
ab-Initio Study of Hydrogen Bond Networks in 1,2,3-Triazole Phases


Abstract
1. Introduction
2. Results
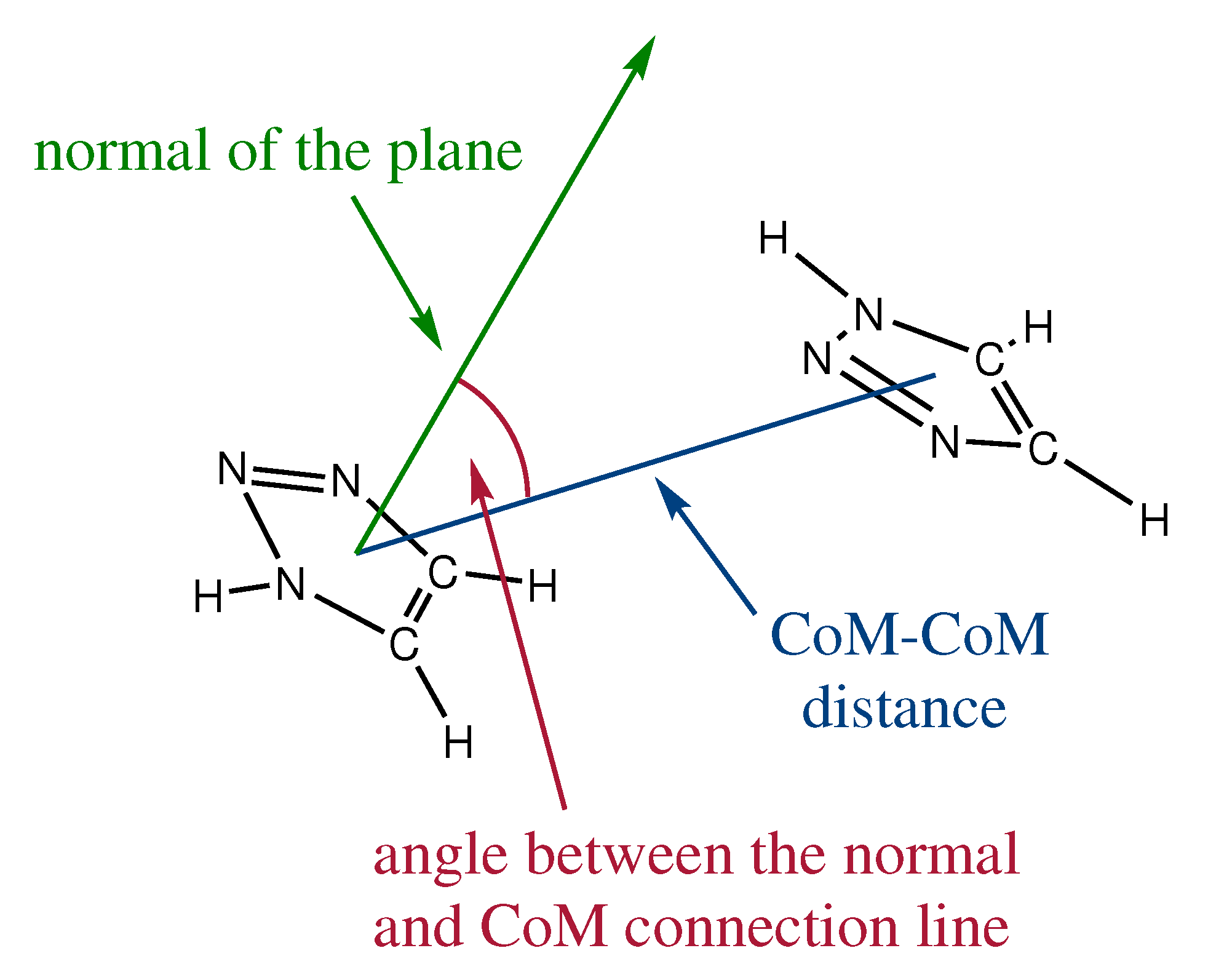
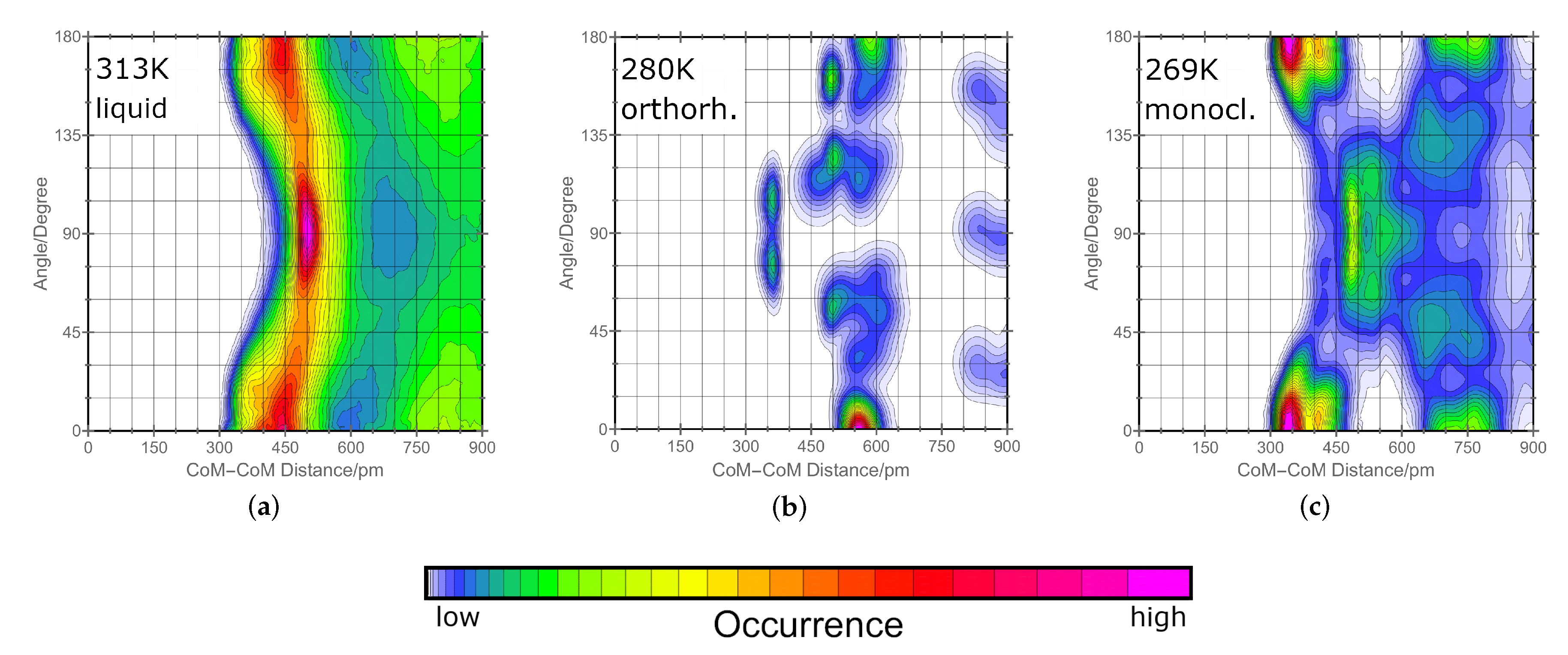
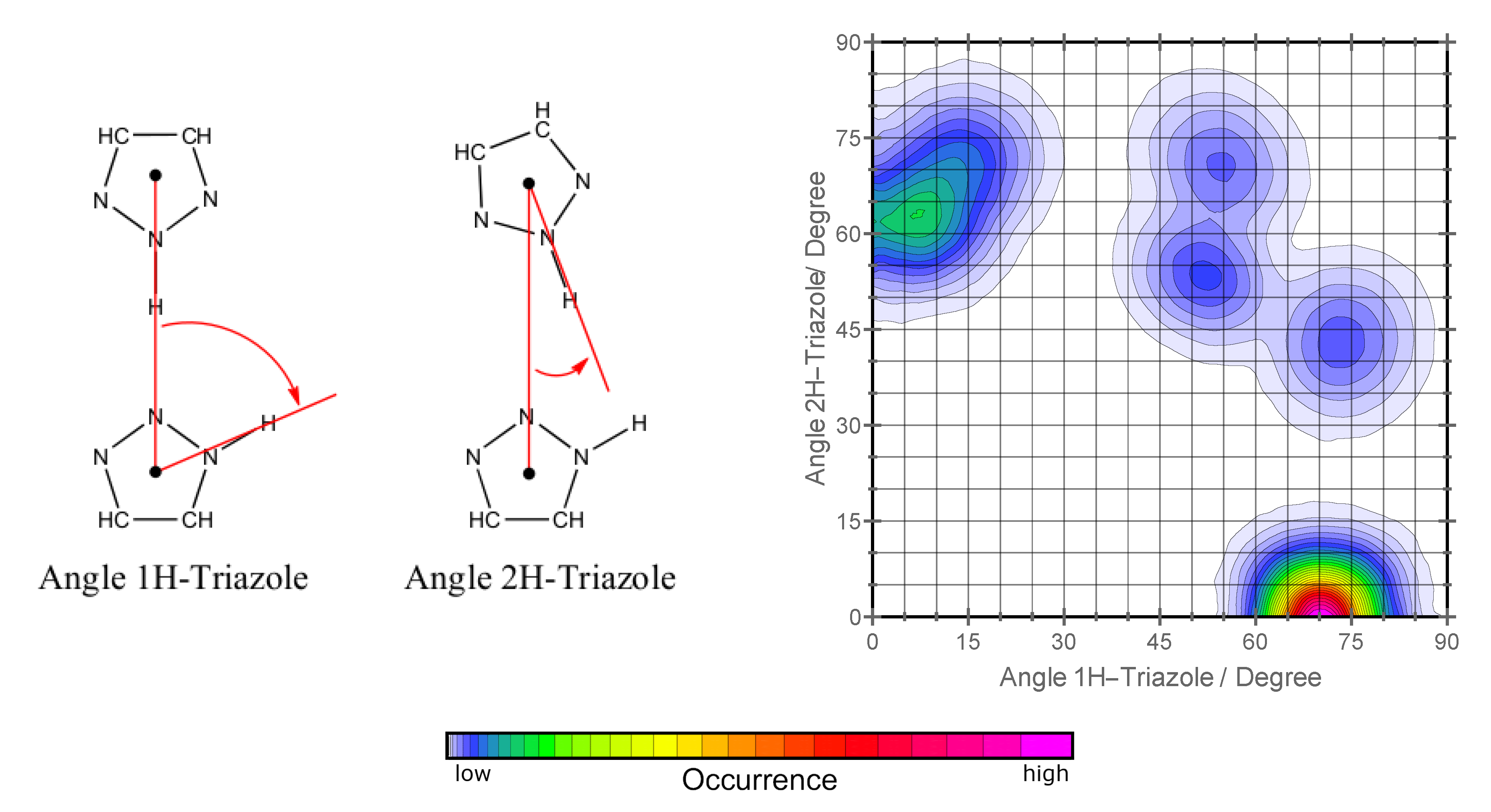
2.1. Orientation in Liquid and Crystalline Phases
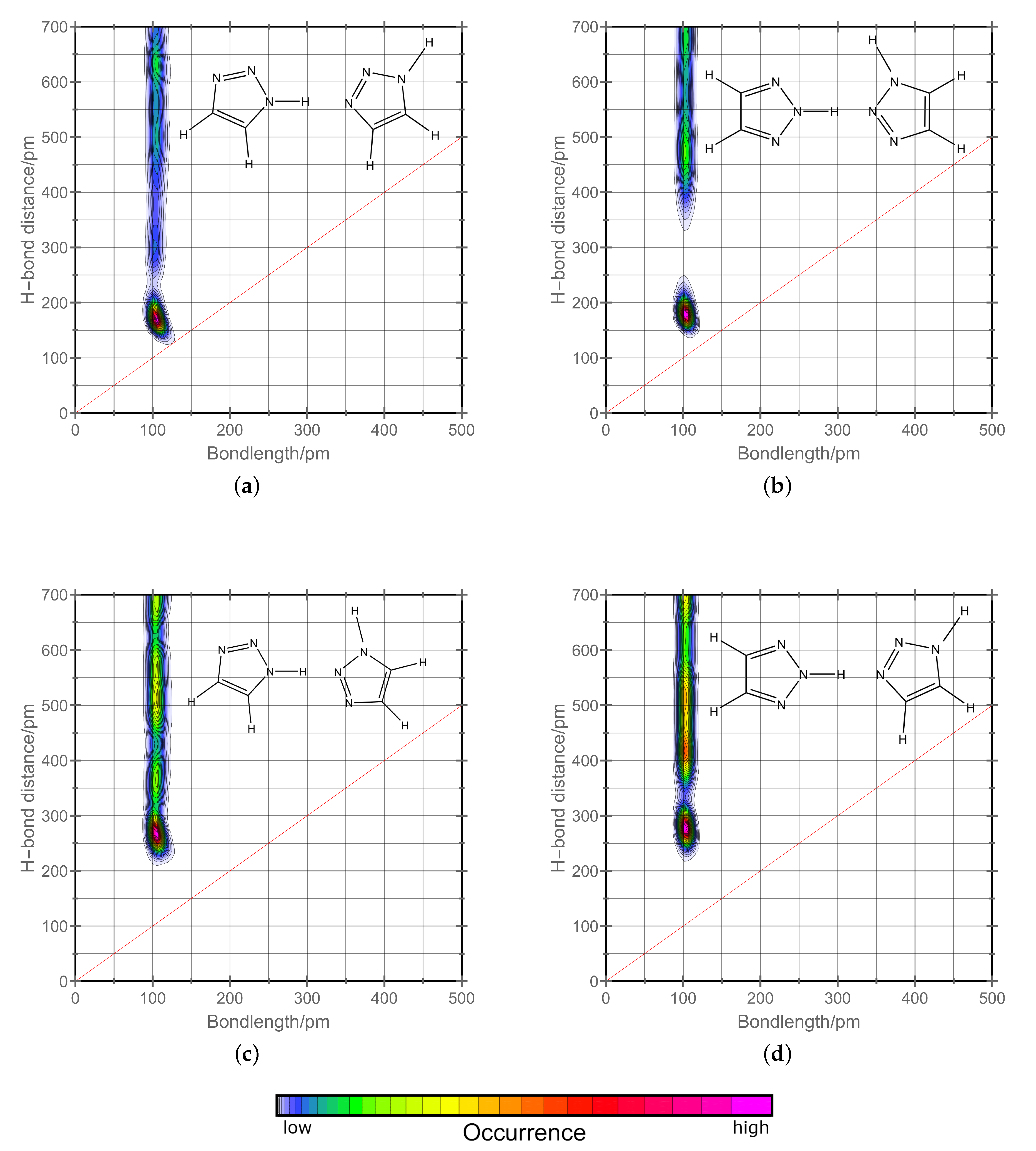
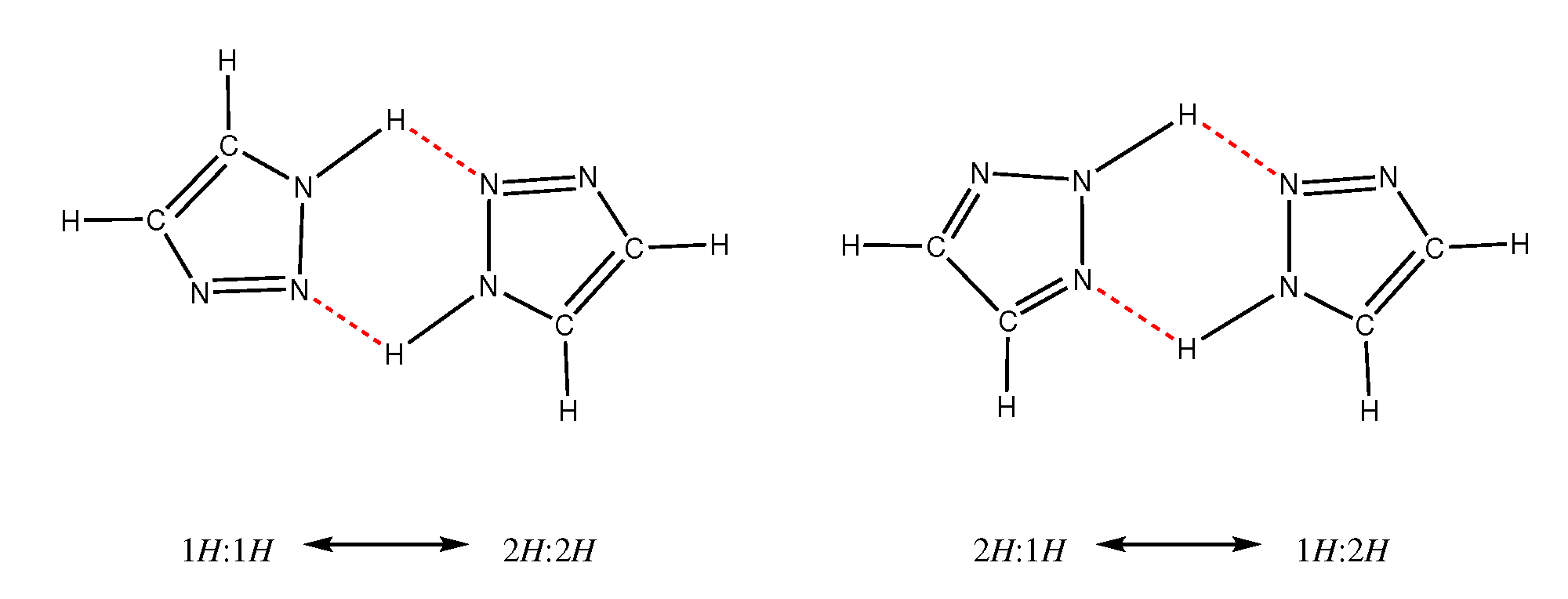
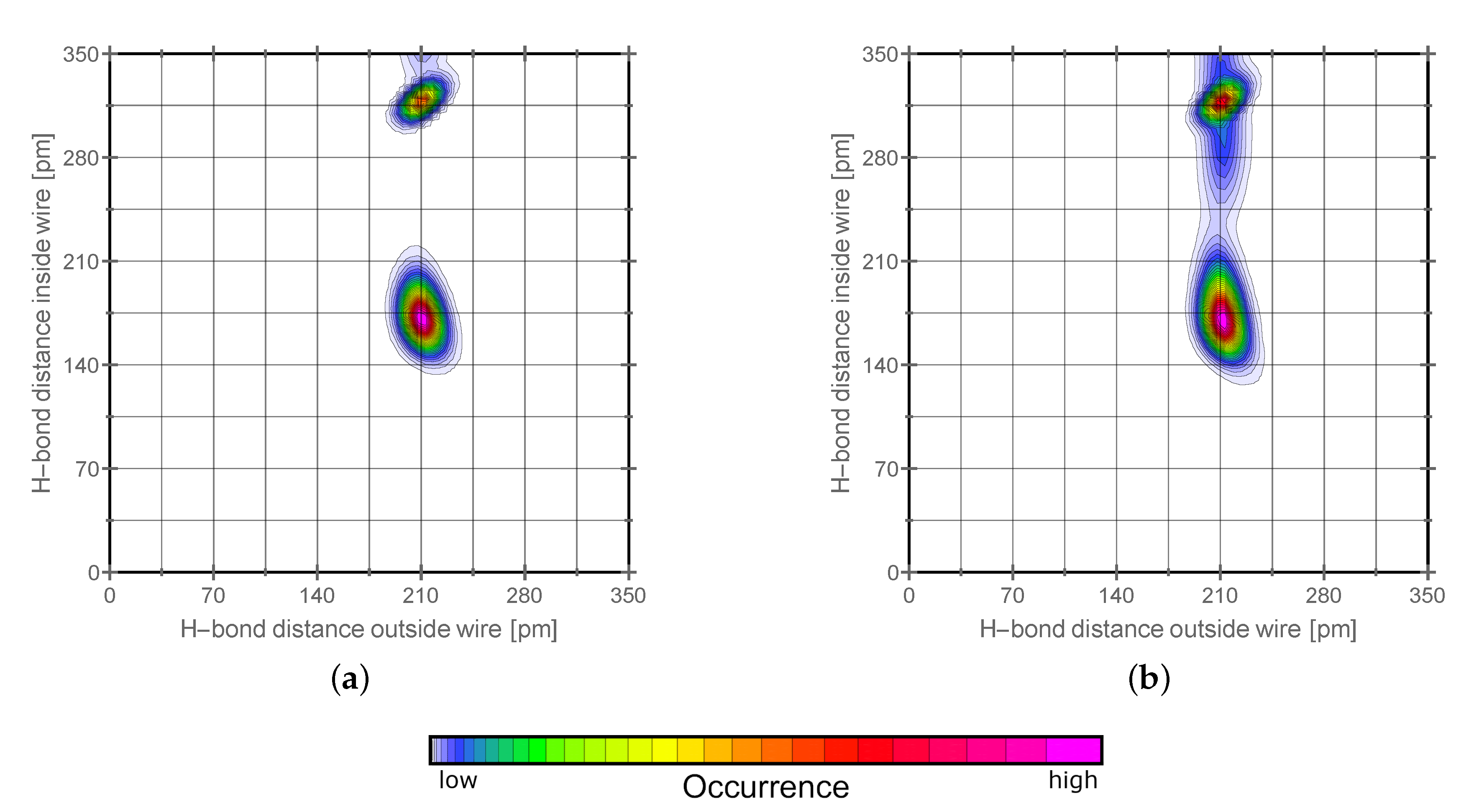
2.2. H-Network and Role of Tautomers
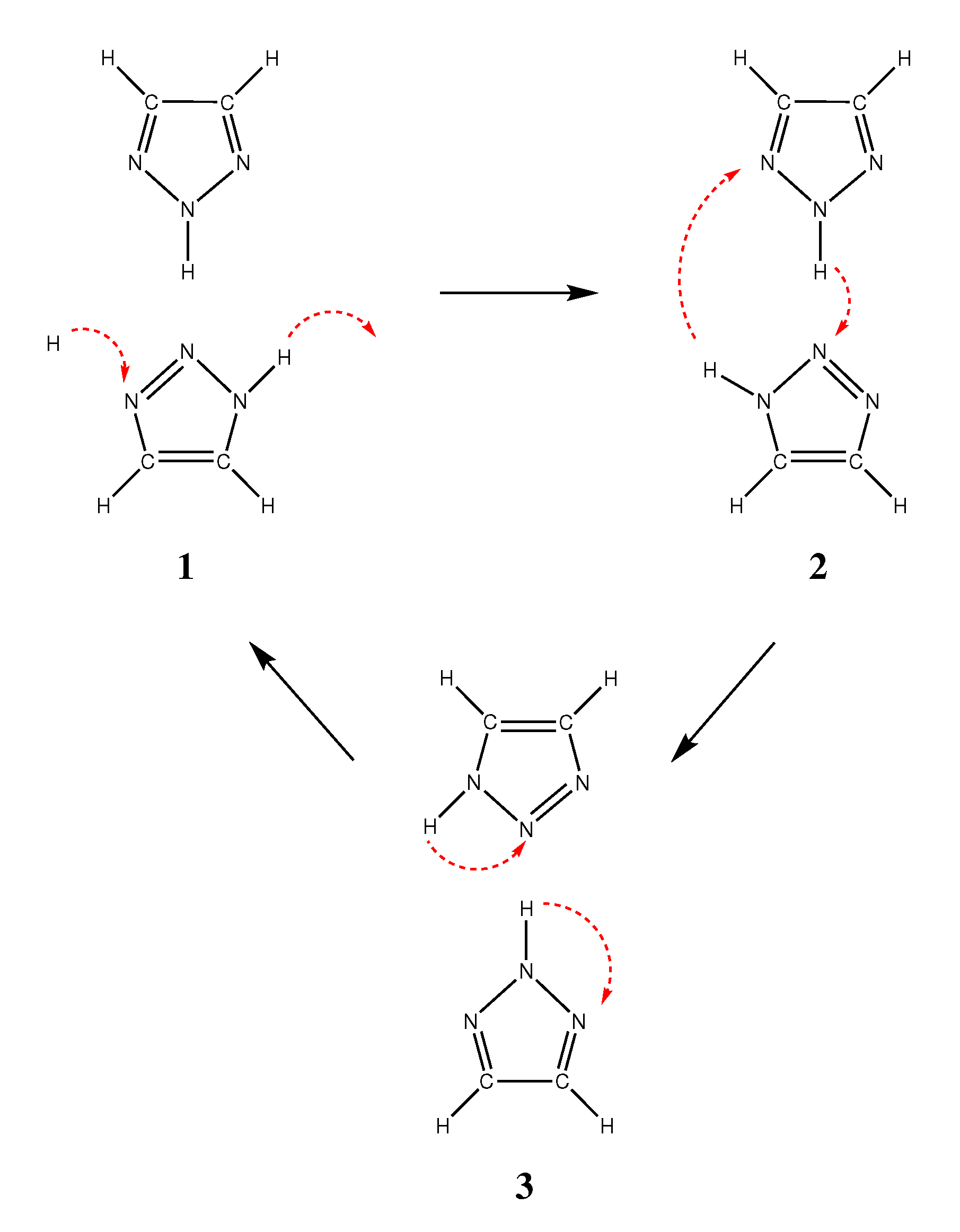
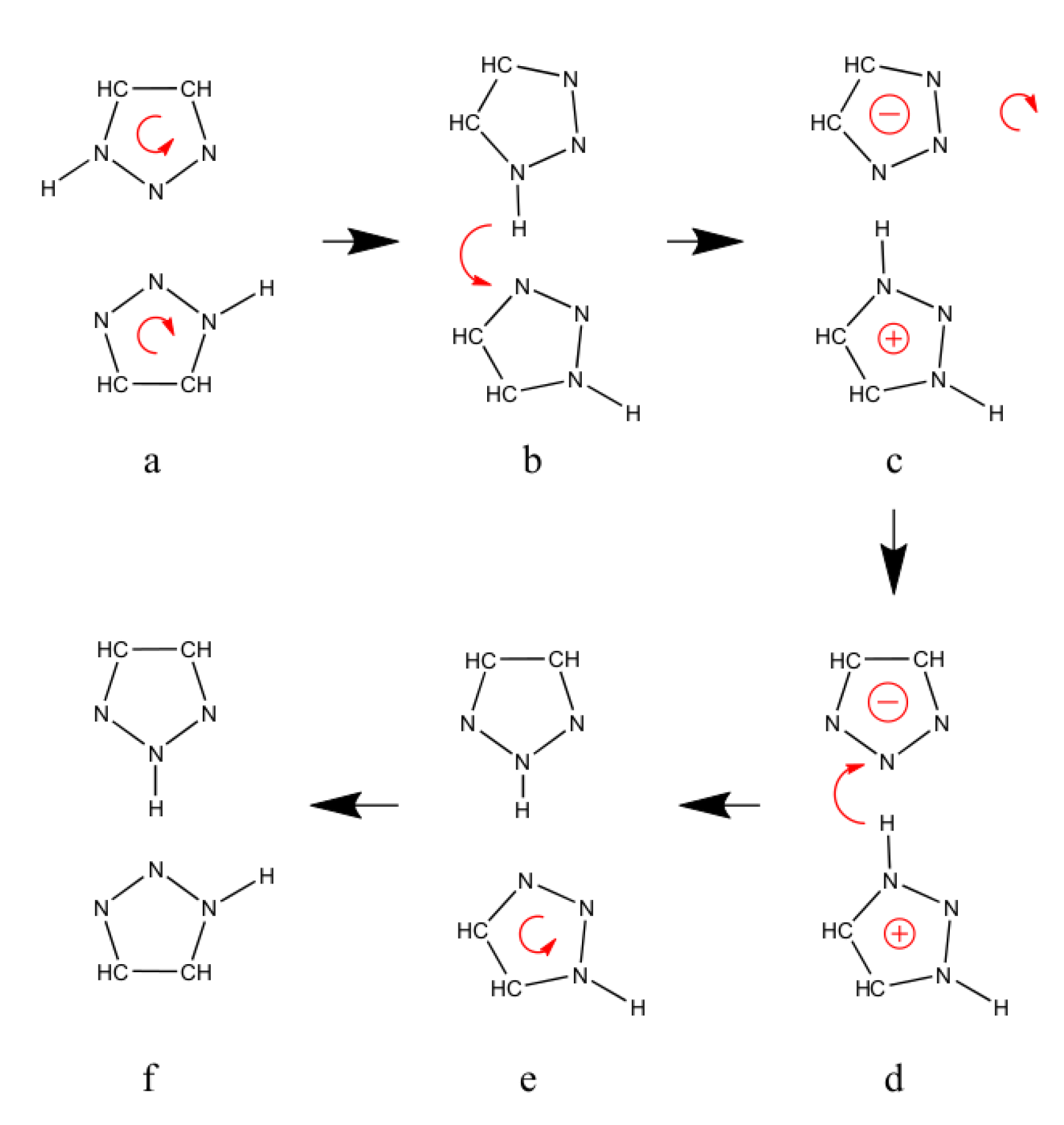
2.3. Reorientation Mechanism
2.4. Diffusion Coefficients
3. Discussion
4. Materials and Methods
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mehta, V.; Cooper, J.S. Review and analysis of PEM fuel cell design and manufacturing. J. Power Sources 2003, 114, 32–53. [Google Scholar] [CrossRef]
- Kreuer, K.D.; Paddison, S.J.; Spohr, E.; Schuster, M. Transport in proton conductors for fuel-cell applications: Simulations, elementary reactions, and phenomenology. Chem. Rev. 2004, 104, 4637–4678. [Google Scholar] [CrossRef] [PubMed]
- Smitha, B.; Sridhar, S.; Khan, A. Solid polymer electrolyte membranes for fuel cell applications—A review. J. Membr. Sci. 2005, 259, 10–26. [Google Scholar] [CrossRef]
- Liu, H.; Song, C.; Zhang, L.; Zhang, J.; Wang, H.; Wilkinson, D.P. A review of anode catalysis in the direct methanol fuel cell. J. Power Sources 2006, 155, 95–110. [Google Scholar] [CrossRef]
- Kirubakaran, A.; Jain, S.; Nema, R. A review on fuel cell technologies and power electronic interface. Renew. Sust. Energ. Rev. 2009, 13, 2430–2440. [Google Scholar] [CrossRef]
- Peighambardoust, S.; Rowshanzamir, S.; Amjadi, M. Review of the proton exchange membranes for fuel cell applications. Int. J. Hydrogen Energ. 2010, 35, 9349–9384. [Google Scholar] [CrossRef]
- Paddison, S.J.; Kreuer, K.D.; Maier, J. About the choice of the protogenic group in polymer electrolyte membranes: Ab initio modelling of sulfonic acid, phosphonic acid, and imidazole functionalized alkanes. Phys. Chem. Chem. Phys. 2006, 8, 4530–4542. [Google Scholar] [CrossRef]
