; Huang, W.D. Determination of red wine flavonoids by HPLC and effect of aging. Food Chem. 2007, 101, 428–433. [Google Scholar] [CrossRef]
- Giovannini, T.; Egidi, F.; Cappelli, C. Molecular spectroscopy of aqueous solutions: A theoretical perspective. Chem. Soc. Rev. 2020, 49, 5664–5677. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, T.; Egidi, F.; Cappelli, C. Theory and algorithms for chiroptical properties and spectroscopies of aqueous systems. Phys. Chem. Chem. Phys. 2020, 22, 22864–22879. [Google Scholar] [CrossRef] [PubMed]
- Loco, D.; Polack, É.; Caprasecca, S.; Lagardere, L.; Lipparini, F.; Piquemal, J.P.; Mennucci, B. A QM/MM approach using the AMOEBA polarizable embedding: From ground state energies to electronic excitations. J. Chem. Theory Comput. 2016, 12, 3654–3661. [Google Scholar] [CrossRef]
- Aidas, K.; Møgelhøj, A.; Nilsson, E.J.; Johnson, M.S.; Mikkelsen, K.V.; Christiansen, O.; Söderhjelm, P.; Kongsted, J. On the performance of quantum chemical methods to predict solvatochromic effects: The case of acrolein in aqueous solution. J. Chem. Phys. 2008, 128, 194503. [Google Scholar] [CrossRef]
- Tomasi, J.; Persico, M. Molecular interactions in solution: An overview of methods based on continuous distributions of the solvent. Chem. Rev. 1994, 94, 2027–2094. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Tomasi, J.; Cammi, R.; Mennucci, B.; Cappelli, C.; Corni, S. Molecular properties in solution described with a continuum solvation model. Phys. Chem. Chem. Phys. 2002, 4, 5697–5712. [Google Scholar] [CrossRef]
- Cappelli, C. Integrated QM/Polarizable MM/Continuum Approaches to Model Chiroptical Properties of Strongly Interacting Solute-Solvent Systems. Int. J. Quantum Chem. 2016, 116, 1532–1542. [Google Scholar] [CrossRef]
- Mennucci, B. Polarizable Continuum Model. WIREs Comput. Mol. Sci. 2012, 2, 386–404. [Google Scholar] [CrossRef]
- Giovannini, T.; Ambrosetti, M.; Cappelli, C. A polarizable embedding approach to second harmonic generation (SHG) of molecular systems in aqueous solutions. Theor. Chem. Acc. 2018, 137, 74. [Google Scholar] [CrossRef]
- Egidi, F.; Russo, R.; Carnimeo, I.; D’Urso, A.; Mancini, G.; Cappelli, C. The Electronic Circular Dichroism of Nicotine in Aqueous Solution: A Test Case for Continuum and Mixed Explicit-Continuum Solvation Approaches. J. Phys. Chem. A 2015, 119, 5396–5404. [Google Scholar] [CrossRef] [PubMed]
- Lipparini, F.; Egidi, F.; Cappelli, C.; Barone, V. The optical rotation of methyloxirane in aqueous solution: A never ending story? J. Chem. Theory Comput. 2013, 9, 1880–1884. [Google Scholar] [CrossRef] [PubMed]
- Cappelli, C.; Mennucci, B.; Monti, S. Environmental effects on the spectroscopic properties of gallic acid: A combined classical and quantum mechanical study. J. Phys. Chem. A 2005, 109, 1933–1943. [Google Scholar] [CrossRef]
- Warshel, A.; Karplus, M. Calculation of ground and excited state potential surfaces of conjugated molecules. I. Formulation and parametrization. J. Am. Chem. Soc. 1972, 94, 5612–5625. [Google Scholar] [CrossRef]
- Warshel, A.; Levitt, M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J. Mol. Biol. 1976, 103, 227–249. [Google Scholar] [CrossRef]
