Synthesis of Polycyclic Ether-Benzopyrans and In Vitro Inhibitory Activity against Leishmania tarentolae

 and
and
Abstract
1. Introduction
2. Results and Discussion
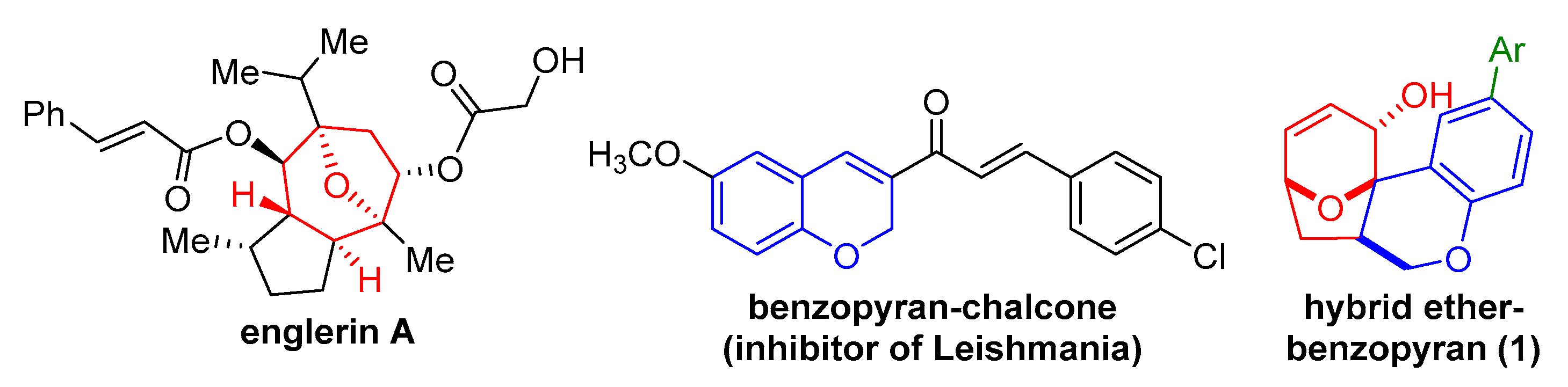
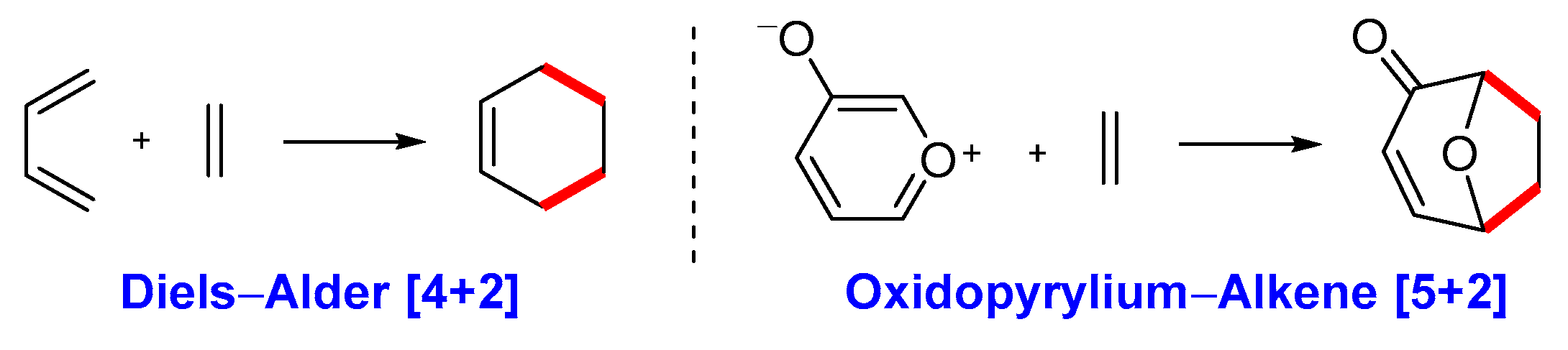
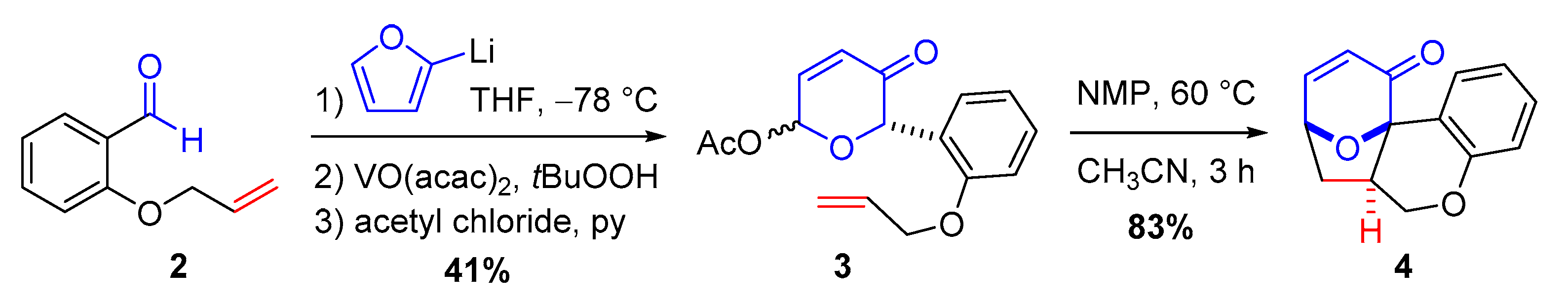
2.1. Previous Synthesis of Polycyclic Ether-Benzopyran
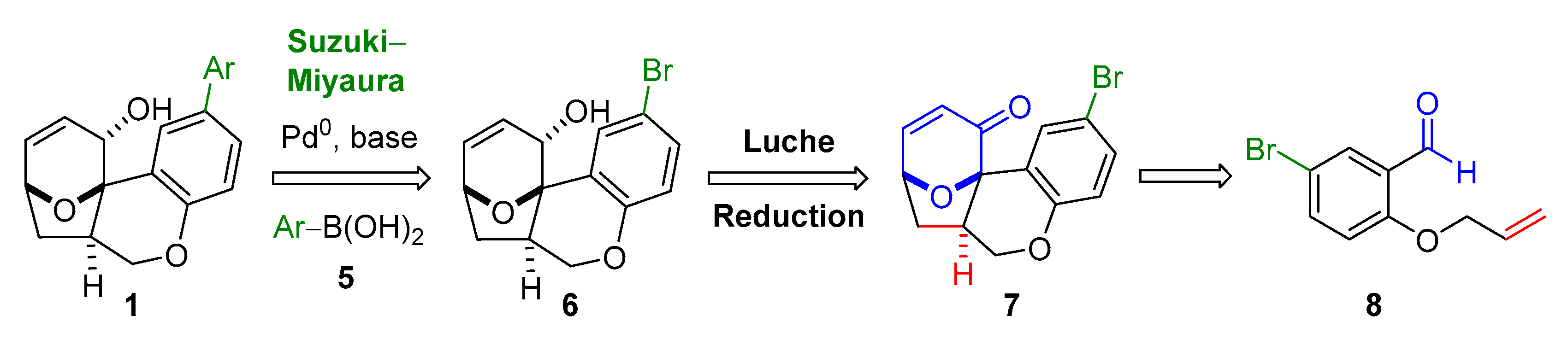
2.2. Proposed Suzuki-Miyaura Pathway toward Construction of Ether-Benzopyran Analogues