- Arslan, A.; Kıralp, S.; Toppare, L.; Bozkurt, A. Novel Conducting Polymer Electrolyte Biosensor Based on Poly(1-vinyl imidazole) and Poly(acrylic acid) Networks. Langmuir 2006, 22, 2912–2915. [Google Scholar] [CrossRef]
- Schechter, A.; Savinell, R.F. Imidazole and 1-methyl imidazole in phosphoric acid doped polybenzimidazole, electrolyte for fuel cells. Solid State Ionics 2002, 147, 181–187. [Google Scholar] [CrossRef]
- Suzuki, K.; Yamaguchi, M.; Hotta, S.; Tanabe, N.; Yanagida, S. A new alkyl-imidazole polymer prepared as an inonic polymer electrolyte by in situ polymerization of dye sensitized solar cells. J. Photoch. Photobio. A 2004, 164, 81–85. [Google Scholar] [CrossRef]
- Wang, J.; Yue, X.; Zhang, Z.; Yang, Z.; Li, Y.; Zhang, H.; Yang, X.; Wu, H.; Jiang, Z. Enhancement of Proton Conduction at Low Humidity by Incorporating Imidazole Microcapsules into Polymer Electrolyte Membranes. Adv. Funct. Mater. 2012, 22, 4539–4546. [Google Scholar] [CrossRef]
- Nagamani, C.; Versek, C.; Thorn, M.; Tuominen, M.T.; Thayumanavan, S. Proton Conduction in 1H-1,2,3-triazole Polymers: Imidazole-Like or Pyrazole-Like? J. Polym. Sci. Pol. Chem. 2010, 48, 1851–1858. [Google Scholar] [CrossRef]
- Nguyen, M.D.T.; Dang, H.S.; Kim, D. Proton exchange membranes based on sulfonated poly(arylene ether ketone) containing triazole group for enhanced proton conductivity. J. Membr. Sci. 2015, 496, 13–20. [Google Scholar] [CrossRef]
- Subbaraman, R.; Ghassemi, H.; Zawodzinski, T. Triazole and triazole derivatives as proton transport facilitators in polymer electrolyte membrane fuel cells. Solid State Ionics 2009, 180, 1143–1150. [Google Scholar] [CrossRef]
- Günday, S.T.; Bozkurt, A.; Meyer, W.H.; Wegner, G. Effects of different acid functional groups on proton conductivity of polymer-1,2,4-triazole blends. J. Polym. Sci. Pol. Phys. 2006, 44, 3315–3322. [Google Scholar] [CrossRef]
