A Novel Purification Procedure for Active Recombinant Human DPP4 and the Inability of DPP4 to Bind SARS-CoV-2

 , , , ,
, , , ,  , , , and
, , , and 
Abstract
1. Introduction
2. Results
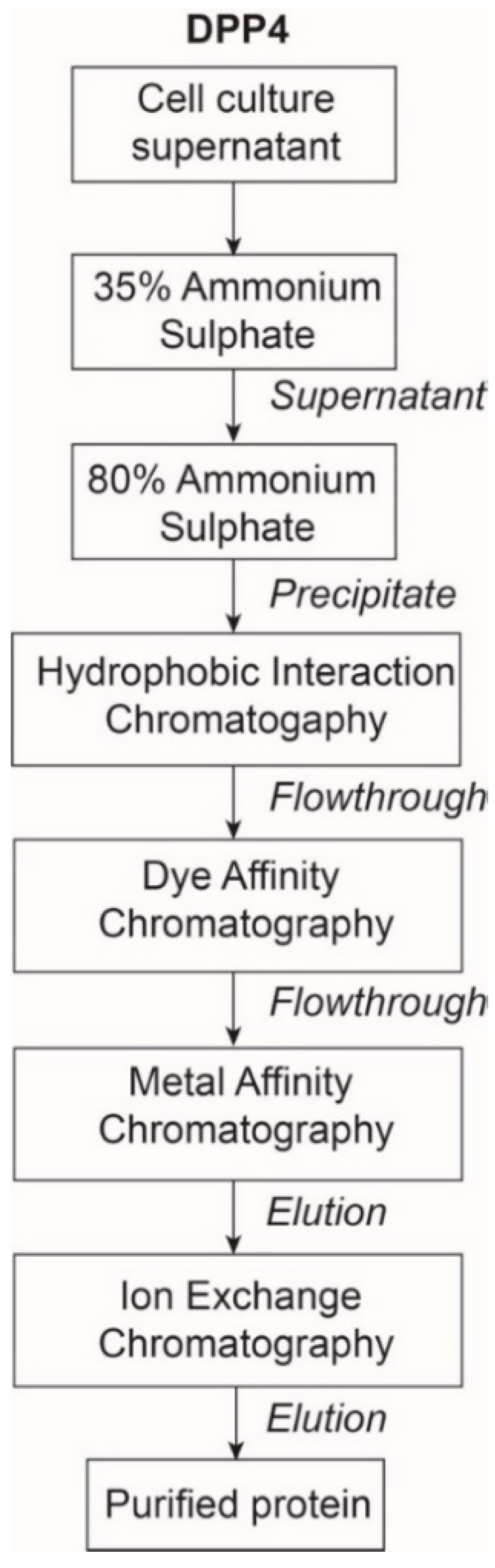
2.1. The Expression and Purification of DPP4
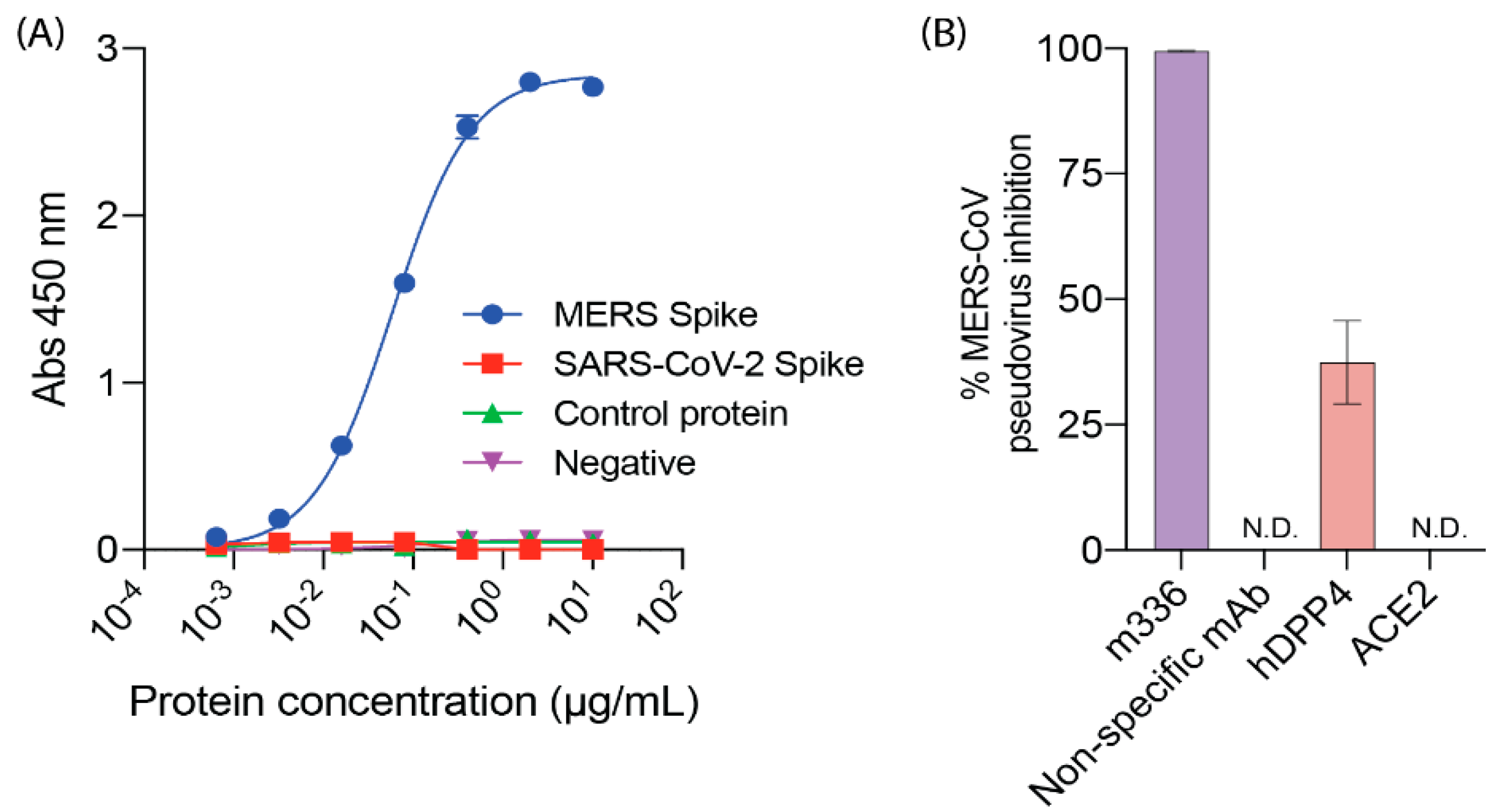
2.2. DPP4, MERS-CoV and SARS-CoV-2
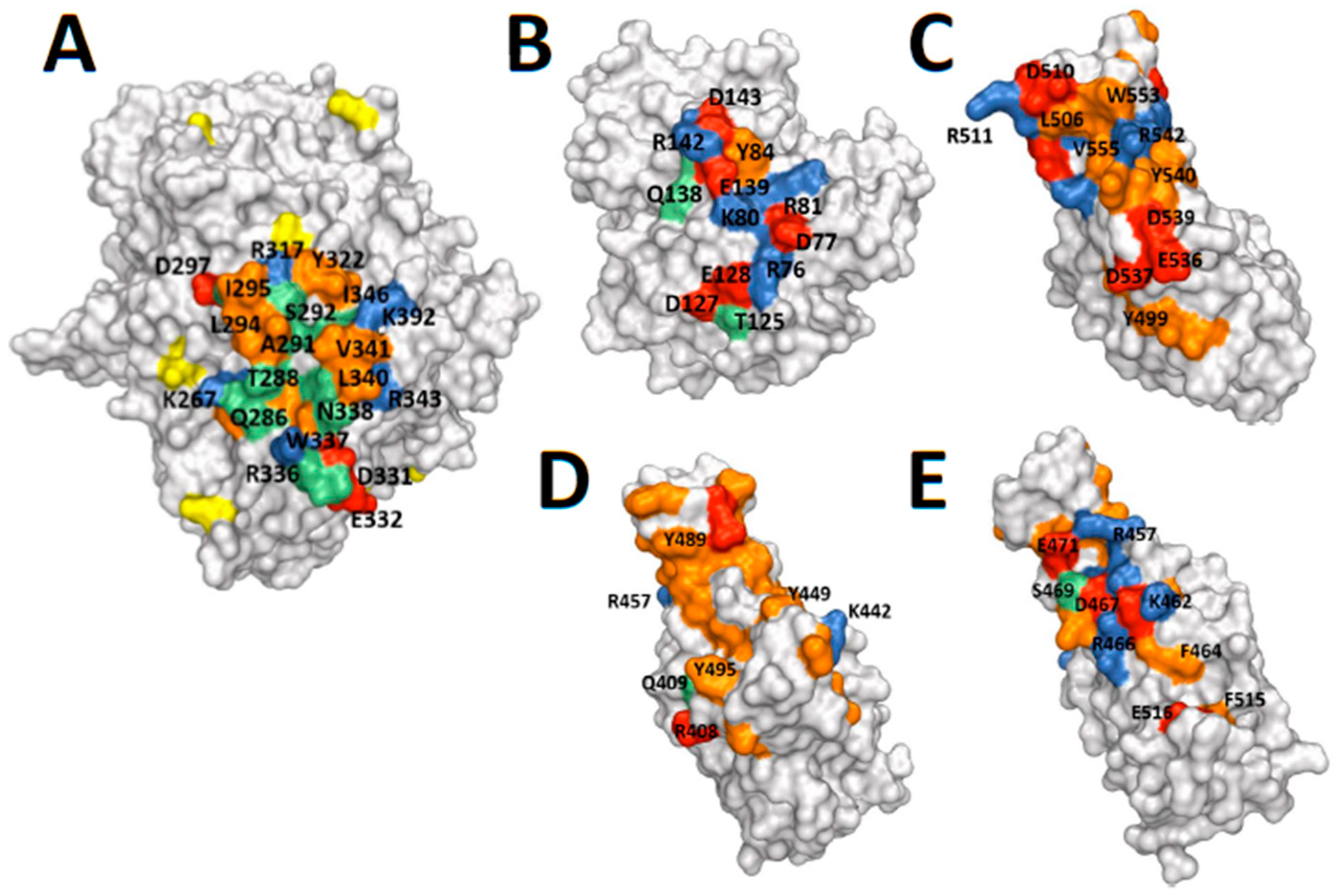
2.3. Protein Structures: Depictions of Binding Surfaces
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Expression and Purification of Active Soluble Human DPP4
4.2.1. Expression of DPP4 in Insect Sf9 Cells
4.2.2. Purification of DPP4
4.3. Expression and Purification of SARS-CoV-2 Full-Length Spike, SARS-CoV-2 RBD, and Human ACE2
4.3.1. Generation of Expression Constructs
4.3.2. Expression and Purification of SARS-CoV-2 Full-Length Spike, SARS-CoV-2 RBD, and Human ACE2
4.4. SDS–PAGE
4.5. Enzyme Assays
4.6. Surface Plasmon Resonance Assay
4.7. ELISA
4.8. MERS-CoV Pseudovirus Assay
4.9. Protein Structure Depictions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dunaevsky, Y.E.; Tereshchenkova, V.F.; Oppert, B.; Belozersky, M.A.; Filippova, I.Y.; Elpidina, E.N. Human proline specific peptidases: A comprehensive analysis. Biochim. Biophys. Acta 2020, 1864, 129636. [Google Scholar] [CrossRef] [PubMed]
- Enz, N.; Vliegen, G.; De Meester, I.; Jungraithmayr, W. CD26/DPP4-a potential biomarker and target for cancer therapy. Pharmacol. Ther. 2019, in press. [Google Scholar] [CrossRef] [PubMed]