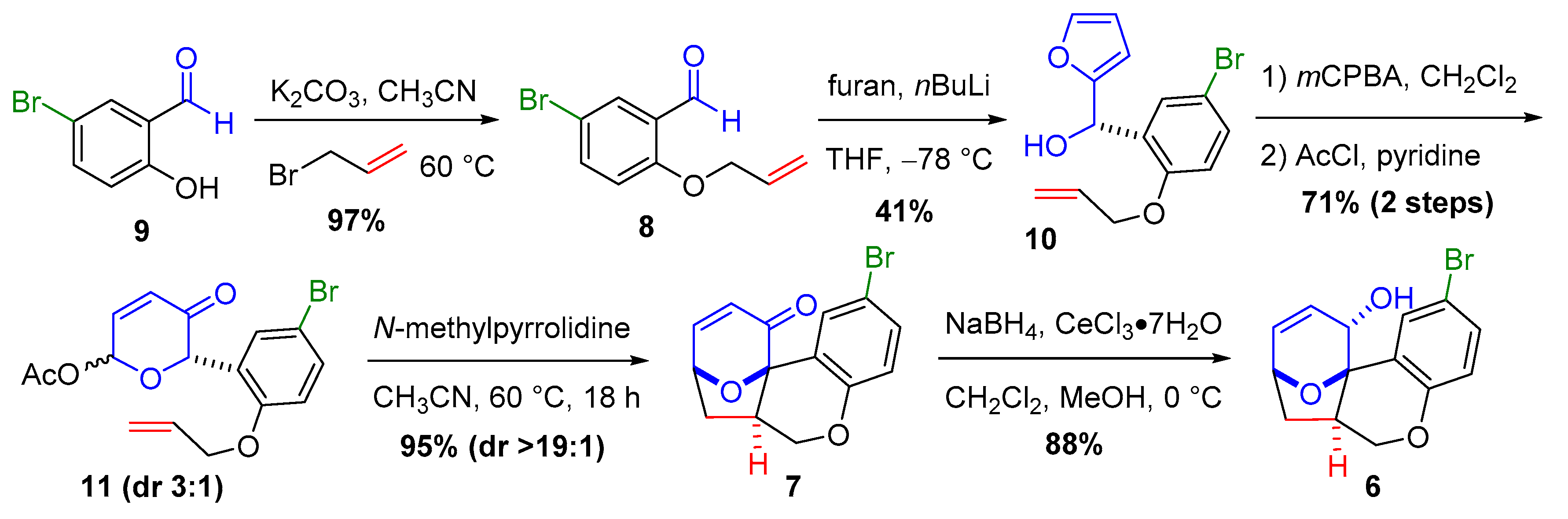
2.3. Construction of the Suzuki-Miyaura Aryl Bromide Precursor
2.4. Palladium Catalyzed Suzuki-Miyaura Cross-Coupling
2.5. Biological Investigations
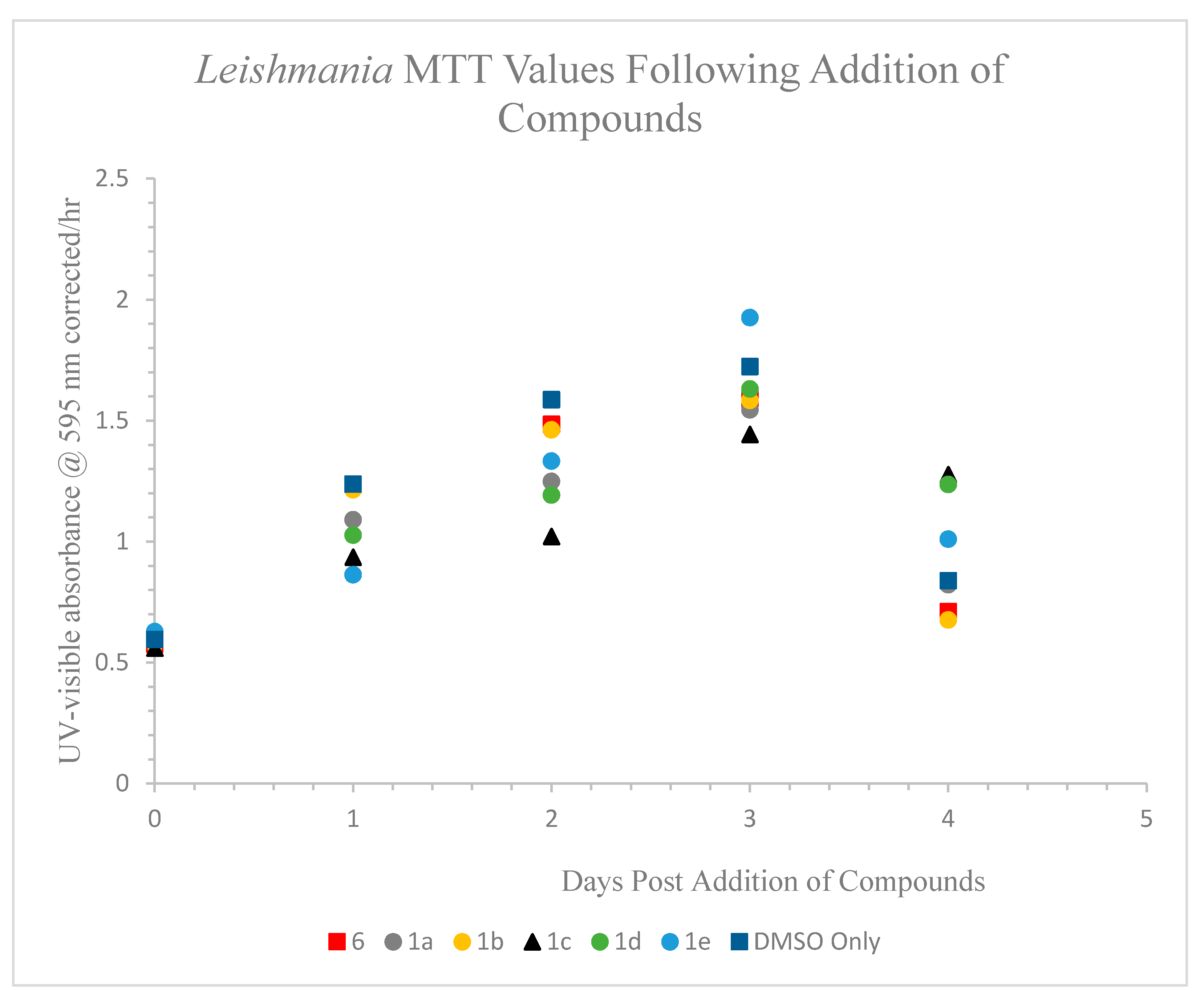
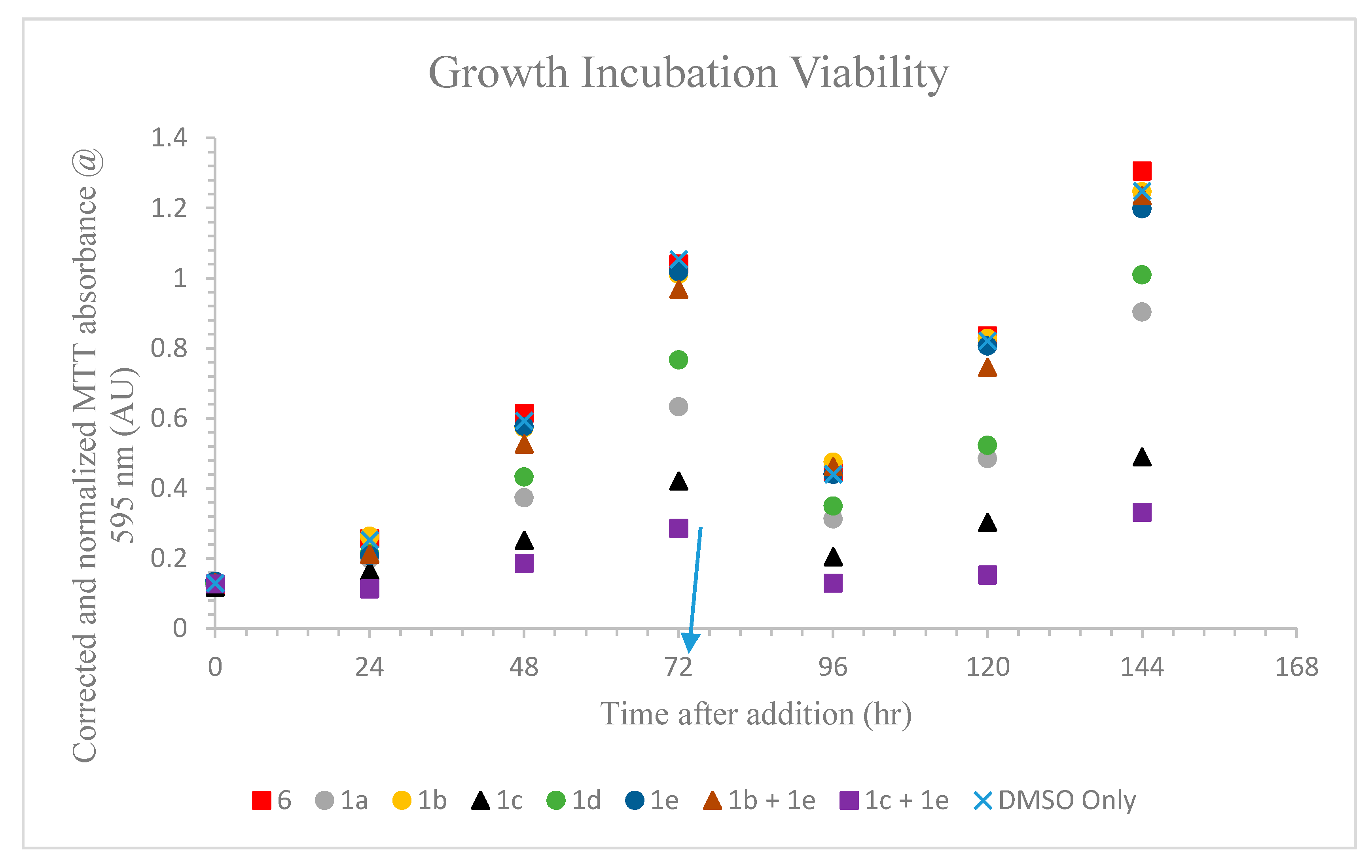
2.5.1. Incubation Effects on L. tarentolae Growth
2.5.2. Test Compound Inhibition and Apparent Recovery by Leishmania after Transfer to New Medium
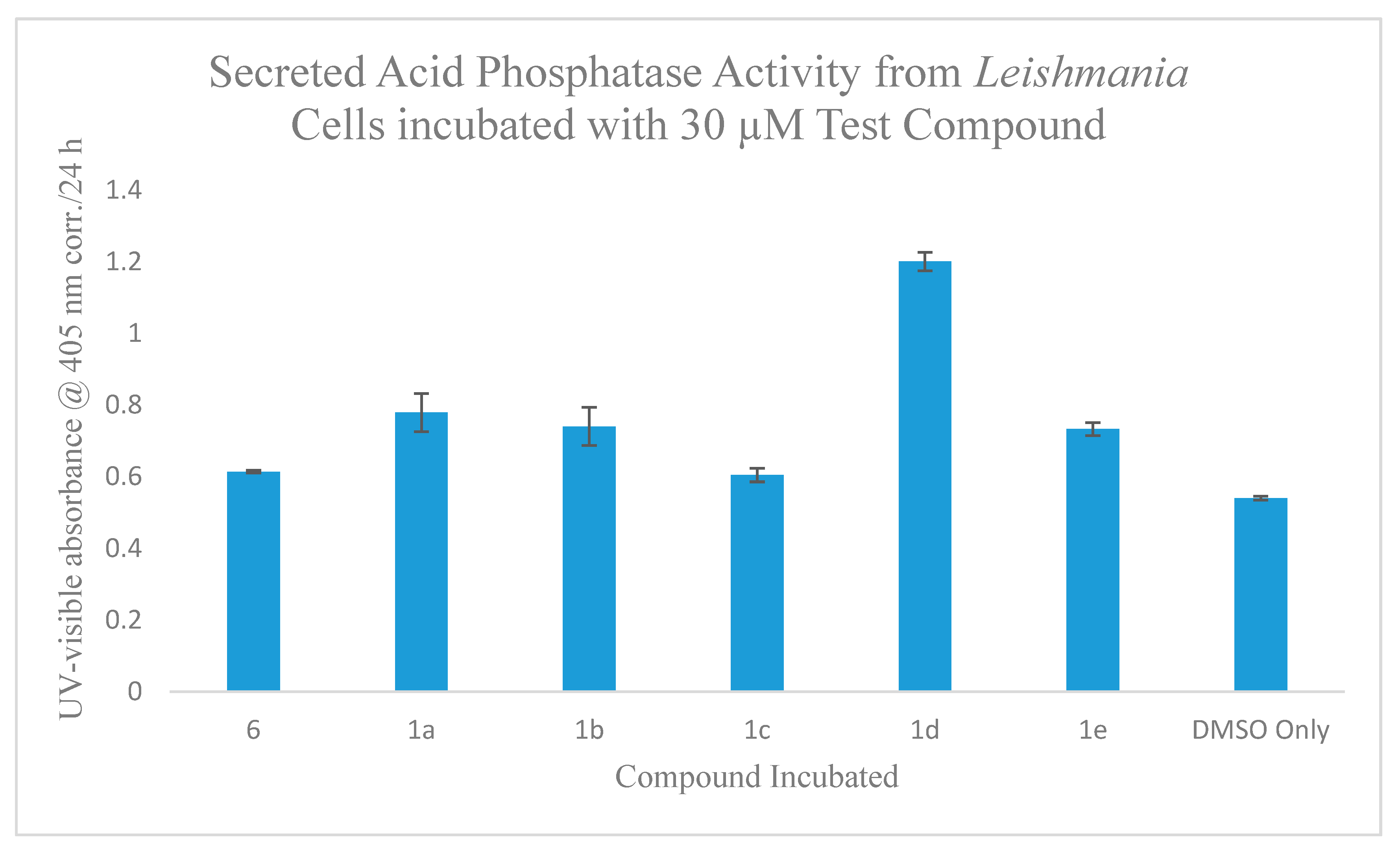