- Li, H.Q.; Liu, X.J.; Xu, J.; Xu, D.; Ni, H.; Wang, S.; Wang, Z. Enhanced proton conductivity of sulfonated poly(arylene ether ketone sulfone) for fuel cells by grafting triazole groups onto polymer chains. J. Membr. Sci. 2016, 509, 173–181. [Google Scholar] [CrossRef]
- Ahn, M.K.; Lee, S.B.; Min, C.M.; Yu, Y.G.; Jang, J.; Gim, M.Y.; Lee, J.S. Enhanced proton conductivity at low humidity of proton exchange membranes with triazole moieties in the side chains. J. Membr. Sci. 2017, 523, 480–486. [Google Scholar] [CrossRef]
- Li, S.; Zhou, Z.; Zhang, Y.; Liu, M.; Li, W. 1H-1,2,4-Triazole: An Effective Solvent for Proton-Conducting Electrolytes. Chem. Mater. 2005, 17, 5884–5886. [Google Scholar] [CrossRef]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click Chemistry: Diverse Chemical Function from a Few Good Reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Schulze, B.; Schubert, U.S. Beyond click chemistry–supramolecular interactions of 1,2,3-triazoles. Chem. Soc. Rev. 2014, 43, 2522–2571. [Google Scholar] [CrossRef]
- Moses, J.E.; Moorhouse, A.D. The growing applications of click chemistry. Chem. Soc. Rev. 2007, 36, 1249–1262. [Google Scholar] [CrossRef] [PubMed]
- Agalave, S.G.; Maujan, S.R.; Pore, V.S. Click Chemistry: 1,2,3-Triazoles as Pharmacophores. Chem. Asian J. 2011, 6, 2696–2718. [Google Scholar] [CrossRef] [PubMed]
- Brüssel, M.; Zahn, S.; Hey-Hawkins, E.; Kirchner, B. Toward the calculations of bioinorganic systems from ab initio molecular dynamics simulations and static quantum chemistry. Adv. Inorg. Chem. 2010, 62, 111. [Google Scholar]
- Brehm, M.; Weber, H.; Pensado, A.S.; Stark, A.; Kirchner, B. Proton transfer and polarity changes in ionic liquid–water mixtures: A perspective on hydrogen bonds from ab initio molecular dynamics at the example of 1-ethyl-3-methylimidazolium acetate–water mixtures—Part 1. Phys. Chem. Chem. Phys. 2012, 14, 5030. [Google Scholar] [CrossRef]
