
Carbon Atoms Speaking Out: How the Geometric Sensitivity of 13C Chemical Shifts Leads to Understanding the Colour Tuning of Phycocyanobilin in Cph1 and AnPixJ


Abstract
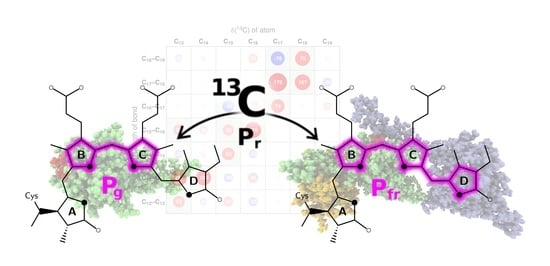
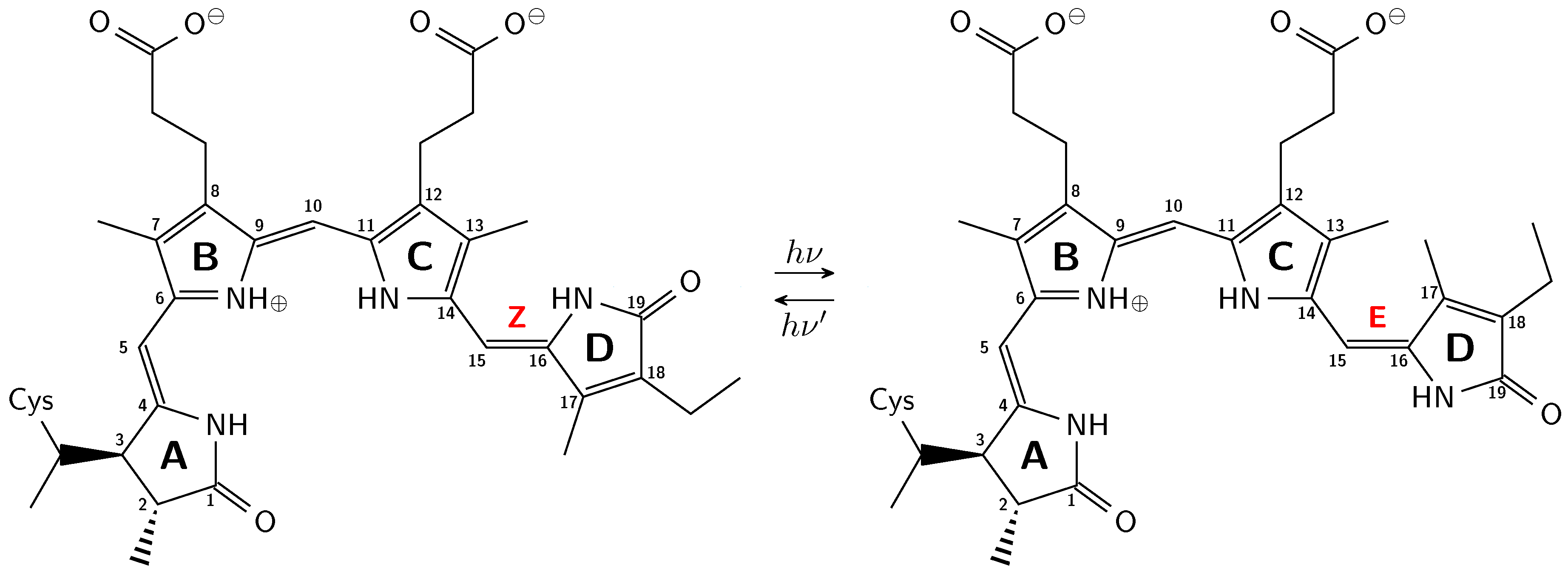
1. Introduction
2. Results
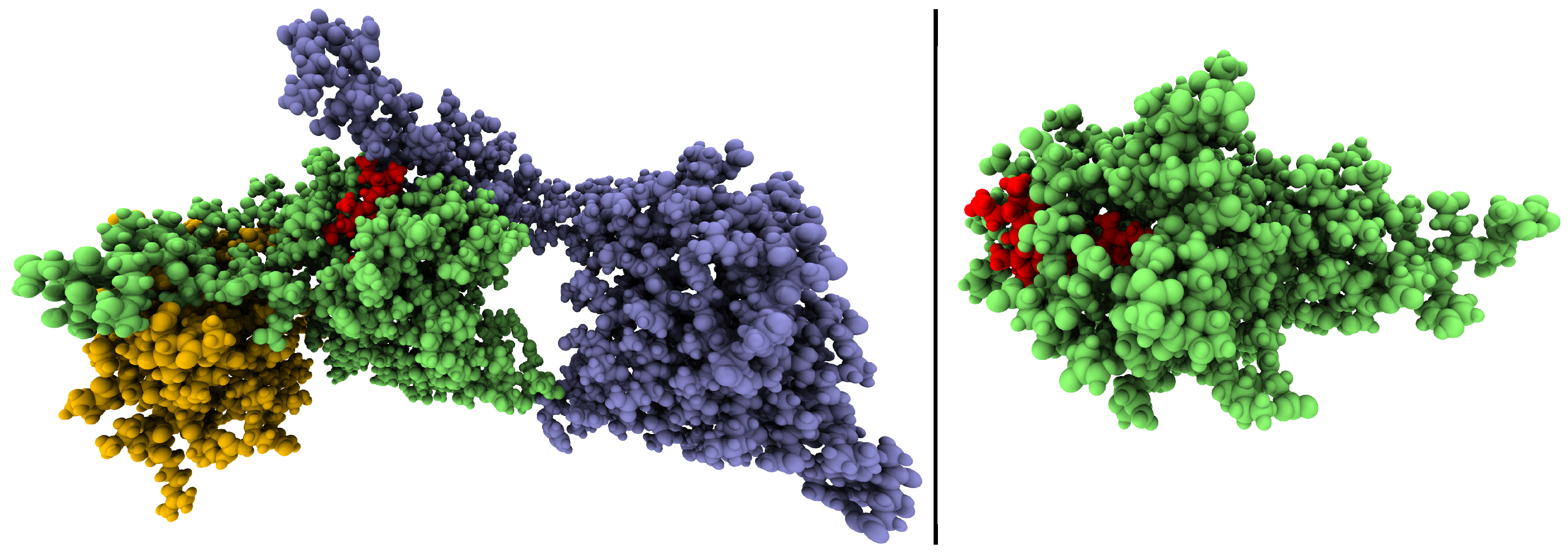
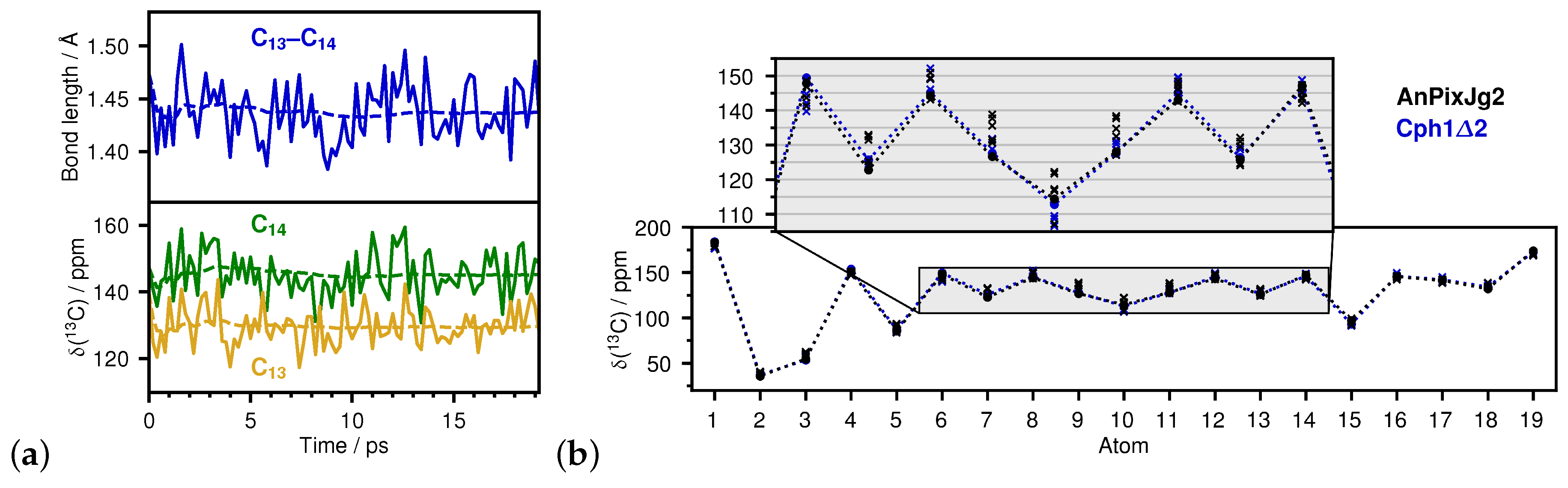
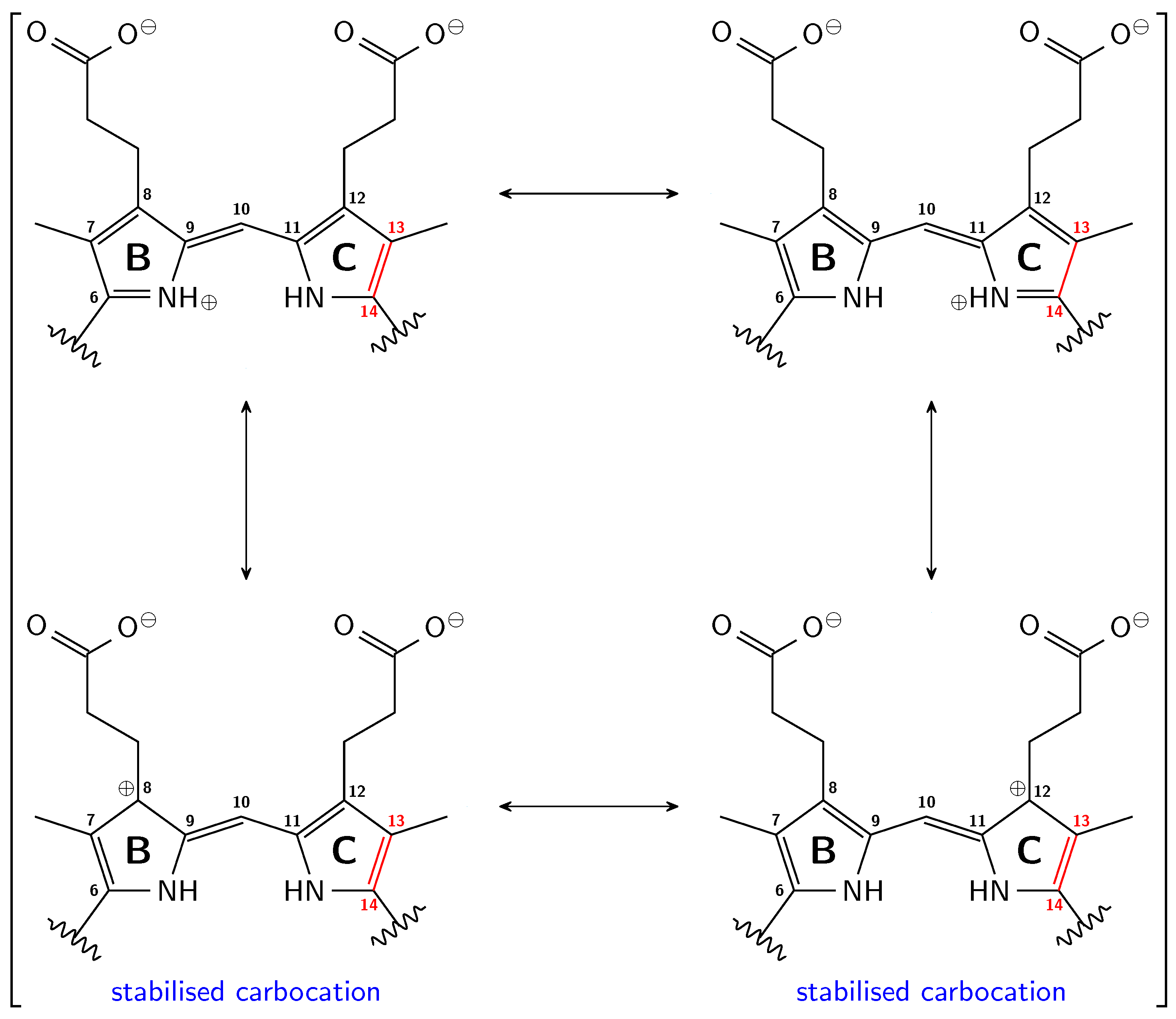
2.1. How Bond Alternation and Thermal Fluctuations Determine 13C-NMR Chemical Shifts
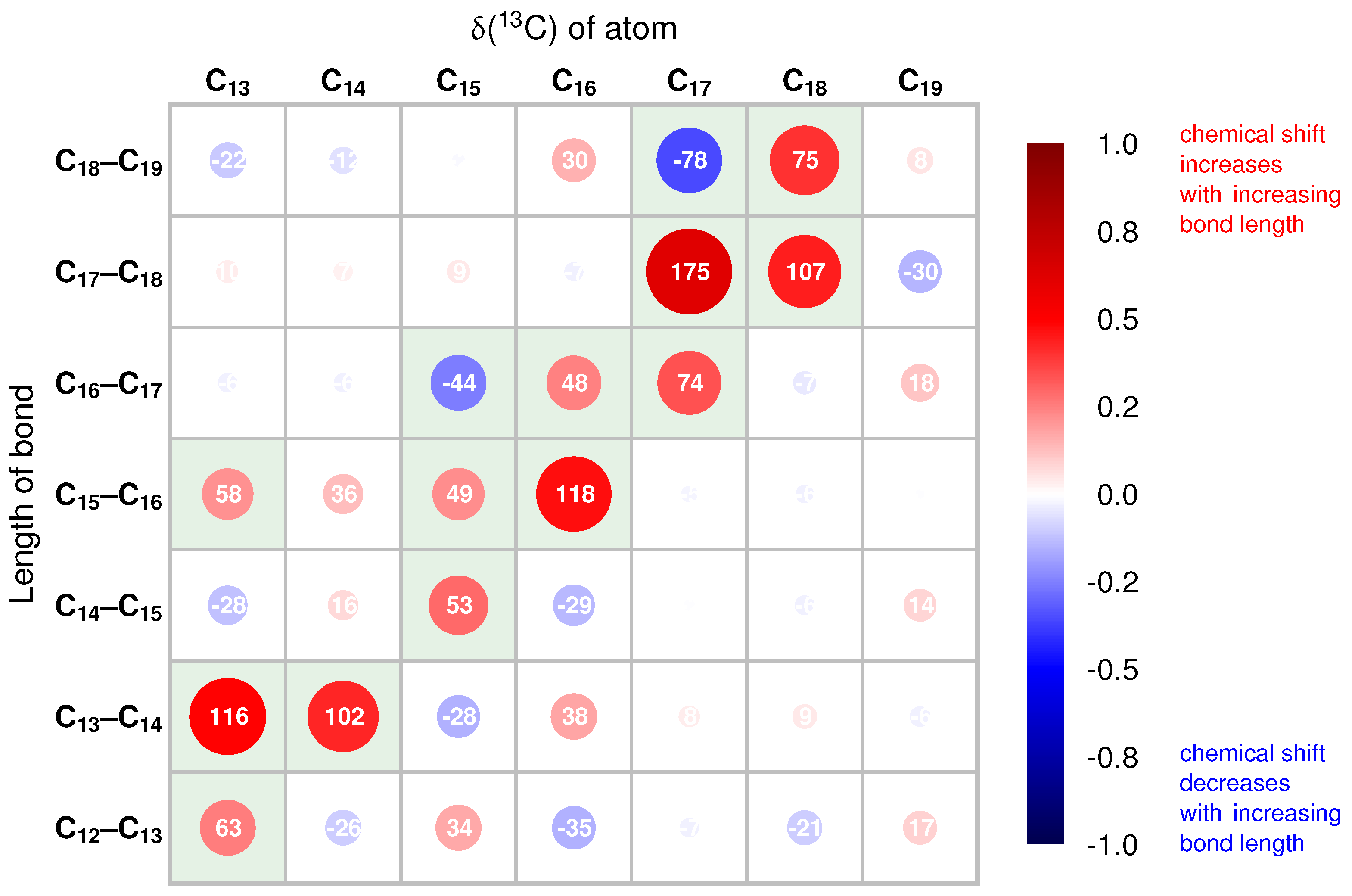
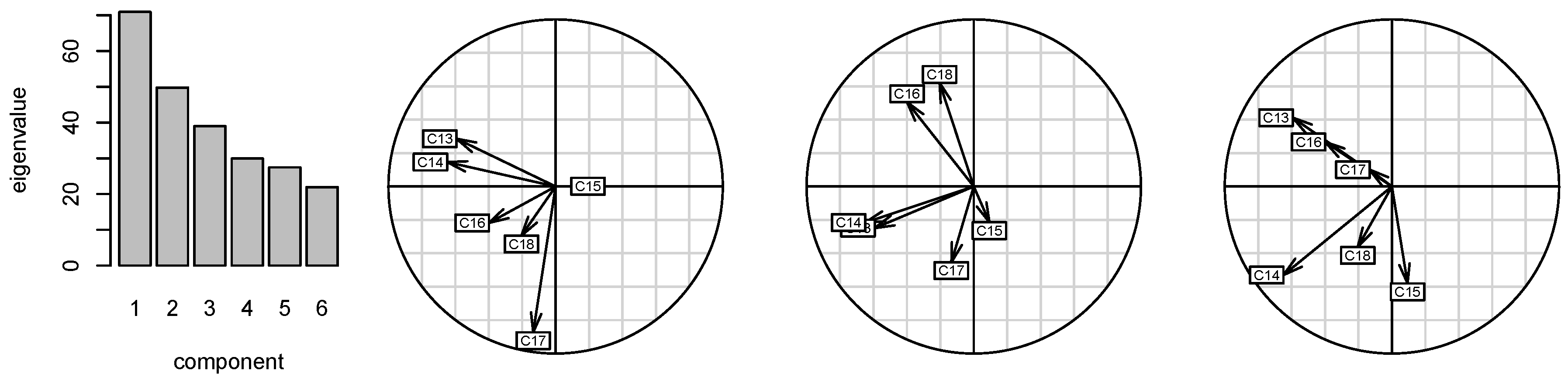
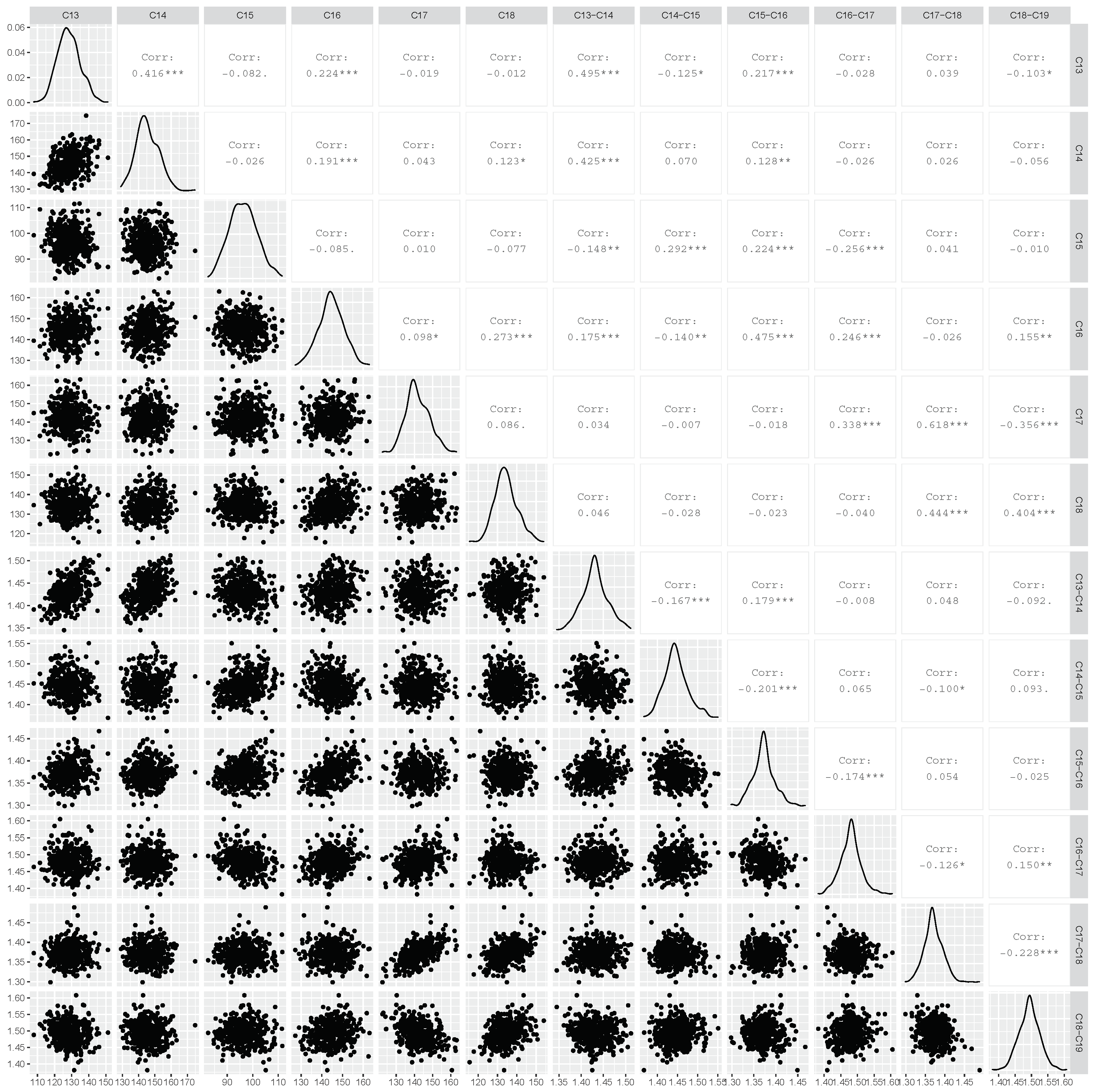
2.2. Correlations of 13C Chemical Shifts and Their Geometric Sensitivity
2.3. Inversion of the Geometric Sensitivity Points to Pg and Pfr States
3. Discussion
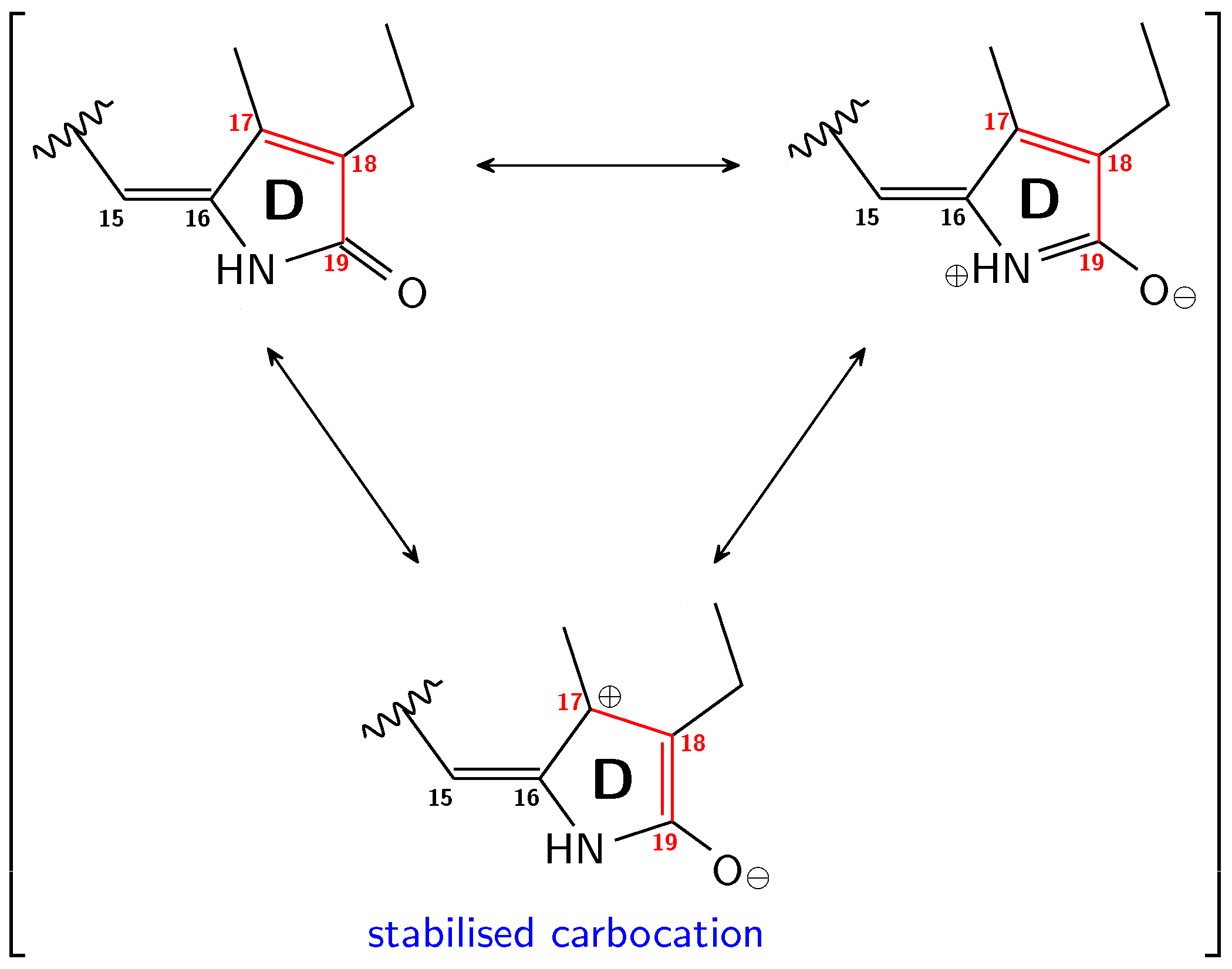
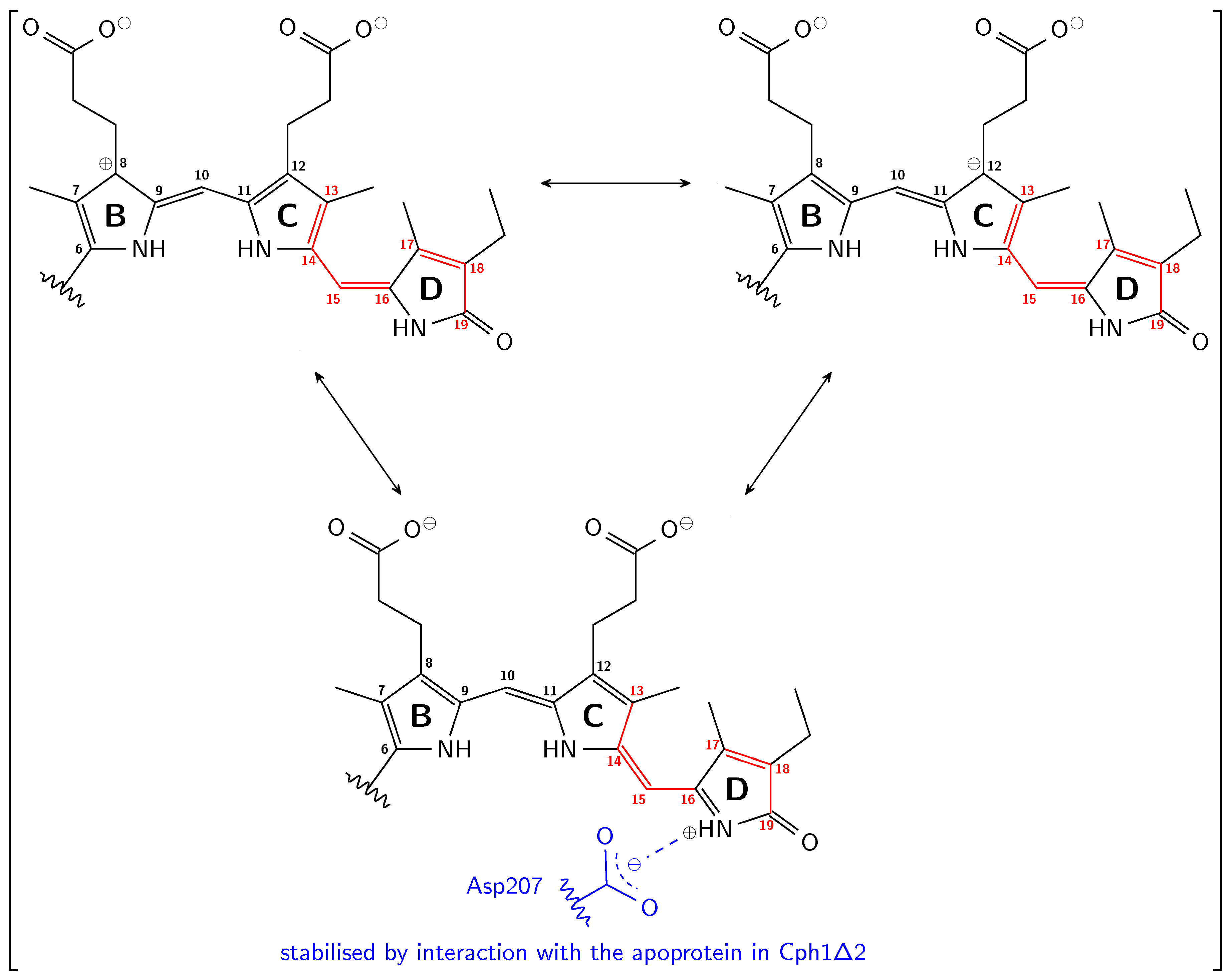
3.1. Pg: Short Conjugation
3.2. Pfr: Long Conjugation
4. Conclusions
5. Materials and Methods
5.1. Preparations
5.2. Force Field Molecular Dynamics Simulations
5.3. QM/MM Molecular Dynamics Simulations
5.4. Calculation of Nuclear Shieldings
5.5. Statistical Analysis
6. Other
 using chemfig 1.56 [109].
using chemfig 1.56 [109].Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AIMD | Ab initio molecular dynamics |
| AnPixJg2 | Second GAF domain (out of four) of a phototaxis regulator found in heterocyst-forming |
| cyanobacteria Anabaena (Nostoc) sp. PCC 7120 (all1069) [13,67]. Pix, originally pis (PhototaxIS), | |
| is a class of regulatory genes required for positive phototactic movement (PositIve phototaXis), | |
| whereas J is a serial letter [15] | |
| BLYP | Becke/Lee-Yang-Parr (exchange/correlation) |
| CBCR | Cyanobacteriochrome |
| CHARMM | Chemistry at Harvard macromolecular mechanics |
| Cph12 | “Cyanobacterial phytochrome 1”, i.e., the first phytochrome found in cyanobacteria, more |
| precisely Synechocystis sp. PCC 6803 (slr0473) [10,46,47]. It functions as light-regulated histidine | |
| protein kinase [10,35]. Δ2 denotes the sensory module of the protein comprising 514 N-terminal | |
| residues; Δ1 comprises 492 N-terminal residues [48] | |
| CSGT | Continuous set of gauge transformations |
| DFT | Density functional theory |
| FFMD | Force field molecular dynamics |
| GAF | cGMP phosphodiesterase/adenylyl cyclase/FhlA |
| GAPW | Gaussian-augmented plane waves |
| GEEP | Gaussian expansion of the electrostatic potential |
| GPW | Gaussian and plane waves |
| GTH | Goedecker-Teter-Hutter (pseudopotential) |
| IGAIM | Individual Gauges for Atoms in Molecules |
| IR | Infrared |
| MAS | Magic angle spinning |
| MD | Molecular dynamics |
| MM | Molecular mechanics |
| NMR | Nuclear magnetic resonance |
| NVT | Canonical (ensemble) |
| PAS | period circadian/aryl hydrocarbon receptor nuclear translocator/single-minded |
| PBC | Periodic boundary conditions |
| PCA | Principal component analysis |
| PCB | Phycocyanobilin |
| PHY | Phytochrome specific |
| PP | Pseudopotential |
| QM | Quantum mechanics |
| QM/MM | (Hybrid) Quantum mechanics/molecular mechanics |
| QMP | QM part |
| RMSE | Root mean square error |
| UV | Ultraviolett |
Appendix A
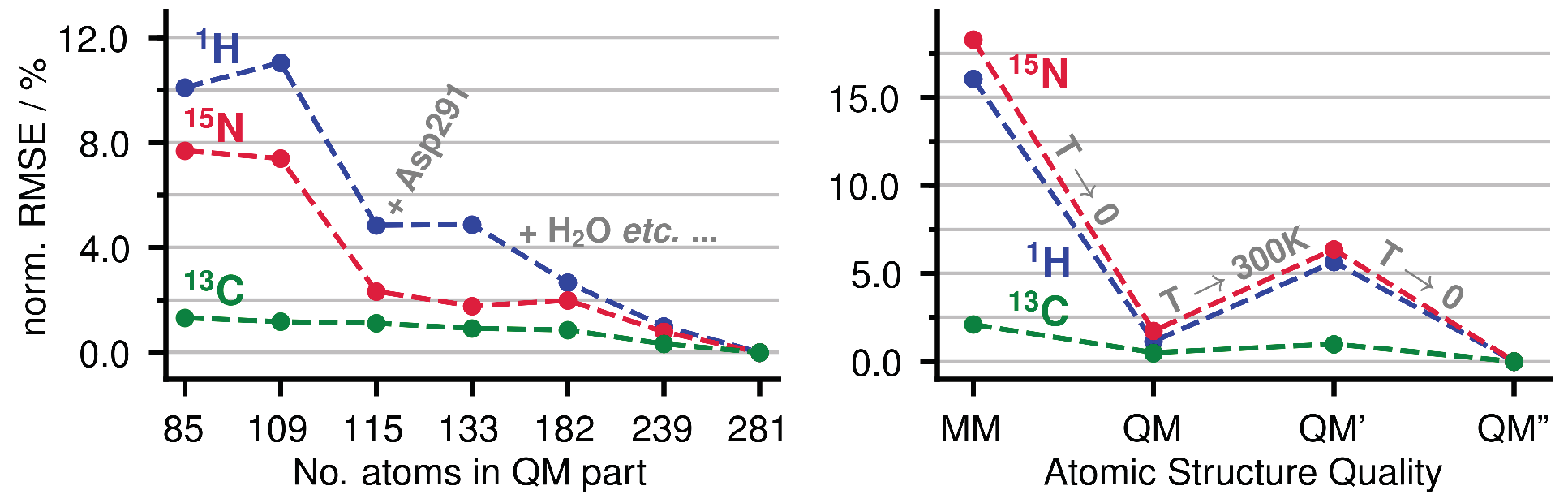
