
Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
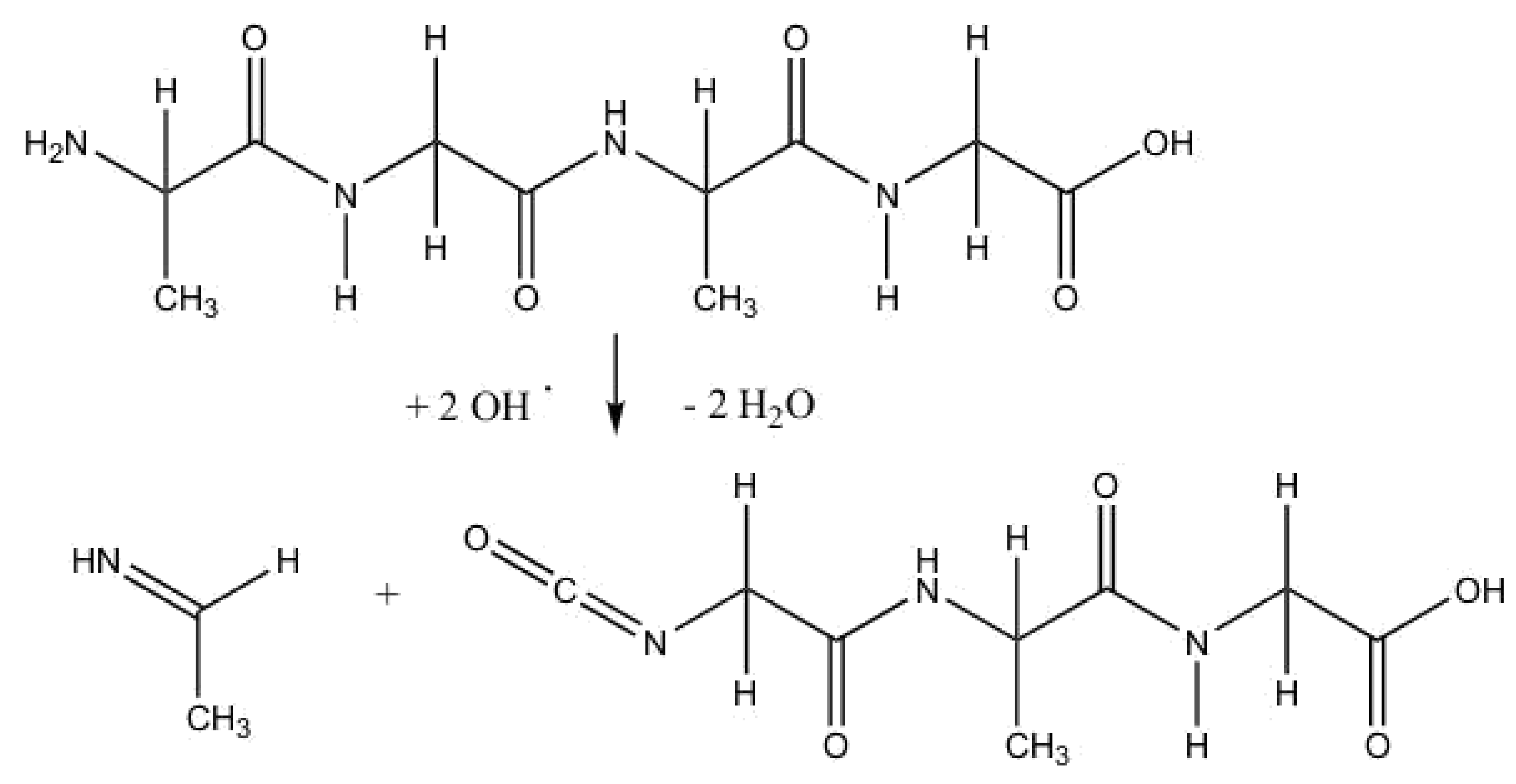{kind=link}
Abstract
1. Introduction
2. Results and Discussion
2.1. Anodic Reaction of Amino Acids
2.2. Anodic Reaction of Peptides
3. Methods
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Holleman, A.F.; Wiberg, E. Lehrbuch der Anorganischen Chemie, 101st ed.; De Gruyter: Berlin, Germany, 1749. [Google Scholar]
- Sternberg, A.; Bardow, A. Power-to-What?–Environmental assessment of energy storage systems. Energy Environ. Sci. 2015, 8, 389–400. [Google Scholar] [CrossRef]
- Car, R.; Parrinello, M. Unified Approach for Molecular Dynamics and Density-Functional Theory. Phys. Rev. Lett. 1985, 55, 2471–2474. [Google Scholar] [CrossRef] [PubMed]
- Hohenberg, P.; Kohn, W. Inhomogeneous electron gas. Phys. Rev. 1964, 136, 864–871. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, 1133–1138. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Marx, D.; Hutter, J. Modern Methods and Algorithms of Quantum Chemistry. In Ab Initio Molecular Dynamics: Theory and Implementation; Grotendorst, J., Ed.; John von Neumann Institute for Computing: New York, NY, USA, 2000; Volume 1, pp. 301–449. [Google Scholar]
- Van Duin, A.C.T.; Dasgupta, S.; Lorant, F.; Iii, W.A.G. ReaxFF: A Reactive Force Field for Hydrocarbons. J. Phys. Chem. 2001, 105, 9396–9409. [Google Scholar] [CrossRef]
- Chenoweth, K.; Van Duin, A.C.T.; Goddard, W.A. ReaxFF Reactive Force Field for Molecular Dynamics Simulations of Hydrocarbon Oxidation. J. Phys. Chem. A 2008, 112, 1040–1053. [Google Scholar] [CrossRef]
- Alonso, J.L.; Andrade, X.; Echenique, P.; Falceto, F.; Prada-Gracia, D.; Rubio, A. Efficient formalism for large-scale ab initio molecular dynamics based on time-dependent density funtional theory. Chem. Phys. Lett. 2008, 101, 1–6. [Google Scholar]
- Parrinello, M. From silicon to RNA: The coming of age of ab initio molecular dynamics. Solid State Commun. 1997, 102, 107–120. [Google Scholar] [CrossRef]
- Fois, E.; Gamba, A.; Tabacchi, G. First-principles simulation of the intracage oxidation of nitrite to nitrate sodalite. Chem. Phys. Lett. 2000, 329, 1–6. [Google Scholar] [CrossRef]