- Senn, H.M.; Thiel, W. QM/MM methods for biomolecular systems. Angew. Chem. Int. Ed. 2009, 48, 1198–1229. [Google Scholar] [CrossRef]
- Lin, H.; Truhlar, D.G. QM/MM: What have we learned, where are we, and where do we go from here? Theor. Chem. Acc. 2007, 117, 185–199. [Google Scholar] [CrossRef]
- Curutchet, C.; Muñoz-Losa, A.; Monti, S.; Kongsted, J.; Scholes, G.D.; Mennucci, B. Electronic energy transfer in condensed phase studied by a polarizable QM/MM model. J. Chem. Theory Comput. 2009, 5, 1838–1848. [Google Scholar] [CrossRef] [PubMed]
- Lipparini, F.; Cappelli, C.; Scalmani, G.; De Mitri, N.; Barone, V. Analytical first and second derivatives for a fully polarizable QM/classical hamiltonian. J. Chem. Theory Comput. 2012, 8, 4270–4278. [Google Scholar] [CrossRef]
- Lipparini, F.; Cappelli, C.; Barone, V. Linear response theory and electronic transition energies for a fully polarizable QM/classical hamiltonian. J. Chem. Theory Comput. 2012, 8, 4153–4165. [Google Scholar] [CrossRef]
- Olsen, J.M.H.; Kongsted, J. Molecular properties through polarizable embedding. Adv. Quantum Chem. 2011, 61, 107–143. [Google Scholar]
- Olsen, J.M.H.; Steinmann, C.; Ruud, K.; Kongsted, J. Polarizable density embedding: A new QM/QM/MM-based computational strategy. J. Phys. Chem. A 2015, 119, 5344–5355. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, E.; Thiel, W. Solvent boundary potentials for hybrid QM/MM computations using classical drude oscillators: A fully polarizable model. J. Chem. Theory Comput. 2012, 8, 4527–4538. [Google Scholar] [CrossRef]
- Giovannini, T.; Puglisi, A.; Ambrosetti, M.; Cappelli, C. Polarizable QM/MM approach with fluctuating charges and fluctuating dipoles: The QM/FQFμ model. J. Chem. Theory Comput. 2019, 15, 2233–2245. [Google Scholar] [CrossRef]
- Rick, S.W.; Stuart, S.J.; Berne, B.J. Dynamical fluctuating charge force fields: Application to liquid water. J. Chem. Phys. 1994, 101, 6141–6156. [Google Scholar] [CrossRef]
- Rick, S.W.; Stuart, S.J.; Bader, J.S.; Berne, B. Fluctuating charge force fields for aqueous solutions. J. Mol. Liq. 1995, 65, 31–40. [Google Scholar] [CrossRef]
- Rick, S.W.; Berne, B.J. Dynamical Fluctuating Charge Force Fields: The Aqueous Solvation of Amides. J. Am. Chem. Soc. 1996, 118, 672–679. [Google Scholar] [CrossRef]
- Giovannini, T.; Olszowka, M.; Cappelli, C. Effective Fully Polarizable QM/MM Approach To Model Vibrational Circular Dichroism Spectra of Systems in Aqueous Solution. J. Chem. Theory Comput. 2016, 12, 5483–5492. [Google Scholar] [CrossRef]
- Egidi, F.; Giovannini, T.; Del Frate, G.; Lemler, P.M.; Vaccaro, P.H.; Cappelli, C. A combined experimental and theoretical study of optical rotatory dispersion for (R)-glycidyl methyl ether in aqueous solution. Phys. Chem. Chem. Phys. 2019, 21, 3644–3655. [Google Scholar] [CrossRef] [PubMed]
- Giovannini, T.; Macchiagodena, M.; Ambrosetti, M.; Puglisi, A.; Lafiosca, P.; Lo Gerfo, G.; Egidi, F.; Cappelli, C. Simulating vertical excitation energies of solvated dyes: From continuum to polarizable discrete modeling. Int. J. Quantum Chem. 2019, 119, e25684. [Google Scholar] [CrossRef]
- Macchiagodena, M.; Mancini, G.; Pagliai, M.; Barone, V. Accurate prediction of bulk properties in hydrogen bonded liquids: Amides as case studies. Phys. Chem. Chem. Phys. 2016, 18, 25342–25354. [Google Scholar] [CrossRef] [PubMed]