Appendix A.1. Size of the QM Part and Convergence

Appendix A.2. Statistical Data

References
- Senge, M.O.; MacGowan, S.A.; O’Brien, J.M. Conformational Control of Cofactors in Nature—The Influence of Protein-Induced Macrocycle Distortion on the Biological Function of Tetrapyrroles. Chem. Commun. 2015, 51, 17031–17063. [Google Scholar] [CrossRef] [PubMed]
- Kadish, K.; Smith, K.; Guilard, R. (Eds.) The Porphyrin Handbook; Academic: San Diego, CA, USA, 2000. [Google Scholar]
- Grossman, A.R.; Schaefer, M.R.; Chiang, G.G.; Collier, J.L. The Phycobilisome, a Light-Harvesting Complex Responsive to Environmental Conditions. Microbiol. Rev. 1993, 57, 725–749. [Google Scholar] [CrossRef] [PubMed]
- Butler, W.L.; Norris, K.H.; Siegelman, H.W.; Hendricks, S.B. Detection, Assay, and Preliminary Purification of the Pigment Controlling Photoresponsive Development of Plants. Proc. Natl. Acad. Sci. USA 1959, 45, 1703–1708. [Google Scholar] [CrossRef] [PubMed]
- Sage, L.C. Pigment of the Imagination: A History of Phytochrome Research; Academic: San Diego, CA, USA, 1992. [Google Scholar]
- Rockwell, N.C.; Su, Y.S.; Lagarias, J.C. Phytochrome Structure and Signaling Mechanisms. Annu. Rev. Plant Biol. 2006, 57, 837–858. [Google Scholar] [CrossRef]
- Tu, S.L.; Lagarias, J.C. The Phytochromes. In Handbook of Photosensory Receptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 121–149. [Google Scholar] [CrossRef]
- Huq, E.; Quail, P.H. Phytochrome Signaling. In Handbook of Photosensory Receptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 151–170. [Google Scholar] [CrossRef]
- Vierstra, R.D.; Karniol, B. Phytochromes in Microorganisms. In Handbook of Photosensory Receptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 171–195. [Google Scholar] [CrossRef]
- Yeh, K.C.; Wu, S.H.; Murphy, J.T.; Lagarias, J.C. A Cyanobacterial Phytochrome Two-Component Light Sensory System. Science 1997, 277, 1505–1508. [Google Scholar] [CrossRef]
- Schmidt, A.; Sauthof, L.; Szczepek, M.; Lopez, M.F.; Escobar, F.V.; Qureshi, B.M.; Michael, N.; Buhrke, D.; Stevens, T.; Kwiatkowski, D.; et al. Structural Snapshot of a Bacterial Phytochrome in Its Functional Intermediate State. Nat. Commun. 2018, 9, 4912. [Google Scholar] [CrossRef]
- Ikeuchi, M.; Ishizuka, T. Cyanobacteriochromes: A New Superfamily of Tetrapyrrole-Binding Photoreceptors in Cyanobacteria. Photochem. Photobiol. Sci. 2008, 7, 1159–1167. [Google Scholar] [CrossRef]
- Narikawa, R.; Fukushima, Y.; Ishizuka, T.; Itoh, S.; Ikeuchi, M. A Novel Photoactive GAF Domain of Cyanobacteriochrome AnPixJ That Shows Reversible Green/Red Photoconversion. J. Mol. Biol. 2008, 380, 844–855. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Martin, S.S.; Lagarias, J.C. Red/Green Cyanobacteriochromes: Sensors of Color and Power. Biochemistry 2012, 51, 9667–9677. [Google Scholar] [CrossRef]
- Suzuki, F.; Fujita, H.; Ikeuchi, M.; Yoshihara, S.; Geng, X.X. Novel Putative Photoreceptor and Regulatory Genes Required for the Positive Phototactic Movement of the Unicellular Motile Cyanobacterium Synechocystis Sp. PCC 6803. Plant Cell Physiol. 2000, 41, 1299–1304. [Google Scholar] [CrossRef]