- Weber, H.; Hollóczki, O.; Pensado, A.S.; Kirchner, B. Side chain fluorination and anion effect on the structure of 1-butyl-3-methylimidazolium ionic liquids. J. Chem. Phys. 2013, 139, 084502. [Google Scholar] [CrossRef]
- Wang, S.; Li, Z.; Zhang, Y.; Liu, X.; Han, J.; Li, X.; Liu, Z.; Liu, S.; Choy, W.C.H. Water-Soluble Triazolium Ionic-Liquid-Induced Surface Self-Assembly to Enhance the Stability and Efficiency of Perovskite Solar Cells. Adv. Funct. Mater. 2019, 29, 1900417. [Google Scholar] [CrossRef]
- Tseng, M.C.; Cheng, H.T.; Shen, M.J.; Chu, Y.H. Bicyclic 1,2,3-Triazolium Ionic Liquids: Synthesis, Characterization, and Application to Rutaecarpine Synthesis. Org. Lett. 2011, 13, 4434–4437. [Google Scholar] [CrossRef]
- Dimitrov-Raytchev, P.; Beghdadi, S.; Serghei, A.; Drockenmuller, E. Main-chain 1,2,3-triazolium-based poly(ionic liquid)s issued from AB + AB click chemistry polyaddition. J. Polym. Sci. Pol. Chem. 2013, 51, 34–38. [Google Scholar] [CrossRef]
- Mudraboyina, B.P.; Obadia, M.M.; Allaoua, I.; Sood, R.; Serghei, A.; Drockenmuller, E. 1,2,3-Triazolium-Based Poly(ionic liquid)s with Enhanced Ion Conducting Properties Obtained through a Click Chemistry Polyaddition Strategy. Chem. Mater. 2014, 26, 1720–1726. [Google Scholar] [CrossRef]
- Rogers, R.D.; Seddon, K.R. Ionic Liquids–Solvents of the Future? Science 2003, 302, 792–793. [Google Scholar] [CrossRef]
- Welton, T. Ionic liquids in Green Chemistry. Green Chem. 2011, 13, 225. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, S.; Deng, Y. Recent advances in ionic liquid catalysis. Green Chem. 2011, 13, 2619–2637. [Google Scholar] [CrossRef]
- Marr, P.C.; Marr, A.C. Ionic liquid gel materials: Applications in green and sustainable chemistry. Green Chem. 2016, 18, 105–128. [Google Scholar] [CrossRef]