- Gorrell, M.D. Dipeptidyl peptidase IV and related enzymes in cell biology and liver disorders. Clin. Sci. 2005, 108, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Rosenstock, J.; Perkovic, V.; Johansen, O.E.; Cooper, M.E.; Kahn, S.E.; Marx, N.; McGuire, D.K. Effect of Linagliptin vs. Placebo on Major Cardiovascular Events in Adults with Type 2 Diabetes and High Cardiovascular and Renal Risk: The CARMELINA Randomized Clinical Trial. JAMA 2019, 321, 69–79. [Google Scholar] [CrossRef]
- Tanaka, T.; Duke-Cohan, J.S.; Kameoka, J.; Yaron, A.; Lee, I.; Schlossman, S.F.; Morimoto, C. Enhancement of antigen-induced T-cell proliferation by soluble CD26/dipeptidyl peptidase IV. Proc. Natl Acad. Sci. USA 1994, 91, 3082–3086. [Google Scholar] [CrossRef]
- Wang, X.M.; Holz, L.E.; Chowdhury, S.; Cordoba, S.P.; Evans, K.A.; Gall, M.G.; Gorrell, M.D. Profibrotic role of dipeptidyl peptidase 4 in a carbon tetrachloride induced experimental liver injury. Immunol. Cell Biol. 2017, 95, 443–453. [Google Scholar] [CrossRef]
- De Meester, I.; Vanham, G.; Kestens, L.; Vanhoof, G.; Bosmans, E.; Gigase, P.; Scharpé, S. Binding of adenosine deaminase to the lymphocyte surface via CD26. Eur. J. Immunol. 1994, 24, 566–570. [Google Scholar] [CrossRef]
- De Meester, I.; Vanhoof, G.; Lambeir, A.M.; Scharpé, S. Use of immobilised adenosine deaminase (EC 3.5.4.4) for the rapid purification of native human CD26 dipeptidyl peptidase IV (EC 3.4.14.5). J. Immunol. Methods 1996, 189, 99–105. [Google Scholar] [CrossRef]