- Karniol, B.; Wagner, J.R.; Walker, J.M.; Vierstra, R.D. Phylogenetic Analysis of the Phytochrome Superfamily Reveals Distinct Microbial Subfamilies of Photoreceptors. Biochem. J. 2005, 392, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Wiebeler, C.; Rao, A.G.; Gärtner, W.; Schapiro, I. The Effective Conjugation Length Is Responsible for the Red/Green Spectral Tuning in the Cyanobacteriochrome Slr1393g3. Angew. Chem. Int. Ed. 2019, 58, 1934–1938. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, N.C.; Martin, S.S.; Feoktistova, K.; Lagarias, J.C. Diverse Two-Cysteine Photocycles in Phytochromes and Cyanobacteriochromes. Proc. Natl. Acad. Sci. USA 2011, 108, 11854–11859. [Google Scholar] [CrossRef] [PubMed]
- Enomoto, G.; Hirose, Y.; Narikawa, R.; Ikeuchi, M. Thiol-Based Photocycle of the Blue and Teal Light-Sensing Cyanobacteriochrome Tlr1999. Biochemistry 2012, 51, 3050–3058. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Martin, S.S.; Gulevich, A.G.; Lagarias, J.C. Phycoviolobilin Formation and Spectral Tuning in the DXCF Cyanobacteriochrome Subfamily. Biochemistry 2012, 51, 1449–1463. [Google Scholar] [CrossRef]
- Schäfer, E.; Nagy, F. Light-Activated Intracellular Movement of Phytochrome. In Handbook of Photosensory Receptors; Wiley-VCH Verlag GmbH: Weinheim, Germany, 2005; pp. 197–210. [Google Scholar] [CrossRef]
- Velazquez Escobar, F.; Hildebrandt, T.; Utesch, T.; Schmitt, F.J.; Seuffert, I.; Michael, N.; Schulz, C.; Mroginski, M.A.; Friedrich, T.; Hildebrandt, P. Structural Parameters Controlling the Fluorescence Properties of Phytochromes. Biochemistry 2014, 53, 20–29. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Ohlendorf, R.; Möglich, A. Cyanobacteriochromes in Full Color and Three Dimensions. Proc. Natl. Acad. Sci. USA 2013, 110, 806–807. [Google Scholar] [CrossRef]
- Rumyantsev, K.A.; Shcherbakova, D.M.; Zakharova, N.I.; Verkhusha, V.V.; Turoverov, K.K. Design of Near-Infrared Single-Domain Fluorescent Protein GAF-FP Based on Bacterial Phytochrome. Cell Tissue Biol. 2017, 11, 16–26. [Google Scholar] [CrossRef]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian Expression of Infrared Fluorescent Proteins Engineered from a Bacterial Phytochrome. Science 2009, 324, 804–807. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, X.J.; Wang, Z.B.; Chen, Y.; Wang, X.; Zhou, M.; Scheer, H.; Zhao, K.H. Fused-Gene Approach to Photoswitchable and Fluorescent Biliproteins. Angew. Chem. Int. Ed. 2010, 49, 5456–5458. [Google Scholar] [CrossRef]
- Simon, J.; Losi, A.; Zhao, K.H.; Gärtner, W. FRET in a Synthetic Flavin- and Bilin-Binding Protein. Photochem. Photobiol. 2017, 93, 1057–1062. [Google Scholar] [CrossRef] [PubMed]
- Piatkevich, K.D.; Subach, F.V.; Verkhusha, V.V. Engineering of Bacterial Phytochromes for Near-Infrared Imaging, Sensing, and Light-Control in Mammals. Chem. Soc. Rev. 2013, 42, 3441–3452. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.N.; Ding, W.L.; Jiang, S.P.; Miao, D.; Tan, Z.Z.; Hu, J.L.; Scheer, H.; Zhao, K.H. Bright Near-Infrared Fluorescence Bio-Labeling with a Biliprotein Triad. Biochim. Biophys. Acta Bioenerg. Mol. Cell Res. 2019, 1866, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Scheer, H.; Yang, X.; Zhao, K.H. Biliproteins and Their Applications in Bioimaging. Procedia Chem. 2015, 14, 176–185. [Google Scholar] [CrossRef][Green Version]