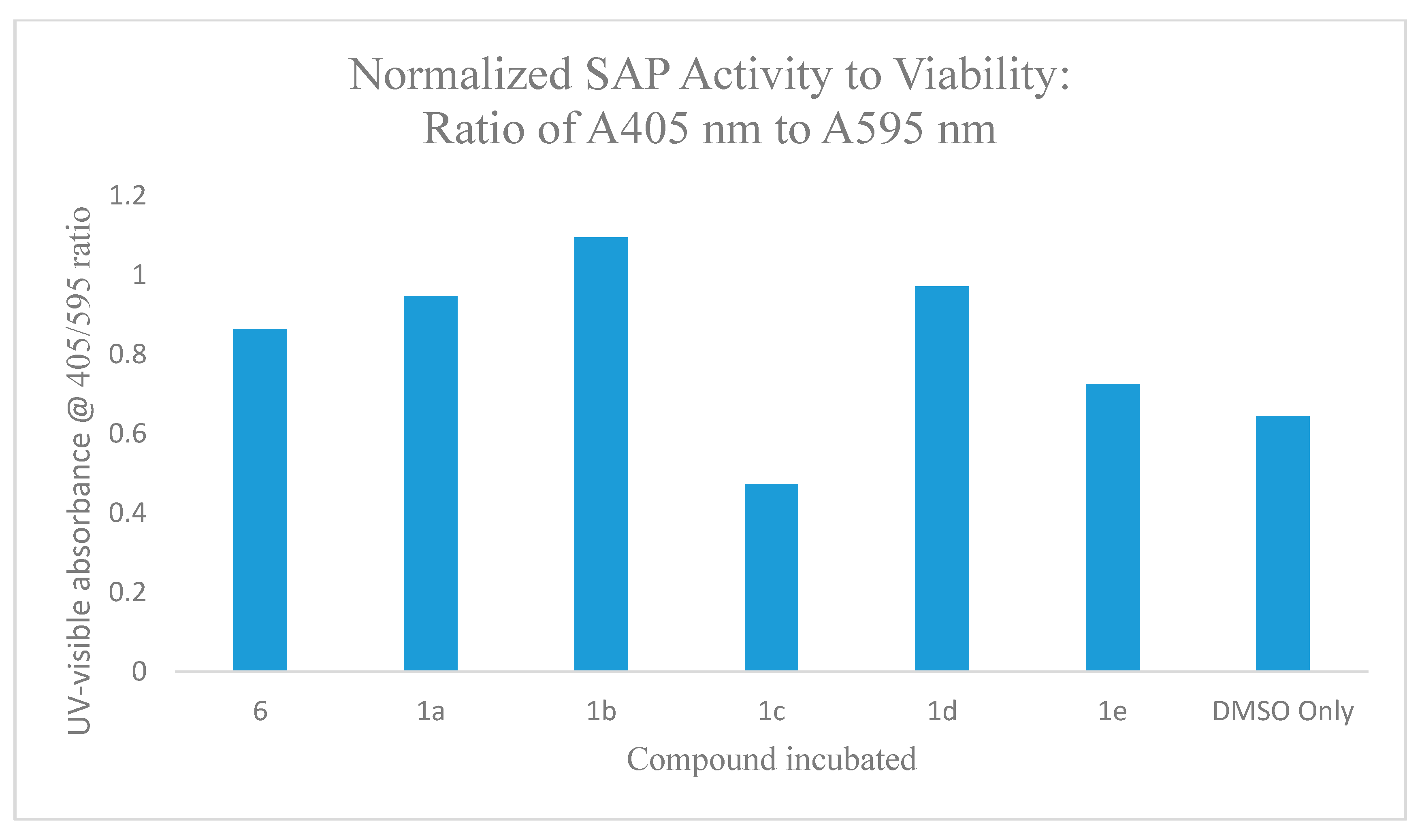
2.5.3. Incubation Effects of Secreted Acid Phosphatase (SAP) Activity
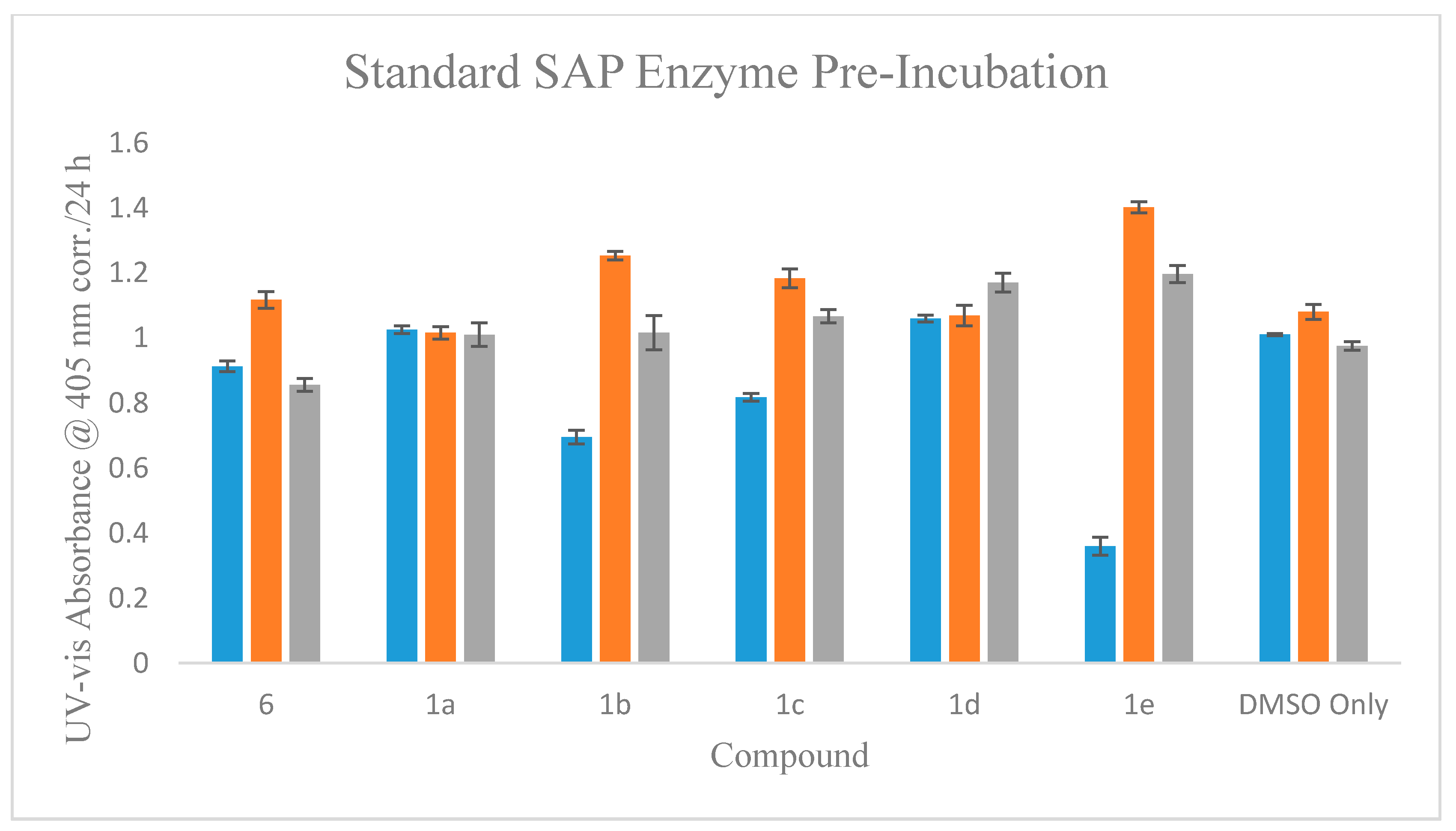
2.5.4. Standard SAP Direct Enzymic Effect Assay
2.5.5. Standard Leishmania SAP Enzyme Kinetic Assay
2.5.6. Detection of Leishmania Nitric Oxide Using a Specific Fluorescent Probe, DAF-FM
2.5.7. Cell Toxicity Screening: C6 Glial Cells
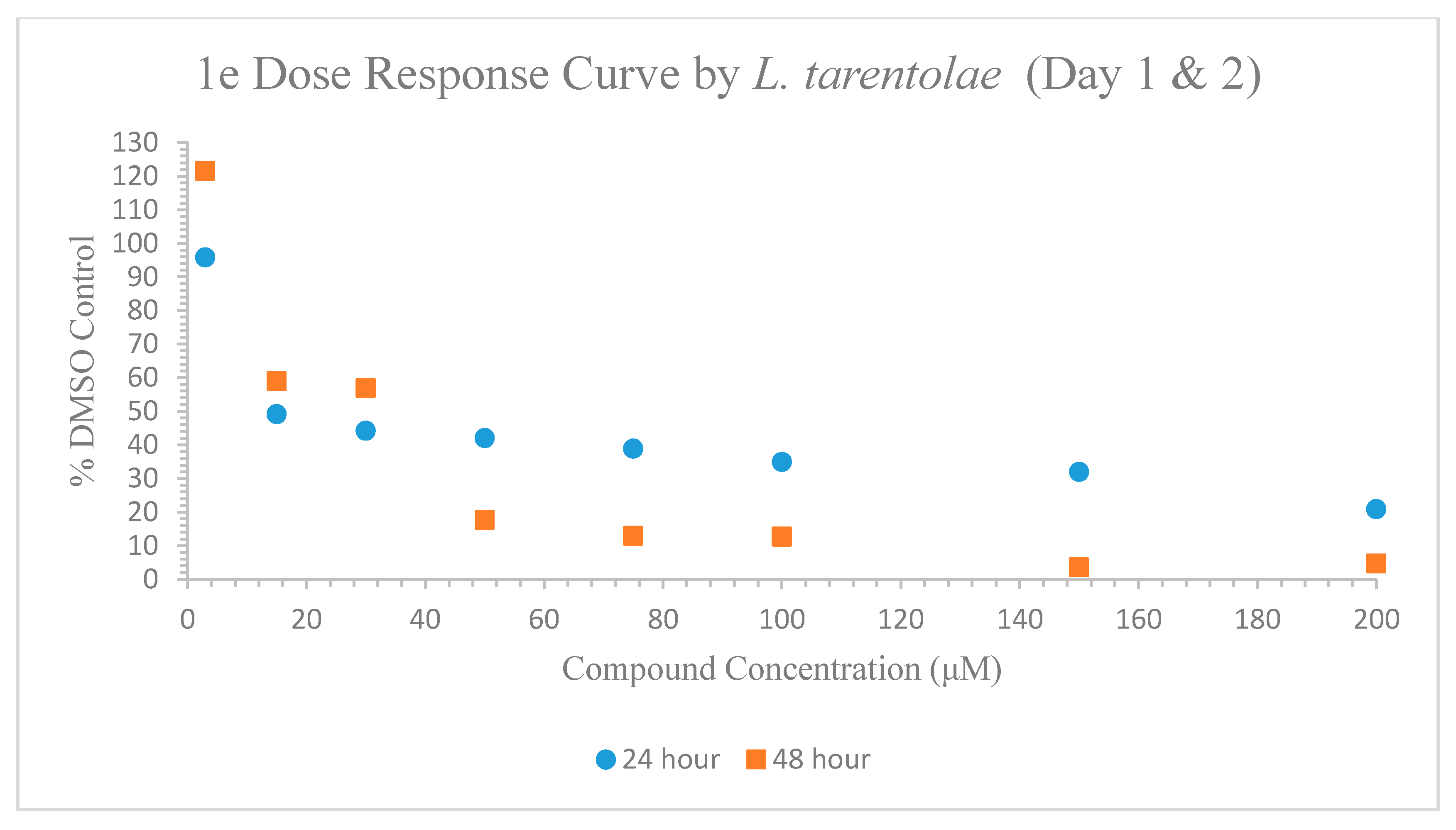
2.5.8. ED-50 (Effective Dose) Study on Leishmania
2.5.9. Photosensitivity study on Leishmania
3. Conclusions
4. Materials and Methods
4.1. General Methods—Organic Synthesis
4.2. Preparation of Aryl Bromide 6
4.3. Preparation of Biaryls 1a–e
4.4. Biological Methods
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Centers for Disease Control and Prevention (CDC). About Leishmaniasis. Available online: https://www.cdc.gov/parasites/leishmaniasis/gen_info/faqs.html (accessed on 17 October 2020).