- Boero, M.; Parrinello, M.; Terakura, K. First Principles Molecular Dynamics Study of Ziegler−Natta Heterogeneous Catalysis. J. Am. Chem. Soc. 1998, 120, 2746–2752. [Google Scholar] [CrossRef]
- Frank, I.; Parrinello, M.; Klamt, A. Insight into Chemical Reactions from First-Principles Simulations: The Mechanism of the Gas-Phase Reaction of OH Radicals with Ketones. J. Phys. Chem. 1998, 102, 3614–3617. [Google Scholar] [CrossRef]
- Reinhardt, S.; Marian, C.; Frank, I. The influence of excess ammonia on the mechanism of the reaction of boron trichloride with ammonia–An ab initio molecular dynamics study. Angew. Chem. 2001, 113, 3683–3685. [Google Scholar] [CrossRef]
- Nonnenberg, C.; Frank, I. Formation and decay of tetrazane derivates–a Car-Parrinello molecular dynamics study. Phys. Chem. Chem. Phys. 2008, 10, 4383–4392. [Google Scholar] [CrossRef] [PubMed]
- Hofbauer, F.; Frank, I. Electrolysis of Water in the Diffusion Layer: First-Principles Molecular Dynamics Simulation. Chem. A Eur. J. 2011, 18, 277–282. [Google Scholar] [CrossRef]
- Frank, I. Ab-Initio Molecular Dynamics Simulation of the Electrolysis of Waste Water. ChemistrySelect 2019, 4, 4376–4381. [Google Scholar] [CrossRef]
- Blumberger, J.; Bernasconi, L.; Tavernelli, I.; Vuilleumier, R.; Sprik, M. Electronic structure and solvation of copper and silver ions: A theoretical picture of a model aqueous redox reaction. J. Am. Chem. Soc. 2004, 126, 3928–3938. [Google Scholar] [CrossRef]
- Blumberger, J.; Tateyama, Y.; Sprik, M. Ab initio molecular dynamics simulation of redox reactions in solution. Comput. Phys. Commun. 2005, 169, 256–261. [Google Scholar] [CrossRef]
- Zhang, C.; Sayer, T.; Hutter, J.; Sprik, M. Modelling electrochemical systems with finite field molecular dynamics. J. Phys. Energy 2020, 2, 032005. [Google Scholar] [CrossRef]
- Tomilov, A.P.; Fioshin, M.Y. Free radical reactions in the electrolysis of organic compounds. Russ. Chem. Rev. 1963, 32, 30–44. [Google Scholar] [CrossRef]
- Wiebe, A.; Gieshoff, T.; Möhle, S.; Rodrigo, E.; Zirbes, M.; Waldvogel, S.R. Electrifying organic synthesis. Angew. Chem. Int. Ed. 2018, 57, 5594–5619. [Google Scholar] [CrossRef] [PubMed]
- CPMD. Copyright IBM Corp, 1990–2008; Copyright MPI für Festkörperforshung Stuttgart 1997–2001. Available online: http://www.cpmd.org/ (accessed on 16 January 2012).