- Filonov, G.S.; Piatkevich, K.D.; Ting, L.M.; Zhang, J.; Kim, K.; Verkhusha, V.V. Bright and Stable Near-Infrared Fluorescent Protein for In Vivo Imaging. Nat. Biotechnol. 2011, 29, 757. [Google Scholar] [CrossRef] [PubMed]
- Ryu, M.H.; Gomelsky, M. Near-Infrared Light Responsive Synthetic c-Di-GMP Module for Optogenetic Applications. ACS Synth. Biol. 2014, 3, 802–810. [Google Scholar] [CrossRef]
- Shimizu-Sato, S.; Huq, E.; Tepperman, J.M.; Quail, P.H. A Light-Switchable Gene Promoter System. Nat. Biotechnol. 2002, 20, 1041. [Google Scholar] [CrossRef]
- Wu, X.J.; Yang, H.; Sheng, Y.; Zhu, Y.L.; Li, P.P. Fluorescence Properties of a Novel Cyanobacteriochrome GAF Domain from Spirulina That Exhibits Moderate Dark Reversion. Int. J. Mol. Sci. 2018, 19, 2253. [Google Scholar] [CrossRef]
- Essen, L.O.; Mailliet, J.; Hughes, J. The Structure of a Complete Phytochrome Sensory Module in the Pr Ground State. Proc. Natl. Acad. Sci. USA 2008, 105, 14709–14714. [Google Scholar] [CrossRef]
- Rüdiger, W.; Thümmler, F.; Cmiel, E.; Schneider, S. Chromophore Structure of the Physiologically Active Form (P(Fr)) of Phytochrome. Proc. Natl. Acad. Sci. USA 1983, 80, 6244–6248. [Google Scholar] [CrossRef]
- Song, C.; Rohmer, T.; Tiersch, M.; Zaanen, J.; Hughes, J.; Matysik, J. Solid-State NMR Spectroscopy to Probe Photoactivation in Canonical Phytochromes. Photochem. Photobiol. 2013, 89, 259–273. [Google Scholar] [CrossRef] [PubMed]
- Rohmer, T.; Lang, C.; Bongards, C.; Gupta, K.B.S.S.; Neugebauer, J.; Hughes, J.; Gärtner, W.; Matysik, J. Phytochrome as Molecular Machine: Revealing Chromophore Action during the Pfr—Pr Photoconversion by Magic-Angle Spinning NMR Spectroscopy. J. Am. Chem. Soc. 2010, 132, 4431–4437. [Google Scholar] [CrossRef] [PubMed]
- Heyes, D.J.; Khara, B.; Sakuma, M.; Hardman, S.J.O.; O’Cualain, R.; Rigby, S.E.J.; Scrutton, N.S. Ultrafast Red Light Activation of Synechocystis Phytochrome Cph1 Triggers Major Structural Change to Form the Pfr Signalling-Competent State. PLoS ONE 2012, 7, e52418. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Psakis, G.; Lang, C.; Mailliet, J.; Gärtner, W.; Hughes, J.; Matysik, J. Two Ground State Isoforms and a Chromophore D-Ring Photoflip Triggering Extensive Intramolecular Changes in a Canonical Phytochrome. Proc. Natl. Acad. Sci. USA 2011, 108, 3842–3847. [Google Scholar] [CrossRef]
- Song, C.; Psakis, G.; Kopycki, J.; Lang, C.; Matysik, J.; Hughes, J. The D-Ring, Not the a-Ring, Rotates in Synechococcus OS-B Phytochrome. J. Biol. Chem. 2014, 289, 2552–2562. [Google Scholar] [CrossRef]
- Hirose, Y.; Rockwell, N.C.; Nishiyama, K.; Narikawa, R.; Ukaji, Y.; Inomata, K.; Lagarias, J.C.; Ikeuchi, M. Green/Red Cyanobacteriochromes Regulate Complementary Chromatic Acclimation via a Protochromic Photocycle. Proc. Natl. Acad. Sci. USA 2013, 110, 4974–4979. [Google Scholar] [CrossRef]