- Burza, S.; Croft, A.I.; Boelaert, M. Leishmaniasis. Lancet 2020, 392, 951–970. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Epidemiology and Risk Factors. Available online: https://www.cdc.gov/parasites/leishmaniasis/epi.html (accessed on 17 October 2020).
- Torres-Guerrero, E.; Quintanilla-Cedillo, M.R.; Ruiz-Esmenjaud, J.; Arenas, R. Leishmaniasis: A review. F1000Research 2017, 6, 750. [Google Scholar] [CrossRef]
- Herrera, G.; Barragan, N.; Luna, N.; Martinez, D.; De Martino, F.; Medina, J.; Nino, S.; Paez, L.; Ramirez, A.; Vega, L.; et al. An interactive database of Leishmania species distribution in the Americas. Sci. Data 2020, 7, 110. [Google Scholar] [CrossRef]
- Clarke, C.F.; Bradley, K.K.; Wright, J.H.; Glowicz, J. Case Report: Emergence of Autochthonous Cutaneous Leishmaniasis in Northeastern Texas and Southeastern Oklahoma. Am. J. Trop. Med. Hyg. 2013, 88, 157–161. [Google Scholar] [CrossRef]
- McHugh, C.P.; Melby, P.C.; LaFon, S.G. Leishmaniasis in Texas: Epidemiology and clinical aspects of human cases. Am. J. Trop. Med. Hyg. 1996, 55, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Aronson, N.; Herwaldt, B.L.; Libman, M.; Pearson, R.; Lopez-Velez, R.; Weina, P.; Carvalho, E.M.; Ephros, M.; Jeronimo, S.; Magill, A. Diagnosis and Treatment of Leishmaniasis: Clinical Practice Guidelines by the Infectious Diseases Society of America (IDSA) and the American Society of Tropical Medicine and Hygiene (ASTMH). Clin. Inf. Dis. 2016, 63, e202–e264. [Google Scholar] [CrossRef] [PubMed]
- Centers for Disease Control and Prevention (CDC). Disease. Available online: https://www.cdc.gov/parasites/leishmaniasis/disease.html (accessed on 17 October 2020).
- Bekhit, A.A.; El-Agroudy, E.; Helmy, A.; Ibrahim, T.M.; Shavandi, A.; Bekhit, A.E.-D.A. Leishmania treatment and prevention: Natural and synthesized drugs. Eur. J. Med. Chem. 2018, 160, 229–244. [Google Scholar] [CrossRef] [PubMed]
- Nagle, A.S.; Khare, S.; Kumar, A.B.; Supek, F.; Buchynskyy, A.; Mathison, C.J.N.; Chennamaneni, N.K.; Pendem, N.; Buckner, F.S.; Gelb, M.H.; et al. Recent Developments in Drug Discovery for Leishmaniasis and Human African Trypanosomiasis. Chem. Rev. 2014, 114, 11305–11347. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention (CDC). Treatments. Available online: https://www.cdc.gov/parasites/leishmaniasis/health_professionals/index.html#tx (accessed on 17 October 2020).
- Ratnayake, R.; Covell, D.; Ransom, T.T.; Gustafson, K.R.; Beutler, J.A. Englerin A, a Selective Inhibitor of Renal Cancer Cell Growth, from Phyllanthus engleri. Org. Lett. 2008, 11, 57–60. [Google Scholar] [CrossRef]
- Pouwer, R.H.; Richard, J.-A.; Tseng, C.C.; Chen, D.Y.-K. Chemical Synthesis of the Englerins. Chem. Asian J. 2012, 7, 22–35. [Google Scholar] [CrossRef]
- Chain, W.J. Synthetic Strategies toward the Guaiane Sesquiterpene Englerin A. Synlett 2011, 2011, 2605–2608. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Kang, Q.; Ng, S.Y.; Chen, D.Y.-K. Total Synthesis of Englerin A. J. Am. Chem. Soc. 2010, 132, 8219–8222. [Google Scholar] [CrossRef] [PubMed]
- Sourbier, C.; Scroggins, B.T.; Ratnayake, R.; Prince, T.L.; Lee, S.; Lee, M.-J.; Nagy, P.L.; Lee, Y.H.; Trepel, J.B.; Buetler, J.A.; et al. Englerin A Stimulates PKCθ to Inhibit Insulin Signaling and to Simultaneously Activate HSF1: Pharmacologically Induced Synthetic Lethality. Cancer Cell 2013, 23, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Tran, K.D.; Sanchez, M.A.; Mezewghi, H.A.; Landfear, S.M. Glucose Transporters and Virulence in Leishmania Mexicana. mSphere 2018, 3, e00349-18. [Google Scholar] [CrossRef] [PubMed]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Roecker, A.J.; Cao, G.-Q.; Barluenga, S.; Mitchell, H.J. Natural Product-like Combinatorial Libraries based on Privileged Structures. 1. General Principles and Solid-Phase Synthesis of Benzopyrans. J. Am. Chem. Soc. 2000, 122, 9939–9953. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Roecker, A.J.; Barluenga, S.; Cao, G.-Q.; Affleck, R.L.; Lillig, J.E. Natural Product-like Combinatorial Libraries based on Privileged Structures. 2. Construction of a 10,000-Membered Benzopyran Library by Directed Split-and-Pool Chemistry Using NanoKans and Optical Encoding. J. Am. Chem. Soc. 2000, 122, 9954–9967. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Pfefferkorn, J.A.; Barluenga, S.; Mitchell, H.J.; Roecker, A.J.; Cao, G.-Q. Natural Product-like Combinatorial Libraries based on Privileged Structures. 3. The “Libraries from Libraries” Principle for Diversity Enhancement of Benzopyran Libraries. J. Am. Chem. Soc. 2020, 122, 9968–9976. [Google Scholar] [CrossRef]
- Duarte, C.D.; Barreiro, E.J.; Fraga, C.A.M. Privileged Structures: A Useful Concept for the Rational Design of New Lead Candidates. Mini Rev. Med. Chem. 2007, 7, 1108–1119. [Google Scholar] [CrossRef]
- Foroumadi, A.; Emami, S.; Sorkhi, M.; Nakhjiri, M.; Nazarian, Z.; Heydari, S.; Ardestani, S.K.; Poorrajab, F.; Shafiee, A. Chromene-Based Synthetic Chalcones as Potent Antileshmanial Agents: Synthesis and Biological Activity. Chem. Biol. Drug Des. 2010, 75, 590–596. [Google Scholar] [CrossRef]
- Lopez, S.P.; Yepes, L.M.; Perez-Castillo, Y.; Robledo, S.M.; de Sousa, D.P. Alkyl and Aryl Derivatives Based on p-Coumaric Acid Modification and Inhibitory Action against Leishmania braziliensis and Plasmodium falciparum. Molecules 2020, 25, 3178. [Google Scholar] [CrossRef]
- Klatt, S.; Simpson, L.; Maslov, D.A.; Konthur, Z. Leishmania tarentolae: Taxonomic classification and its application as a promising biotechnological expression host. PLoS Negl. Trop. Dis. 2019, 13, e0007424. [Google Scholar] [CrossRef] [PubMed]
- Taylor, V.M.; Muñoz, D.L.; Cedeño, D.L.; Vélez, I.D.; Jones, M.A.; Robledo, S.M. Leishmania tarentolae: Utility as an in vitro model for screening of antileishmanial agents. Exp. Parasitol. 2010, 126, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Belzowski, A.J.; Hooker, J.D.; Young, A.M.; Cedeño, D.L.; Peters, S.J.; Lash, T.D.; Jones, M.A. Nitric Oxide Production within Leishmania tarentolae Axenic Promastigotes and Amastigotes Is Induced by Carbaporphyrin Ketals. JSM Trop. Med. Res. 2016, 1, 1004. [Google Scholar]
- Dorsey, B.M.; Cass, C.L.; Cedeño, D.L.; Vallejo, R.; Jones, M.A. Effects of Specific Electric Field Stimulation on the Kinetics of Secreted Acid Phosphatases from Leishmania tarentolae and Implications for Therapy. Pathogens 2018, 7, 77. [Google Scholar] [CrossRef] [PubMed]
- Kunz, H.; Mullen, K. Natural Products and Materials Chemistry-Separated Forever? J. Am. Chem. Soc. 2013, 135, 8764–8769. [Google Scholar] [CrossRef] [PubMed]
- Kesavan, S.; Marcaurelle, L.A. Translational synthetic chemistry. Nat. Chem. Biol. 2013, 9, 210–213. [Google Scholar] [CrossRef]
- Hoffmann, R.W. Natural Products Synthesis: Changes over Time. Angew. Chem. Int. Ed. 2013, 52, 123–130. [Google Scholar] [CrossRef]
- Mohr, J.T.; Krout, M.R.; Stoltz, B.M. Natural products as inspiration for the development of asymmetric catalysis. Nature 2008, 455, 323–332. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The Evolving Role of Natural Products in Drug Discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef]
- Butler, M. The Role of Natural Product Chemistry in Drug Discovery. J. Nat. Prod. 2004, 67, 2141–2153. [Google Scholar] [CrossRef]
- Lesney, M.S. Nature’s Pharmaceuticals. Today’s Chemist at Work, July 2004; 26–32. Available online: http://www.scripps.edu/baran/pdfExtras/bothernp.pdf (accessed on 17 October 2020).