- Macchiagodena, M.; Mancini, G.; Pagliai, M.; Cardini, G.; Barone, V. New atomistic model of pyrrole with improved liquid state properties and structure. Int. J. Quantum Chem. 2017. [Google Scholar] [CrossRef]
- Naseem, B.; Sabri, A.; Hasan, A.; Shah, S.S. Interaction of flavonoids within organized molecular assemblies of anionic surfactant. Colloids Surfaces B Biointerfaces 2004, 35, 7–13. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Revision A.03; Gaussian Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Abrahama, M.J.; Murtola, T.; Schulz, R.; Pálla, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Mark, P.; Nilsson, L. Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K. J. Phys. Chem. A 2001, 105, 9954–9960. [Google Scholar] [CrossRef]
- Bayly, C.I.; Cieplak, P.; Cornell, W.; Kollman, P.A. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 1993, 97, 10269–10280. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Boys, S. Quantum Theory of Atoms, Molecules, and the Solid State; Academic Press: New York, NY, USA, 1966; p. 253. [Google Scholar]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [PubMed]
- Brehm, M.; Kirchner, B. TRAVIS-A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef] [PubMed]
- Puglisi, A.; Giovannini, T.; Antonov, L.; Cappelli, C. Interplay between conformational and solvent effects in UV-visible absorption spectra: Curcumin tautomers as a case study. Phys. Chem. Chem. Phys. 2019, 21, 15504–15514. [Google Scholar] [CrossRef] [PubMed]
- Gómez, S.; Giovannini, T.; Cappelli, C. Absorption spectra of xanthines in aqueous solution: A computational study. Phys. Chem. Chem. Phys. 2020, 22, 5929–5941. [Google Scholar] [CrossRef] [PubMed]
- Di Remigio, R.; Giovannini, T.; Ambrosetti, M.; Cappelli, C.; Frediani, L. Fully polarizable QM/fluctuating charge approach to two-photon absorption of aqueous solutions. J. Chem. Theory Comput. 2019, 15, 4056–4068. [Google Scholar] [CrossRef] [PubMed]
- Reichardt, C. Solvatochromism, thermochromism, piezochromism, halochromism, and chiro-solvatochromism of pyridinium N-phenoxide betaine dyes. Chem. Soc. Rev. 1992, 21, 147–153. [Google Scholar] [CrossRef]
- Reichardt, C. Solvatochromic dyes as solvent polarity indicators. Chem. Rev. 1994, 94, 2319–2358. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G.; Guido, C.A.; Mennucci, B.; Scalmani, G.; Frisch, M.J. Practical computation of electronic excitation in solution: Vertical excitation model. Chem. Sci. 2011, 2, 2143–2161. [Google Scholar] [CrossRef]
- Duchemin, I.; Guido, C.A.; Jacquemin, D.; Blase, X. The Bethe–Salpeter formalism with polarisable continuum embedding: Reconciling linear-response and state-specific features. Chem. Sci. 2018, 9, 4430–4443. [Google Scholar] [CrossRef]
- Biczysko, M.; Bloino, J.; Brancato, G.; Cacelli, I.; Cappelli, C.; Ferretti, A.; Lami, A.; Monti, S.; Pedone, A.; Prampolini, G.; et al. Integrated computational approaches for spectroscopic studies of molecular systems in the gas phase and in solution: Pyrimidine as a test case. Theor. Chem. Acc. 2012, 131, 1201. [Google Scholar] [CrossRef]