- Scarbath-Evers, L.K.; Jähnigen, S.; Elgabarty, H.; Song, C.; Narikawa, R.; Matysik, J.; Sebastiani, D. Structural Heterogeneity in a Parent Ground-State Structure of AnPixJg2 Revealed by Theory and Spectroscopy. Phys. Chem. Chem. Phys. 2017, 19, 13882–13894. [Google Scholar] [CrossRef]
- Peng, P.P.; Dong, L.L.; Sun, Y.F.; Zeng, X.L.; Ding, W.L.; Scheer, H.; Yang, X.; Zhao, K.H. The Structure of Allophycocyanin B from Synechocystis PCC 6803 Reveals the Structural Basis for the Extreme Redshift of the Terminal Emitter in Phycobilisomes. Acta Crystallogr. Sect. D 2014, 70, 2558–2569. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Martin, S.S.; Gulevich, A.G.; Lagarias, J.C. Conserved Phenylalanine Residues Are Required for Blue-Shifting of Cyanobacteriochrome Photoproducts. Biochemistry 2014, 53, 3118–3130. [Google Scholar] [CrossRef]
- Kaneko, T.; Sato, S.; Kotani, H.; Tanaka, A.; Asamizu, E.; Nakamura, Y.; Miyajima, N.; Hirosawa, M.; Sugiura, M.; Sasamoto, S.; et al. Sequence Analysis of the Genome of the Unicellular Cyanobacterium Synechocystis Sp. Strain PCC6803. II. Sequence Determination of the Entire Genome and Assignment of Potential Protein-Coding Regions. DNA Res. 1996, 3, 109–136. [Google Scholar] [CrossRef]
- Hughes, J.; Lamparter, T.; Mittmann, F.; Hartmann, E.; Gärtner, W.; Wilde, A.; Börner, T. A Prokaryotic Phytochrome. Nature 1997, 386, 663. [Google Scholar] [CrossRef] [PubMed]
- Hahn, J.; Strauss, H.M.; Landgraf, F.T.; Gimenèz, H.F.; Lochnit, G.; Schmieder, P.; Hughes, J. Probing Protein– Chromophore Interactions in Cph1 Phytochrome by Mutagenesis. FEBS J. 2006, 273, 1415–1429. [Google Scholar] [CrossRef] [PubMed]
- Takeda, K.; Terazima, M. Photoinduced Orientation Change of the Dimer Structure of the Pr-i State of Cph1Δ2. Biochemistry 2018, 57, 5058–5071. [Google Scholar] [CrossRef] [PubMed]
- Velazquez Escobar, F.; Lang, C.; Takiden, A.; Schneider, C.; Balke, J.; Hughes, J.; Alexiev, U.; Hildebrandt, P.; Mroginski, M.A. Protonation-Dependent Structural Heterogeneity in the Chromophore Binding Site of Cyanobacterial Phytochrome Cph1. J. Phys. Chem. B 2017, 121, 47–57. [Google Scholar] [CrossRef]
- Stöppler, D.; Song, C.; van Rossum, B.J.; Geiger, M.A.; Lang, C.; Mroginski, M.A.; Jagtap, A.P.; Sigurdsson, S.T.; Matysik, J.; Hughes, J.; et al. Dynamic Nuclear Polarization Provides New Insights into Chromophore Structure in Phytochrome Photoreceptors. Angew. Chem. Int. Ed. 2016, 55, 16017–16020. [Google Scholar] [CrossRef]
- Yang, Y.; Heyne, K.; Mathies, R.A.; Dasgupta, J. Non-Bonded Interactions Drive the Sub-Picosecond Bilin Photoisomerization in the Pfr State of Phytochrome Cph1. ChemPhysChem 2016, 17, 369–374. [Google Scholar] [CrossRef]
- Kim, P.W.; Rockwell, N.C.; Martin, S.S.; Lagarias, J.C.; Larsen, D.S. Heterogeneous Photodynamics of the Pfr State in the Cyanobacterial Phytochrome Cph1. Biochemistry 2014, 53, 4601–4611. [Google Scholar] [CrossRef]
- Kim, P.W.; Pan, J.; Rockwell, N.C.; Chang, C.W.; Taylor, K.C.; Clark Lagarias, J.; Larsen, D.S. Ultrafast E to Z Photoisomerization Dynamics of the Cph1 Phytochrome. Chem. Phys. Lett. 2012, 549, 86–92. [Google Scholar] [CrossRef][Green Version]