- MacCoss, M.; Baillie, T.A. Organic Chemistry in Drug Discovery. Science 2004, 303, 1810–1813. [Google Scholar] [CrossRef] [PubMed]
- Alvim-Gaston, M.; Grese, T.; Mahoui, A.; Palkowitz, A.D.; Pineiro-Nunez, M.; Watson, I. Open-Innovation Drug Discovery (OIDD): A Potential Path to Novel Therapeutic Chemical Space. Curr. Top. Med. Chem. 2014, 14, 294–303. [Google Scholar] [CrossRef] [PubMed]
- O’Conner, C.J.; Beckmann, H.S.G.; Spring, D.R. Diversity-oriented synthesis: Producing chemical tools for dissecting biology. Chem. Soc. Rev. 2012, 41, 4444–4456. [Google Scholar] [CrossRef] [PubMed]
- Anagnostaki, E.E.; Zografos, A.L. Common synthetic scaffolds in the synthesis of structurally diverse natural products. Chem. Soc. Rev. 2012, 41, 5613–5625. [Google Scholar] [CrossRef]
- Dandapani, S.; Marcaurelle, L.A. Accessing new chemical space for ‘undruggable’ targets. Nat. Chem. Biol. 2010, 6, 861–863. [Google Scholar] [CrossRef]
- Spandl, R.J.; Diaz-Gavilan, M.; O’Connell, K.M.G.; Thomas, G.L.; Spring, D.R. Diversity-Oriented Synthesis. Chem. Rec. 2008, 8, 129–142. [Google Scholar] [CrossRef]
- Tan, D.S. Diversity-oriented synthesis: Exploring the intersections between chemistry and biology. Nat. Chem. Biol. 2005, 1, 74–84. [Google Scholar] [CrossRef]
- Ulaczyk-Lesanko, A.; Hall, D.G. Wanted: New multicomponent reactions for generating libraries of polycyclic natural products. Curr. Opin. Chem. Biol. 2005, 9, 266–276. [Google Scholar] [CrossRef]
- Burke, M.; Schreiber, S.L. A Planning Strategy for Diversity-Oriented Synthesis. Angew. Chem. Int. Ed. 2004, 43, 46–58. [Google Scholar] [CrossRef]
- Schreiber, S. Target-Oriented and Diversity-Oriented Organic Synthesis in Drug Discovery. Science 2000, 287, 1964–1969. [Google Scholar] [CrossRef]
- Dakas, P.-Y.; Parga, J.A.; Hoing, S.; Scholer, H.R.; Sterneckert, J.; Kumar, K.; Waldmann, H. Discovery of Neuritogenic Compound Classes Inspired by Natural Products. Angew. Chem. Int. Ed. 2013, 52, 9576–9581. [Google Scholar] [CrossRef] [PubMed]
- Voigt, T.; Gerding-Reimers, C.; Tran, T.T.N.; Bergmann, S.; Lachance, H.; Scholermann, B.; Brockmeyer, A.; Janning, P.; Ziegler, S.; Waldmann, H. A Natural Product Inspired Tetrahydropyran Collection Yields Mitosis Modulators that Synergistically Target CSE1L and Tubulin. Angew. Chem. Int. Ed. 2012, 51, 410–414. [Google Scholar]
- Heidebrecht, R.W., Jr.; Mulrooney, C.; Austin, C.P.; Barker, R.H., Jr.; Beaudoin, J.A.; Cheng, K.C.-C.; Comer, E.; Dandapani, S.; Dick, J.; Duvall, J.R.; et al. Diversity-Oriented Synthesis Yields a Novel Lead for the Treatment of Malaria. ACS Med. Chem. Lett. 2012, 3, 112–117. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.B., III; Kim, W.-S. Diversity-oriented synthesis leads to an effective class of bifunctional linchpins uniting anion relay chemistry (ARC) with benzyne reactivity. Proc. Natl. Acad. Sci. USA 2011, 108, 6787–6792. [Google Scholar] [CrossRef] [PubMed]
- Morton, D.; Leach, S.; Cordier, C.; Warriner, S.; Nelson, A. Synthesis of Natural-Product-Like Molecules with Eighty Distinct Scaffolds. Angew. Chem. Int. Ed. 2009, 48, 104–109. [Google Scholar] [CrossRef]
- Itami, K.; Kamei, T.; Yoshida, J. Diversity-Oriented Synthesis of Tamifoxen-type Tetrasubstituted Olefins. J. Am. Chem. Soc. 2003, 125, 14670–14671. [Google Scholar] [CrossRef]
- Cordier, C.; Morton, D.; Murrison, S.; Nelson, A.; O’Leary-Steele, C. Natural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries. Nat. Prod. Rep. 2008, 25, 719–737. [Google Scholar] [CrossRef]
- Nishiwaki, N. Methods and Applications of Cycloaddition Reactions in Organic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Kobayashi, S.; Jørgensen, K.A. Cycloaddition Reactions in Organic Chemistry; Wiley-VCH: Weinheim, Germany, 2002. [Google Scholar]
- Carruthers, W. Cycloaddition Reactions in Organic Chemistry, Volume 8 (Tetrahedron Organic Chemistry); Pergamon: Oxford, UK, 1990. [Google Scholar]
- Diels, O.; Alder, K. Synthesen in der hydroaromatischen Reihe. Justus Liebigs Ann. Chem. 1928, 460, 98–122. [Google Scholar] [CrossRef]
- Nicolaou, K.C.; Snyder, S.A.; Montagnon, T.; Vassilikogiannakis, G. The Diels-Alder Reaction in Total Synthesis. Angew. Chem. Int. Ed. 2002, 41, 1668–1698. [Google Scholar] [CrossRef]
- Corey, E.J. Catalytic Enantioselective Diels-Alder Reactions: Methods, Mechanistic Fundamentals, Pathways, and Applications. Angew. Chem. Int. Ed. 2002, 41, 1650–1667. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in the [5+2] Cycloaddition. Adv. Synth. Catal. 2018, 360, 1551–1583. [Google Scholar] [CrossRef]
- Pellissier, H. Recent Developments in the [5+2] Cycloaddition. Adv. Synth. Catal. 2011, 353, 189–218. [Google Scholar] [CrossRef]
- Nakata, T. Total Synthesis of Marine Polycyclic Ethers. Chem. Rev. 2005, 105, 4314–4317. [Google Scholar] [CrossRef]
- Bejcek, L.P.; Murelli, R.P. Oxidopyrylium [5+2] cycloaddition chemistry: Historical Perspective and recent advances (2008–2018). Tetrahedron 2018, 74, 2501–2521. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.; Murali Krishna, U.; Trivedi, G.K. Cycloaddition of oxidopyrylium species in organic synthesis. Tetrahedron 2008, 64, 3405–3428. [Google Scholar] [CrossRef]
- Mascareñas, J.L. The [5+2] Cycloaddition Chemistry of β-Alkoxy-γ-pyrones. Adv. Cycloaddit. 1999, 6, 1–54. [Google Scholar]
- Min, L.; Liu, X.; Li, C.-C. Total Synthesis of Natural Products with Bridged Bicyclo[m.n.1] Ring Systems via Type II [5 + 2] Cycloaddition. Acc. Chem. Res. 2020, 53, 703–718. [Google Scholar] [CrossRef]
- Gao, K.; Zhang, Y.-G.; Wang, Z.; Ding, H. Recent development on the [5+2] cycloadditions and their application in natural product synthesis. Chem. Commun. 2019, 55, 1859–1878. [Google Scholar] [CrossRef]