- Osada, I.; de Vries, H.; Scrosati, B.; Passerini, S. Ionic-Liquid-Based Polymer Electrolytes for Battery Applications. Angew. Chem. Int. Edit. 2016, 55, 500–513. [Google Scholar] [CrossRef]
- Somers, A.E.; Howlett, P.C.; MacFarlane, D.R.; Forsyth, M. A Review of Ionic Liquid Lubricants. Lubricants 2013, 1, 3–21. [Google Scholar] [CrossRef]
- Zhou, F.; Liang, Y.; Liu, W. Ionic liquid lubricants: Designed chemistry for engineering applications. Chem. Soc. Rev. 2009, 38, 2590–2599. [Google Scholar] [CrossRef]
- Yan, F.; Yu, S.; Zhang, X.; Qiu, L.; Chu, F.; You, J.; Lu, J. Enhanced Proton Conduction in Polymer Electrolyte Membranes as Synthesized by Polymerization of Protic Ionic Liquid-Based Microemulsions. Chem. Mater. 2009, 21, 1480–1484. [Google Scholar] [CrossRef]
- Kreuer, K.D. Proton Conductivity: Materials and Applications. Chem. Mater. 1996, 8, 610–641. [Google Scholar] [CrossRef]
- Kreuer, K.; Fuchs, A.; Ise, M.; Spaeth, M.; Maier, J. Imidazole and pyrazole-based proton conducting polymers and liquids. Electrochim. Acta 1998, 43, 1281–1288. [Google Scholar] [CrossRef]
- Kreuer, K. On the complexity of proton conduction phenomena. Solid State Ionics 2000, 136-137, 149–160. [Google Scholar] [CrossRef]
- Vilčiauskas, L.; De Araujo, C.C.; Kreuer, K.D. Proton conductivity and diffusion in molten phosphinic acid (H3PO2): The last member of the phosphorus oxoacid proton conductor family. Solid State Ionics 2012, 212, 6–9. [Google Scholar] [CrossRef]
- Wehmeyer, C.; Schrader, M.; Andrienko, D.; Sebastiani, D. Water-Free Proton Conduction in Hexakis(p-Phosphonatophenyl)benzene Nanochannels. J. Phys. Chem. C 2013, 117, 12366–12372. [Google Scholar] [CrossRef]
- Pulst, M.; Balko, J.; Golitsyn, Y.; Reichert, D.; Busse, K.; Kressler, J. Proton conductivity and phase transitions in 1,2,3-triazole. Phys. Chem. Chem. Phys. 2016, 18, 6153–6163. [Google Scholar] [CrossRef] [PubMed]
- Billes, F.; Endredi, H.; Keresztury, G. Vibrational spectroscopy of triazoles and tetrazole. J. Mol. Struct. Theochem. 2000, 530, 183–200. [Google Scholar] [CrossRef]