- Spillane, K.M.; Dasgupta, J.; Mathies, R.A. Conformational Homogeneity and Excited-State Isomerization Dynamics of the Bilin Chromophore in Phytochrome Cph1 from Resonance Raman Intensities. Biophys. J. 2012, 102, 709–717. [Google Scholar] [CrossRef][Green Version]
- Mailliet, J.; Psakis, G.; Feilke, K.; Sineshchekov, V.; Essen, L.O.; Hughes, J. Spectroscopy and a High-Resolution Crystal Structure of Tyr263 Mutants of Cyanobacterial Phytochrome Cph1. J. Mol. Biol. 2011, 413, 115–127. [Google Scholar] [CrossRef]
- Song, C.; Psakis, G.; Lang, C.; Mailliet, J.; Zaanen, J.; Gärtner, W.; Hughes, J.; Matysik, J. On the Collective Nature of Phytochrome Photoactivation. Biochemistry 2011, 50, 10987–10989. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, S.; Mroginski, M.A. Molecular Dynamics of Phycocyanobilin Binding Bacteriophytochromes: A Detailed Study of Structural and Dynamic Properties. J. Phys. Chem. B 2010, 114, 16677–16686. [Google Scholar] [CrossRef] [PubMed]
- Mroginski, M.A.; von Stetten, D.; Escobar, F.V.; Strauss, H.M.; Kaminski, S.; Scheerer, P.; Günther, M.; Murgida, D.H.; Schmieder, P.; Bongards, C.; et al. Chromophore Structure of Cyanobacterial Phytochrome Cph1 in the Pr State: Reconciling Structural and Spectroscopic Data by QM/MM Calculations. Biophys. J. 2009, 96, 4153–4163. [Google Scholar] [CrossRef] [PubMed]
- Rockwell, N.C.; Shang, L.; Martin, S.S.; Lagarias, J.C. Distinct Classes of Red/Far-Red Photochemistry within the Phytochrome Superfamily. Proc. Natl. Acad. Sci. USA 2009, 106, 6123–6127. [Google Scholar] [CrossRef]
- Hahn, J.; Strauss, H.M.; Schmieder, P. Heteronuclear NMR Investigation on the Structure and Dynamics of the Chromophore Binding Pocket of the Cyanobacterial Phytochrome Cph1. J. Am. Chem. Soc. 2008, 130, 11170–11178. [Google Scholar] [CrossRef]
- Matute, R.A.; Contreras, R.; Pérez-Hernández, G.; González, L. The Chromophore Structure of the Cyanobacterial Phytochrome Cph1 as Predicted by Time-Dependent Density Functional Theory. J. Phys. Chem. B 2008, 112, 16253–16256. [Google Scholar] [CrossRef]
- Rohmer, T.; Lang, C.; Hughes, J.; Essen, L.O.; Gärtner, W.; Matysik, J. Light-Induced Chromophore Activity and Signal Transduction in Phytochromes Observed by 13C and 15N Magic-Angle Spinning NMR. Proc. Natl. Acad. Sci. USA 2008, 105, 15229–15234. [Google Scholar] [CrossRef]
- Murgida, D.H.; von Stetten, D.; Hildebrandt, P.; Schwinté, P.; Siebert, F.; Sharda, S.; Gärtner, W.; Mroginski, M.A. The Chromophore Structures of the Pr States in Plant and Bacterial Phytochromes. Biophys. J. 2007, 93, 2410–2417. [Google Scholar] [CrossRef]
- Rockwell, N.C.; Lagarias, J.C. The Structure of Phytochrome: A Picture Is Worth a Thousand Spectra. Plant Cell 2006, 18, 4–14. [Google Scholar] [CrossRef]
- Rohmer, T.; Strauss, H.; Hughes, J.; de Groot, H.; Gärtner, W.; Schmieder, P.; Matysik, J. 15N MAS NMR Studies of Cph1 Phytochrome: Chromophore Dynamics and Intramolecular Signal Transduction. J. Phys. Chem. B 2006, 110, 20580–20585. [Google Scholar] [CrossRef]