- Weihofen, W.A.; Liu, J.; Reutter, W.; Saenger, W.; Fan, H. Crystal structure of CD26/Dipeptidyl-peptidase IV in complex with adenosine deaminase reveals a highly amphiphilic interface. J. Biol. Chem. 2004, 41, 43330–43335. [Google Scholar] [CrossRef]
- Yu, D.M.T.; Slaitini, L.; Gysbers, V.; Riekhoff, A.G.M.; Kähne, T.; Knott, H.M.; Gorrell, M.D. Soluble CD26/dipeptidyl peptidase IV enhances human lymphocyte proliferation in vitro independent of dipeptidyl peptidase enzyme activity and adenosine deaminase binding. Scand. J. Immunol. 2011, 73, 102–111. [Google Scholar] [CrossRef]
- Ghorpade, D.S.; Ozcan, L.; Zheng, Z.; Nicoloro, S.M.; Shen, Y.; Chen, E.; Tabas, I. Hepatocyte-secreted DPP4 in obesity promotes adipose inflammation and insulin resistance. Nature 2018, 555, 673. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, L.R.; Bilandzic, M.; Wilson, A.L.; Chen, Y.; Gorrell, M.D.; Oehler, M.K.; Stephens, A.N. Hypoxia regulates DPP4 expression, proteolytic inactivation and shedding from ovarian cancer cells. Int. J. Mol. Sci. 2020, 21, 8110. [Google Scholar] [CrossRef] [PubMed]
- Aertgeerts, K.; Ye, S.; Tennant, M.G.; Collins, B.; Rogers, J.; Sang, B.; Prasad, G.S. Crystal structure of human Dipeptidyl Peptidase IV in complex with a decapeptide reveals details on substrate specificity and tetrahedral intermediate formation. Protein Sci. 2004, 13, 412–421. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, H.B.; Branner, S.; Wiberg, F.C.; Wagtmann, N. Crystal structure of human DPP-IV/CD26 in complex with a substrate analogue. Nat. Struct. Mol. Biol. 2003, 10, 19–25. [Google Scholar] [CrossRef]