- Rauhut, G. Modulation of reaction barriers by generating reactive intermediates: Double proton transfer reactions. Phys. Chem. Chem. Phys. 2003, 5, 791–800. [Google Scholar] [CrossRef]
- Shi, Y.; Chen, Y.; Liang, Y.; Andrews, J.; Dong, H.; Yuan, M.; Ding, W.; Banerjee, S.; Ardebili, H.; Robertson, M.L.; et al. Chemically inert covalently networked triazole-based solid polymer electrolytes for stable all-solid-state lithium batteries. J. Mater. Chem. A 2019, 7, 19691–19695. [Google Scholar] [CrossRef]
- Kirsch, C.; Pulst, M.; Samiullah, M.H.; Ruda, P.; Hasan, N.; Kressler, J. 1,2,3-Triazole mediated Li+-ion conductivity in poly(ethylene oxide) based electrolytes. Solid State Ionics 2017, 309, 163–169. [Google Scholar] [CrossRef]
- Johansson, P.; Béranger, S.; Armand, M.; Nilsson, H.; Jacobsson, P. Spectroscopic and theoretical study of the 1,2,3-Triazole-4,5-dicarbonitrile anion and its lithium ion pairs. Solid State Ionics 2003, 156, 129–139. [Google Scholar] [CrossRef]
- Mauret, P.; Fuyet, J.P.; Fabre, M.; Elgouero, J.; Pardo, M.d.C. Etude de la Tautomerie du V-triazole a Partier des Mesures des Moments Dipolares. J. Chim. Phys. Physicochim. Bio 1973, 70, 1483–1485. [Google Scholar] [CrossRef]
- Bellagamba, M.; Bencivenni, L.; Gontrani, L.; Guidoni, L.; Sadun, C. Tautomerism in liquid 1,2,3-triazole: A combined energy-dispersive X-ray diffraction, molecular dynamics, and FTIR study. Struct. Chem. 2013, 24, 933–943. [Google Scholar] [CrossRef]
- Zhou, Z.; Li, S.; Zhang, Y.; Liu, M.; Li, W. Promotion of proton conduction in polymer electrolyte membranes by 1H-1,2,3-triazole. J. Am. Chem. Soc. 2005, 127, 10824–10825. [Google Scholar] [CrossRef] [PubMed]
- Goward, G.R.; Schuster, M.F.H.; Sebastiani, D.; Schnell, I.; Spiess, H.W. High-Resolution Solid-State NMR Studies of Imidazole-Based Proton Conductors: Structure Motifs and Chemical Exchange from 1H NMR. J. Phys. Chem. 2002, 106, 9322–9334. [Google Scholar] [CrossRef]