- Narikawa, R.; Ishizuka, T.; Muraki, N.; Shiba, T.; Kurisu, G.; Ikeuchi, M. Structures of Cyanobacteriochromes from Phototaxis Regulators AnPixJ and TePixJ Reveal General and Specific Photoconversion Mechanism. Proc. Natl. Acad. Sci. USA 2013, 110, 918–923. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Narikawa, R.; Ikeuchi, M.; Gärtner, W.; Matysik, J. Color Tuning in Red/Green Cyanobacteriochrome AnPixJ: Photoisomerization at ‘5 Causes an Excited-State Destabilization. J. Phys. Chem. B 2015, 119, 9688–9695. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.L.; Gutt, A.; Mechelke, J.; Raffelberg, S.; Tang, K.; Miao, D.; Valle, L.; Borsarelli, C.D.; Zhao, K.H.; Gärtner, W. Combined Mutagenesis and Kinetics Characterization of the Bilin-Binding GAF Domain of the Protein Slr1393 from the Cyanobacterium Synechocystis PCc6803. ChemBioChem 2014, 15, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
- Velazquez Escobar, F.; Utesch, T.; Narikawa, R.; Ikeuchi, M.; Mroginski, M.A.; Gärtner, W.; Hildebrandt, P. Photoconversion Mechanism of the Second GAF Domain of Cyanobacteriochrome AnPixJ and the Cofactor Structure of Its Green-Absorbing State. Biochemistry 2013, 52, 4871–4880. [Google Scholar] [CrossRef]
- Fukushima, Y.; Iwaki, M.; Narikawa, R.; Ikeuchi, M.; Tomita, Y.; Itoh, S. Photoconversion Mechanism of a Green/Red Photosensory Cyanobacteriochrome AnPixJ: Time-Resolved Optical Spectroscopy and FTIR Analysis of the AnPixJ-GAF2 Domain. Biochemistry 2011, 50, 6328–6339. [Google Scholar] [CrossRef] [PubMed]
- Song, C.; Velazquez Escobar, F.; Xu, X.L.; Narikawa, R.; Ikeuchi, M.; Siebert, F.; Gärtner, W.; Matysik, J.; Hildebrandt, P. A Red/Green Cyanobacteriochrome Sustains Its Color despite a Change in the Bilin Chromophores Protonation State. Biochemistry 2015, 54, 5839–5848. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.G.; Wiebeler, C.; Sen, S.; Cerutti, D.S.; Schapiro, I. Histidine Protonation Controls Structural Heterogeneity in the Cyanobacteriochrome AnPixJg2. bioRxiv 2020. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Phillips, J.C.; Braun, R.; Wang, W.; Gumbart, J.; Tajkhorshid, E.; Villa, E.; Chipot, C.; Skeel, R.D.; Kalé, L.; Schulten, K. Scalable Molecular Dynamics with NAMD. J. Comput. Chem. 2005, 26, 1781–1802. [Google Scholar] [CrossRef]
- MacKerell, A.D.; Bashford, D.; Bellott, M.; Dunbrack, R.L.; Evanseck, J.D.; Field, M.J.; Fischer, S.; Gao, J.; Guo, H.; Ha, S.; et al. All-Atom Empirical Potential for Molecular Modeling and Dynamics Studies of Proteins. J. Phys. Chem. B 1998, 102, 3586–3616. [Google Scholar] [CrossRef] [PubMed]
- Vanommeslaeghe, K.; Hatcher, E.; Acharya, C.; Kundu, S.; Zhong, S.; Shim, J.; Darian, E.; Guvench, O.; Lopes, P.; Vorobyov, I.; et al. CHARMM General Force Field: A Force Field for Drug-like Molecules Compatible with the CHARMM All-Atom Additive Biological Force Fields. J. Comput. Chem. 2010, 31, 671–690. [Google Scholar] [CrossRef] [PubMed]
- Feller, S.E.; Zhang, Y.; Pastor, R.W.; Brooks, B.R. Constant Pressure Molecular Dynamics Simulation: The Langevin Piston Method. J. Chem. Phys. 1995, 103, 4613–4621. [Google Scholar] [CrossRef]
- Nosé, S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J. Chem. Phys. 1984, 81, 511–519. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- The CP2K Developers Group. 2001–2020. Available online: www.cp2k.org (accessed on 20 November 2020).