- Liu, X.; Hu, Y.J.; Fan, J.H.; Zhao, J.; Li, S.; Li, C.C. Recent synthetic studies toward natural products via [5+2] cycloaddition reactions. Org. Chem. Front. 2018, 5, 1217–1228. [Google Scholar] [CrossRef]
- Ylijoki, K.E.O.; Stryker, J.M. [5+2] Cycloaddition Reactions in Organic and Natural Product Synthesis. Chem. Rev. 2013, 113, 2244–2266. [Google Scholar] [CrossRef]
- Nguyen, T.V.; Hartmann, J.M.; Enders, D. Recent Synthetic Strategies to Access Seven-Membered Carbocycles in Natural Product Synthesis. Synthesis 2013, 45, 845–873. [Google Scholar] [CrossRef]
- Battiste, M.A.; Pelphrey, P.M.; Wright, D.L. The Cycloaddition Strategy for the Synthesis of Natural Products Containing Carbocyclic Seven-Membered Rings. Chem. Eur. J. 2006, 12, 3438–3447. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Soisson, S.M.; Young, K.; Shoop, W.; Kodali, S.; Galgoci, A.; Painter, R.; Parthasarathy, G.; Tang, Y.S.; Cummings, R.; et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature 2006, 441, 358–361. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Ondeyka, J.; Herath, K.; Jayasuriya, H.; Guan, Z.; Zink, D.L.; Dietrich, L.; Burgess, B.; Ha, S.N.; Wang, J.; et al. Platensimycin and Platencin Congeners from Streptomyces platensis. J. Nat. Prod. 2011, 74, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Aoki, S.; Watanabe, Y.; Sanagawa, M.; Setiawan, A.; Kotoku, N.; Kobayashi, M. Cortistatins A, B, C, and D, Anti-angiogenic Steroidal Alkaloids, from the Marine Sponge Corticium simplex. J. Am. Chem. Soc. 2006, 128, 3148–3149. [Google Scholar] [CrossRef]
- Chen, D.Y.-K.; Tseng, C.C. Chemistry of the cortistatins—A novel class of anti-angiogenic agents. Org. Biomol. Chem. 2010, 8, 2900–2911. [Google Scholar] [CrossRef]
- Li, Y.; Pattenden, G. Perspectives on the structural and biosynthetic interrelationships between oxygenated furanocembranoids and their polycyclic congeners found in corals. Nat. Prod. Rep. 2011, 28, 1269–1310. [Google Scholar] [CrossRef]
- Tang, B.; Bray, C.D.; Pattenden, G. Total synthesis of (+)-intricarene using a biogenetically patterned pathway from (-)-bipinnatin J, involving a novel transannular [5+2] (1,3-dipolar) cycloaddition. Org. Biomol. Chem. 2009, 7, 4448–4457. [Google Scholar] [CrossRef]
- Roethle, P.A.; Hernandez, P.T.; Trauner, D. Exploring Biosynthetic Relationships among Furanocembranoids: Synthesis of (−)-Bipinnatin J, (+)-Intricarene, (+)-Rubifolide, and (+)-Isoepilophodione, B. Org. Lett. 2006, 8, 5901–5904. [Google Scholar] [CrossRef]
- Rokey, S.N.; Simanis, J.A.; Law, C.M.; Pohani, S.; Behrends, S.W.; Bulandr, J.J.; Ferrence, G.M.; Goodell, J.R.; Mitchell, T.A. Intramolecular asymmetric oxidopyrylium-based [5 + 2] cycloadditions. Tetrahedron Lett. 2020, 61, 152377. [Google Scholar] [CrossRef]
- Grabowski, J.P.; Ferrence, G.M.; Mitchell, T.A. Efforts toward the total synthesis of (±)-toxicodenane A utilizing an oxidopyrylium-based [5+2] cycloaddition of a silicon-tethered BOC-pyranone. Tetrahedron Lett. 2020, 61, 152324. [Google Scholar] [CrossRef]
- Bulandr, J.J.; Grabowski, J.P.; Law, C.M.; Shaw, J.L.; Goodell, J.R.; Mitchell, T.A. Investigation of Transfer Group, Tether Proximity, and Alkene Substitution for Intramolecular Silyloxypyrone-Based [5 + 2] Cycloadditions. J. Org. Chem. 2019, 84, 10306–10320. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, R.H.; Law, C.M.; Simanis, J.A.; Woodall, E.L.; Zwick, C.R., III; Wedler, H.B.; Wendelboe, P.; Hamaker, C.G.; Goodell, J.R.; Tantillo, D.J.; et al. Oxidopyrylium-Alkene [5 + 2] Cycloaddition Conjugate Addition Cascade (C3) Sequences: Scope, Limitation, and Computational Investigations. J. Org. Chem. 2018, 83, 9818–9838. [Google Scholar] [CrossRef] [PubMed]
- Simanis, J.A.; Zwick III, C.R.; Woodall, E.L.; Goodell, J.R.; Mitchell, T.A. Further Investigation of Pyranone Activation. Heterocycles 2015, 91, 149–156. [Google Scholar]
- Simanis, J.A.; Law, C.M.; Woodall, E.L.; Hamaker, C.G.; Goodell, J.R.; Mitchell, T.A. Investigation of oxidopyrylium-alkene [5+2] cycloaddition conjugate addition cascade (C3) sequences. Chem. Commun. 2014, 50, 9130–9133. [Google Scholar] [CrossRef] [PubMed]
- Woodall, E.L.; Simanis, J.A.; Hamaker, C.G.; Goodell, J.R.; Mitchell, T.A. Unique Reactivity of anti- and syn-Acetoxypyranones en Route to Oxidopyrylium Intermediates Leading to a Cascade Process. Org. Lett. 2013, 15, 3270–3273. [Google Scholar] [CrossRef]
- Ji, Y.; Benkovics, T.; Beutner, G.L.; Sfouggatakis, C.; Eastgate, M.D.; Blackmond, D.G. Mechanistic Insights into the Vanadium-Catalyzed Achmatowicz Rearrangement of Furfurol. J. Org. Chem. 2015, 80, 1696–1702. [Google Scholar] [CrossRef]
- Georgiadis, M.P.; Albizati, K.F.; Georgiadis, T.M. Oxidative Rearrangement of Furylcarbinols to 6-Hydroxy-2H-Pyran-3(6H)-ones, A Useful Synthon for the Preparation of a Variety of Heterocyclic Compounds. A Review. Org. Prep. Proc. Int. 1992, 24, 95–118. [Google Scholar] [CrossRef]
- Achmatowicz, O.; Szechner, P.B.B.; Zwierzchowska, Z.; Zamojski, A. Synthesis of Methyl 2,3-Dideoxy-DL-alk-2-enonpyranosides from Furan Compounds. Tetrahedron 1971, 27, 1973–1996. [Google Scholar] [CrossRef]
- Ciufolini, M.A.; Hermann, C.Y.W.; Dong, Q.; Shimizu, T.; Swaminathan, S.; Xi, N. Nitrogen Heterocycles from Furans: The Aza-Achmatowicz Reaction. Synlett 1998, 1998, 105–114. [Google Scholar] [CrossRef]
- Torborg, C.; Beller, M. Recent Applications of Palladium-Catalyzed Cross-Coupling Reactions in the Pharmaceutical, Agrochemical, and Fine Chemical Industries. Adv. Synth. Cat. 2009, 351, 3027–3043. [Google Scholar] [CrossRef]