- Pulst, M.; Elgabarty, H.; Sebastiani, D.; Kressler, J. The annular tautomerism of lithium 1,2,3-triazolate. New J. Chem. 2017, 41, 1430–1435. [Google Scholar] [CrossRef]
- Ludueña, G.A.; Wegner, M.; Bjålie, L.; Sebastiani, D. Local Disorder in Hydrogen Storage Compounds: The Case of Lithium Amide/Imide. ChemPhysChem 2010, 11, 2353–2360. [Google Scholar] [CrossRef] [PubMed]
- Hutter, J.; Iannuzzi, M.; Schiffmann, F.; VandeVondele, J. cp2k: Atomistic simulations of condensed matter systems. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2014, 4, 15–25. [Google Scholar] [CrossRef]
- Kühne, T.D.; Iannuzzi, M.; Del Ben, M.; Rybkin, V.V.; Seewald, P.; Stein, F.; Laino, T.; Khaliullin, R.Z.; Schütt, O.; Schiffmann, F.; et al. CP2K: An electronic structure and molecular dynamics software package-Quickstep: Efficient and accurate electronic structure calculations. J. Chem. Phys. 2020, 152, 194103. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. Quickstep: Fast and accurate density functional calculations using a mixed Gaussian and plane waves approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- VandeVondele, J.; Hutter, J. Gaussian basis sets for accurate calculations on molecular systems in gas and condensed phases. J. Chem. Phys. 2007, 127, 114105. [Google Scholar] [CrossRef]
- Nosé, S. A unified formulation of the constant temperature molecular dynamics methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable dual-space Gaussian pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic separable dual-space Gaussian pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [PubMed]
- VandeVondele, J.; Hutter, J. An efficient orbital transformation method for electronic structure calculations. J. Chem. Phys. 2003, 118, 4365–4369. [Google Scholar] [CrossRef]
- Available online: http://lammps.sandia.gov (accessed on 25 January 2016).