- Schiffmann, C.; Sebastiani, D. Artificial Bee Colony Optimization of Capping Potentials for Hybrid Quantum Mechanical/Molecular Mechanical Calculations. J. Chem. Theory Comput. 2011, 7, 1307–1315. [Google Scholar] [CrossRef]
- Ihrig, A.C.; Schiffmann, C.; Sebastiani, D. Specific Quantum Mechanical/Molecular Mechanical Capping-Potentials for Biomolecular Functional Groups. J. Chem. Phys. 2011, 135, 214107. [Google Scholar] [CrossRef]
- Becke, A.D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- VandeVondele, J.; Krack, M.; Mohamed, F.; Parrinello, M.; Chassaing, T.; Hutter, J. QUICKSTEP: Fast and Accurate Density Functional Calculations Using a Mixed Gaussian and Plane Waves Approach. Comput. Phys. Commun. 2005, 167, 103–128. [Google Scholar] [CrossRef]
- Goedecker, S.; Teter, M.; Hutter, J. Separable Dual-Space Gaussian Pseudopotentials. Phys. Rev. B 1996, 54, 1703–1710. [Google Scholar] [CrossRef] [PubMed]
- Hartwigsen, C.; Goedecker, S.; Hutter, J. Relativistic Separable Dual-Space Gaussian Pseudopotentials from H to Rn. Phys. Rev. B 1998, 58, 3641–3662. [Google Scholar] [CrossRef]
- Krack, M. Pseudopotentials for H to Kr Optimized for Gradient-Corrected Exchange-Correlation Functionals. Theor. Chem. Acc. 2005, 114, 145–152. [Google Scholar] [CrossRef]
- Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An Efficient Real Space Multigrid QM/MM Electrostatic Coupling. J. Chem. Theory Comput. 2005, 1, 1176–1184. [Google Scholar] [CrossRef] [PubMed]
- Laino, T.; Mohamed, F.; Laio, A.; Parrinello, M. An Efficient Linear-Scaling Electrostatic Coupling for Treating Periodic Boundary Conditions in QM/MM Simulations. J. Chem. Theory Comput. 2006, 2, 1370–1378. [Google Scholar] [CrossRef]
- Tuckerman, M.E. Statistical Mechanics: Theory and Molecular Simulation; Oxford University Press Inc.: New York, NY, USA, 2010. [Google Scholar]
- Weber, V.; Iannuzzi, M.; Giani, S.; Hutter, J.; Declerck, R.; Waroquier, M. Magnetic Linear Response Properties Calculations with the Gaussian and Augmented-Plane-Wave Method. J. Chem. Phys. 2009, 131, 014106. [Google Scholar] [CrossRef]
- Jensen, F. Polarization Consistent Basis Sets: Principles. J. Chem. Phys. 2001, 115, 9113–9125. [Google Scholar] [CrossRef]
- Keith, T.A.; Bader, R.F. Calculation of Magnetic Response Properties Using a Continuous Set of Gauge Transformations. Chem. Phys. Lett. 1993, 210, 223–231. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020; Available online: https://www.r-project.org/ (accessed on 20 November 2020).
- Gowers, R.J.; Linke, M.; Barnoud, J.; Reddy, T.J.E.; Melo, M.N.; Seyler, S.L.; Domański, J.; Dotson, D.L.; Buchoux, S.; Kenney, I.M.; et al. MDAnalysis: A Python Package for the Rapid Analysis of Molecular Dynamics Simulations. In Proceedings of the 15th Python in Science Conference, Austin, TX, USA, 11–17 July 2016; pp. 98–105. Available online: http://conference.scipy.org/proceedings/scipy2016/pdfs/proceedings.pdf (accessed on 20 November 2020). [CrossRef]
- Michaud-Agrawal, N.; Denning, E.J.; Woolf, T.B.; Beckstein, O. MDAnalysis: A Toolkit for the Analysis of Molecular Dynamics Simulations. J. Comput. Chem. 2011, 32, 2319–2327. [Google Scholar] [CrossRef]
- Oliphant, T.E. Python for Scientific Computing. Comput. Sci. Eng. 2007, 9, 10–20. [Google Scholar] [CrossRef]
- Millman, K.J.; Aivazis, M. Python for Scientists and Engineers. Comput. Sci. Eng. 2011, 13, 9–12. [Google Scholar] [CrossRef]
- Perez, F.; Granger, B.E. IPython: A System for Interactive Scientific Computing. Comput. Sci. Eng. 2007, 9, 21–29. [Google Scholar] [CrossRef]
- Python Software Foundation, Version 3.6. Available online: www.python.org (accessed on 20 November 2020).
- Harris, C.R.; Jarrod Millman, K.; van der Walt, S.J.; Gommers, R.; Virtanen, P.; Cournapeau, D.; Wieser, E.; Taylor, J.; Berg, S.; Smith, N.J.; et al. Array programming with NumPy. Nature 2020, 585, 357–362. [Google Scholar] [CrossRef]
- Virtanen, P.; Gommers, R.; Oliphant, T.E.; Haberland, M.; Reddy, T.; Cournapeau, D.; Burovski, E.; Peterson, P.; Weckesser, W.; Bright, J.; et al. SciPy 1.0: Fundamental algorithms for scientific computing in Python. Nat. Methods 2020, 17, 261–272. [Google Scholar] [CrossRef]
- Hunter, J.D. Matplotlib: A 2D Graphics Environment. Comput. Sci. Eng. 2007, 9, 90–95. [Google Scholar] [CrossRef]
- Stone, J. Tachyon. An Efficient Library for Parallel Ray Tracing and Animation. Available online: jedi.ks.uiuc.edu/~johns/raytracer (accessed on 20 November 2020).
- Chemfig—Draw Molecules with Easy Syntax, Version 1.56. 2010–2020 Christian Tellechea. Available online: https://ctan.org/tex-archive/macros/generic/chemfig (accessed on 20 November 2020).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C–C | C–C | C–C | C–C | C–C | C–C | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| C | C | C | C | C | C | |||||||
| (P–P) * | ||||||||||||
| (P–P) * | ||||||||||||
| (P–P) * | ||||||||||||
| (P–P) * | ||||||||||||
| d(P) ** | 143 | 144 | 137 | 148 | 137 | 149 | ||||||
| (P–P) ** | ||||||||||||
| (P–P) ** | ||||||||||||
| (P–P) ** | ||||||||||||
| (P–P) ** |
| C–C | C–C | C–C | C–C | C–C | C–C | |
|---|---|---|---|---|---|---|
| double | single | double | (single) | single | double | |
| single | double | single | (single) | double | single |
| Parameter | FFMD | AIMD | NMR |
|---|---|---|---|
| Ensemble | NPT | NVT | NVT |
| No. of atoms | 37,651/104,960 * | ≈100 | ≈280 |
| Temperature | 300 K | 300 K | – |
| Pressure | 1.01325 bar | – | – |
| Barostat | Langevin | – | – |
| Thermostat | Nosé–Hoover | Nosé–Hoover | – |
| ABC (Å) | – ** | 30.0, 30.0, 30.0 | 35.0, 35.0, 35.0 |
| Force Field/Functional | CHARMM22 | BLYP-D3 | BLYP-D3 |
| Basis Set | – | TZVP-GTH | pcS2/3 |
| Density Cutoff | – | 320 Ry | 400 Ry |
| No. of Snapshots | – | – | 405 |
| / | ≈1 s | >20 ps | – |
| Other | SHAKE [81] |
| AnPixJg2 | Cph12 | ||||||
|---|---|---|---|---|---|---|---|
| Residue | AIMD | NMR | Capping Atom | Residue | AIMD | NMR | Capping Atom |
| PCB | x | x | – | PCB | x | x | – |
| Trp289 | x | x | CA | Asp207 | x | x | CB/CA ** |
| Asp291 | x | x | CA | Ile208 | x | x | C/C,CB ** |
| His293 | – | x | CA | Arg222 | – | x | CB |
| Arg301 | – | x | CA | Arg254 | – | x | CB |
| Tyr302 | – | x | CA | Ala256 | – | x | CA |
| Phe308 | – | x | CA | Tyr257 | – | x | – |
| His318 | – | x | CA | His258 | x | x | CA/CB ** |
| Phe319 | – | x | CA | Cys259 | x | x | – |
| Ser320 | x | x | CA | His260 | x | x | C |
| Cys321 | x | x | – | Tyr263 | – | x | CA |
| His322 | x | x | C | His290 | – | x | CA |
| Tyr334 | – | x | CA | Water * | x | x | – |
| Water * | – | x | – | ||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jähnigen, S.; Sebastiani, D. Carbon Atoms Speaking Out: How the Geometric Sensitivity of 13C Chemical Shifts Leads to Understanding the Colour Tuning of Phycocyanobilin in Cph1 and AnPixJ. Molecules 2020, 25, 5505. https://doi.org/10.3390/molecules25235505
Jähnigen S, Sebastiani D. Carbon Atoms Speaking Out: How the Geometric Sensitivity of 13C Chemical Shifts Leads to Understanding the Colour Tuning of Phycocyanobilin in Cph1 and AnPixJ. Molecules. 2020; 25(23):5505. https://doi.org/10.3390/molecules25235505
Chicago/Turabian StyleJähnigen, Sascha, and Daniel Sebastiani. 2020. "Carbon Atoms Speaking Out: How the Geometric Sensitivity of 13C Chemical Shifts Leads to Understanding the Colour Tuning of Phycocyanobilin in Cph1 and AnPixJ" Molecules 25, no. 23: 5505. https://doi.org/10.3390/molecules25235505
APA StyleJähnigen, S., & Sebastiani, D. (2020). Carbon Atoms Speaking Out: How the Geometric Sensitivity of 13C Chemical Shifts Leads to Understanding the Colour Tuning of Phycocyanobilin in Cph1 and AnPixJ. Molecules, 25(23), 5505. https://doi.org/10.3390/molecules25235505