- Hodecker, M.; Biczysko, M.; Dreuw, A.; Barone, V. Simulation of Vacuum UV Absorption and Electronic Circular Dichroism Spectra of Methyl Oxirane: The Role of Vibrational Effects. J. Chem. Theory Comput. 2016, 12, 2820–2833. [Google Scholar] [CrossRef] [PubMed]
- Improta, R.; Santoro, F.; Barone, V.; Lami, A. Vibronic model for the quantum dynamical study of the competition between bright and charge-transfer excited states in single-strand polynucleotides: The adenine dimer case. J. Phys. Chem. A 2009, 113, 15346–15354. [Google Scholar] [CrossRef] [PubMed]
- Lamoureux, G.; Harder, E.; Vorobyov, I.V.; Roux, B.; MacKerell, A.D., Jr. A polarizable model of water for molecular dynamics simulations of biomolecules. Chem. Phys. Lett. 2006, 418, 245–249. [Google Scholar] [CrossRef]
- Wu, J.C.; Piquemal, J.P.; Chaudret, R.; Reinhardt, P.; Ren, P. Polarizable molecular dynamics simulation of Zn (II) in water using the AMOEBA force field. J. Chem. Theory Comput. 2010, 6, 2059–2070. [Google Scholar] [CrossRef]
- Giovannini, T.; Riso, R.R.; Ambrosetti, M.; Puglisi, A.; Cappelli, C. Electronic transitions for a fully polarizable qm/mm approach based on fluctuating charges and fluctuating dipoles: Linear and corrected linear response regimes. J. Chem. Phys. 2019, 151, 174104. [Google Scholar] [CrossRef]
- Giovannini, T.; Grazioli, L.; Ambrosetti, M.; Cappelli, C. Calculation of ir spectra with a fully polarizable qm/mm approach based on fluctuating charges and fluctuating dipoles. J. Chem. Theory Comput. 2019, 15, 5495–5507. [Google Scholar] [CrossRef]
- Giovannini, T.; Lafiosca, P.; Cappelli, C. A General Route to Include Pauli Repulsion and Quantum Dispersion Effects in QM/MM Approaches. J. Chem. Theory Comput. 2017, 13, 4854–4870. [Google Scholar] [CrossRef]
- Giovannini, T.; Ambrosetti, M.; Cappelli, C. Quantum Confinement Effects on Solvatochromic Shifts of Molecular Solutes. J. Phys. Chem. Lett. 2019, 10, 5823–5829. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Site | L | K | Q | M |
|---|---|---|---|---|
| O1 | 1.1 (1.5) | 1.4 (1.6) | 1.0 (1.6) | 1.2 (1.5) |
| O2 | 1.3 (1.9) | 1.6 (1.7) | 1.2 (1.8) | 1.0 (1.6) |
| O3 | 1.8 (1.6) | 1.4 (1.1) | 1.4 (1.2) | 1.3 (1.2) |
| O4 | – (–) | 1.1 (1.3) | 1.1 (1.4) | 0.8 (1.1) |
| O5 | 1.0 (1.4) | – (–) | – (–) | 0.9 (1.2) |
| O6 | 0.9 (1.3) | 1.3 (1.7) | 0.9 (1.0) | 0.2 (0.5) |
| O7 | – (–) | – (–) | 1.0 (1.4) | 1.1 (1.3) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Skoko, S.; Ambrosetti, M.; Giovannini, T.; Cappelli, C. Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study. Molecules 2020, 25, 5853. https://doi.org/10.3390/molecules25245853
Skoko S, Ambrosetti M, Giovannini T, Cappelli C. Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study. Molecules. 2020; 25(24):5853. https://doi.org/10.3390/molecules25245853
Chicago/Turabian StyleSkoko, Sulejman, Matteo Ambrosetti, Tommaso Giovannini, and Chiara Cappelli. 2020. "Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study" Molecules 25, no. 24: 5853. https://doi.org/10.3390/molecules25245853
APA StyleSkoko, S., Ambrosetti, M., Giovannini, T., & Cappelli, C. (2020). Simulating Absorption Spectra of Flavonoids in Aqueous Solution: A Polarizable QM/MM Study. Molecules, 25(24), 5853. https://doi.org/10.3390/molecules25245853