- Grimme, S. Semiempirical GGA-type density functional constructed with a long-range dispersion correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Troullier, N.; Martins, J.L. Efficient pseudopotentials for plane-wave calculations. Phys. Rev. B 1991, 43, 1993–2006. [Google Scholar] [CrossRef] [PubMed]
- Boero, M.; Parrinello, M.; Terakura, K.; Weiss, H. Car—Parrinello study of Ziegler—Natta heterogeneous catalysis: Stability and destabilization problems of the active site models. Mol. Phys. 2002, 100, 2935–2940. [Google Scholar] [CrossRef]
- Gunnarsson, O.; Lundqvist, B.I. Exchange and correlation in atoms, molecules, and solids by the spin-density-functional formalism. Phys. Rev. B 1976, 13, 4274–4298. [Google Scholar] [CrossRef]
- Harada, K.; Suzuki, S.; Harada, S.S.K. Formation of amino acids from elemental carbon by contact glow discharge electrolysis. Nat. Cell Biol. 1977, 266, 275–276. [Google Scholar] [CrossRef]
- Harada, K.; Nomoto, M.M.; Gunji, H. Formation of amino acids from aliphatic amines by contact glow discharge electrolysis. Tetrahedron Lett. 1981, 22, 769–772. [Google Scholar] [CrossRef]
- Gupta, S.K.S. Contact Glow Discharge Electrolysis: A Novel Tool for Manifold Applications. Plasma Chem. Plasma Process. 2017, 37, 897–945. [Google Scholar] [CrossRef]







Sample Availability: Samples of the compounds are not available from the authors. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kiakojouri, A.; Nadimi, E.; Frank, I. Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides. Molecules 2020, 25, 5415. https://doi.org/10.3390/molecules25225415
Kiakojouri A, Nadimi E, Frank I. Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides. Molecules. 2020; 25(22):5415. https://doi.org/10.3390/molecules25225415
Chicago/Turabian StyleKiakojouri, Ali, Ebrahim Nadimi, and Irmgard Frank. 2020. "Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides" Molecules 25, no. 22: 5415. https://doi.org/10.3390/molecules25225415
APA StyleKiakojouri, A., Nadimi, E., & Frank, I. (2020). Ab-Initio Molecular Dynamics Simulation of Condensed-Phase Reactivity: The Electrolysis of Amino Acids and Peptides. Molecules, 25(22), 5415. https://doi.org/10.3390/molecules25225415