- Wang, J.M.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and Testing of a General Amber Force Field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Brehm, M.; Kirchner, B. TRAVIS—A Free Analyzer and Visualizer for Monte Carlo and Molecular Dynamics Trajectories. J. Chem. Inf. Model. 2011, 51, 2007–2023. [Google Scholar] [CrossRef]
- Wolfram Research, Inc. Mathematica, Version 10.0; Wolfram Research, Inc.: Champaign, IL, USA, 2020. [Google Scholar]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phase | Temperature | D/ | D/ |
|---|---|---|---|
| liquid | 413 K | (1.2 ± 2.8) | (1.6 ± 1.6) |
| liquid | 313 K | (2.0 ± 3.1) | (3.3 ± 6.8) |
| Trajectory | 269 K | 280 K | 313 K | 413 K |
|---|---|---|---|---|
| No. triazoles | 48 | 32 | 32 | 32 |
| Tautomer ratio 1H:2H | 32:16 | 16:16 | 5:27 | 5:27 |
| Physical time/ps | 75 | 148 | 205 | 101 |
| Boxvector x/Å | 19.726 | 18.662 | 18.3291 | 18.3291 |
| Boxvector y/Å | 10.910 | 19.327 | 18.3291 | 18.3291 |
| Boxvector z/Å | 18.572 | 7.396 | 9.1646 | 9.1646 |
| Crystal system | monoclinic | orthorhombic | ||
| = 93.6085 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peschel, C.; Dreßler, C.; Sebastiani, D. ab-Initio Study of Hydrogen Bond Networks in 1,2,3-Triazole Phases. Molecules 2020, 25, 5722. https://doi.org/10.3390/molecules25235722
Peschel C, Dreßler C, Sebastiani D. ab-Initio Study of Hydrogen Bond Networks in 1,2,3-Triazole Phases. Molecules. 2020; 25(23):5722. https://doi.org/10.3390/molecules25235722
Chicago/Turabian StylePeschel, Christopher, Christian Dreßler, and Daniel Sebastiani. 2020. "ab-Initio Study of Hydrogen Bond Networks in 1,2,3-Triazole Phases" Molecules 25, no. 23: 5722. https://doi.org/10.3390/molecules25235722
APA StylePeschel, C., Dreßler, C., & Sebastiani, D. (2020). ab-Initio Study of Hydrogen Bond Networks in 1,2,3-Triazole Phases. Molecules, 25(23), 5722. https://doi.org/10.3390/molecules25235722