- Sasaki, M.; Fuwa, H. Total Synthesis of Polycyclic Ether Natural Products Based on Suzuki-Miyaura Cross-Coupling. Synlett 2004, 2004, 1851–1874. [Google Scholar] [CrossRef]
- Vitaku, E.; Smith, D.T.; Njardarson, J.T. Analysis of the Structural Diversity, Substitution Patterns, and Frequency of Nitrogen Heterocycles among U.S. FDA Approved Pharmaceuticals. J. Med. Chem. 2014, 57, 10257–10274. [Google Scholar] [CrossRef] [PubMed]
- Luche, J.L. Lanthanides in organic chemistry. 1. Selective 1,2-reductions of conjugated ketones. J. Am. Chem. Soc. 1978, 100, 2226–2227. [Google Scholar] [CrossRef]
- Baell, J.; Walters, M.A. Chemical con artists foil drug discovery. Nature 2014, 514, 481–483. [Google Scholar] [CrossRef] [PubMed]
- Calder, E.D.D.; Sharif, S.A.I.; McGonagle, F.I.; Sutherland, A. One-Pot Synthesis of 5-Amino-2,5-dihydro-1-benzoxepines: Access to Pharmacologically Active Heterocyclic Scaffolds. J. Org. Chem. 2015, 80, 4683–4696. [Google Scholar] [CrossRef] [PubMed]
- Mosmann, T. Rapid Colorimetric Assay for Cellular Growth and Survival: Application to Proliferation and Cytotoxicity Assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Huffman, S.E.; Yawson, G.K.; Fisher, S.S.; Bothwell, P.J.; Platt, D.C.; Jones, M.A.; Hamaker, C.G.; Webb, M.I. Ruthenium(III) Complexes Containing Thiazole-Based Ligands That Modulate Amyloid-β Aggregation. Metallomics 2020, 12, 491–503. [Google Scholar] [CrossRef]
- Morgenthaler, J.B.; Peters, S.J.; Cedeño, D.L.; Constantino, M.H.; Edwards, K.A.; Kamowski, E.M.; Passini, J.C.; Butkus, B.E.; Young, A.M.; Lash, T.D.; et al. Carbaporphyrin ketals as potential agents for a new photodynamic therapy treatment of leishmaniasis. Biorg. Med. Chem. 2008, 14, 7033–7038. [Google Scholar] [CrossRef]
- Vallejo, R.; Platt, D.C.; Rink, J.A.; Cass, C.; Eichenberg, K.; Jones, M.A.; Kelley, C.A.; Gupta, A.; Vallejo, A.; Smith, W.J.; et al. Electrical Stimulation of C6 Glioma Cells in vitro Differentially Modulates Gene Expression Related to Chronic Pain Pathways. Brain Sci. 2019, 9, 303. [Google Scholar] [CrossRef]
- Mendez, R.S.; Dorsey, B.M.; McLauchlan, C.C.; Beio, M.; Turner, T.L.; Nguyen, V.H.; Su, A.; Beynon, W.; Friesen, J.A.; Jones, M.A. Vanadium Complexes Are in vitro Inhibitors of Leishmania Secreted Acid Phosphatases. Int. J. Chem. 2014, 6, 35–49. [Google Scholar] [CrossRef]
- Stockert, J.C.; Horobin, R.W.; Colombo, L.L.; Blázquez-Castro, A. Tetrazolium salts and formazan products in Cell Biology: Viability assessment, fluorescence imaging, and labeling perspectives. Acta Histochem. 2018, 120, 159–167. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Entry | Ar-B (5) | % Yield (1) a |
|---|---|---|
| 1 |  | 69 (1a) |
| 2 |  | 38 (1b) |
| 3 |  | 19 (1c) |
| 4 |  | 69 (1d) |
| 5 |  | 40 (1e) |
| Compound(s) Added | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Condition | 1% DMSO | 6 | 1a | 1b | 1c | 1d | 1e | 1b/1e | 1c/1e |
| Growth phase (AU/hr) | 0.013 | 0.013 | 0.0071 | 0.012 | 0.0042 | 0.0089 | 0.013 | 0.012 | 0.0023 |
| Recovery phase (AU/hr) | 0.017 | 0.018 | 0.012 | 0.016 | 0.0059 | 0.014 | 0.016 | 0.016 | 0.0042 |
| Recovery/Growth Phase Ratio | 1.30 | 1.37 | 1.73 | 1.30 | 1.43 | 1.55 | 1.26 | 1.37 | 1.85 |
| 6 | 1c | 1e | DMSO Control | |
|---|---|---|---|---|
| KM (µM) | 386 | 421 | 364 | 396 |
| Vmax (A 405 nm /24 hr) | 1.07 | 1.15 | 1.06 | 1.08 |
| Compound(s) Added | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Cell counts | 1% DMSO | 6 | 1a | 1b | 1c | 1d | 1e | 1b +1e | 1c +1e |
| cells green | 488 | 344 | 423 | 227 | 525 | 438 | 56 | 69 | 282 |
| cells not green | 248 | 494 | 674 | 669 | 850 | 703 | 315 | 323 | 334 |
| Total cells counted | 736 | 838 | 1097 | 896 | 1375 | 1141 | 371 | 392 | 616 |
| % total green | 66.3 | 41.1 | 38.6 | 25.3 | 38.2 | 38.4 | 15.1 | 17.64 | 45.8 |
| 6 | 1a | 1b | 1c | 1d | 1e | DMSO Control | |
|---|---|---|---|---|---|---|---|
| % DMSO Control | 95.4 | 129.6 | 114.8 | 116.9 | 94.4 | 97.1 | 100 |
| Time after Addition | 6 ED-50 (µM) | 1c ED-50 (µM) | 1e ED-50 (µM) |
|---|---|---|---|
| 24 h | >200 | 72.4 | 23.1 |
| 48 h | >200 | 24.9 | 31.5 |
| Time after Addition | D-N | D-F | D-UV | 6-N | 6-F | 6-UV | 1c-N | 1c-F | 1c-UV | 1e-N | 1e-F | 1e-UV |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 h | 100 | 99.7 | 99.8 | 99.2 | 101.5 | 102.7 | 104.9 | 102.7 | 98.2 | 98.2 | 95.0 | 95.8 |
| 24 h | 100 | 91.1 | 96.6 | 98.0 | 103.3 | 105.1 | 73.8 | 75.0 | 70.9 | 98.0 | 97.1 | 94.9 |
| 48 h | 100 | 104.0 | 100.6 | 99.6 | 105.5 | 100.3 | 64.0 | 67.6 | 61.6 | 99.3 | 100.0 | 100.1 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Singh, S.; Grabowski, J.P.; Pohani, S.; Apuzzo, C.F.; Platt, D.C.; Jones, M.A.; Mitchell, T.A. Synthesis of Polycyclic Ether-Benzopyrans and In Vitro Inhibitory Activity against Leishmania tarentolae. Molecules 2020, 25, 5461. https://doi.org/10.3390/molecules25225461
Singh S, Grabowski JP, Pohani S, Apuzzo CF, Platt DC, Jones MA, Mitchell TA. Synthesis of Polycyclic Ether-Benzopyrans and In Vitro Inhibitory Activity against Leishmania tarentolae. Molecules. 2020; 25(22):5461. https://doi.org/10.3390/molecules25225461
Chicago/Turabian StyleSingh, Sarita, Jacob P. Grabowski, Shilpa Pohani, C. Fiore Apuzzo, David C. Platt, Marjorie A. Jones, and T. Andrew Mitchell. 2020. "Synthesis of Polycyclic Ether-Benzopyrans and In Vitro Inhibitory Activity against Leishmania tarentolae" Molecules 25, no. 22: 5461. https://doi.org/10.3390/molecules25225461
APA StyleSingh, S., Grabowski, J. P., Pohani, S., Apuzzo, C. F., Platt, D. C., Jones, M. A., & Mitchell, T. A. (2020). Synthesis of Polycyclic Ether-Benzopyrans and In Vitro Inhibitory Activity against Leishmania tarentolae. Molecules, 25(22), 5461. https://doi.org/10.3390/molecules25225461