- Bishnoi, R.; Hong, Y.R.; Shah, C.; Ali, A.; Skelton, W.P.T.; Huo, J.; Dang, L.H. Dipeptidyl peptidase 4 inhibitors as novel agents in improving survival in diabetic patients with colorectal cancer and lung cancer: A Surveillance Epidemiology and Endpoint Research Medicare study. Cancer Med. 2019, 8, 3918–3927. [Google Scholar] [CrossRef]
- Deacon, C.F. Physiology and Pharmacology of DPP-4 in Glucose Homeostasis and the Treatment of Type 2 Diabetes. Front. Endocrinol. 2019, 10, 80. [Google Scholar] [CrossRef]
- Kirby, M.S.; Yu, D.M.T.; O’Connor, S.P.; Gorrell, M.D. Inhibitor selectivity in the clinical application of dipeptidyl peptidase-4 inhibition. Clin. Sci. 2010, 118, 31–41. [Google Scholar] [CrossRef]
- Mentlein, R.; Dahms, P.; Grandt, D.; Kruger, R. Proteolytic processing of neuropeptide Y and peptide YY by dipeptidyl peptidase IV. Regul. Pept. 1993, 49, 133–144. [Google Scholar] [CrossRef]
- Abbott, C.A.; McCaughan, G.W.; Levy, M.T.; Church, W.B.; Gorrell, M.D. Binding to human dipeptidyl peptidase IV by adenosine deaminase and antibodies that inhibit ligand binding involves overlapping, discontinuous sites on a predicted β propeller domain. Chem. Eur. J. 1999, 266, 798–810. [Google Scholar] [CrossRef]
- Aertgeerts, K.; Ye, S.; Shi, L.; Prasad, S.G.; Witmer, D.; Chi, E.; Swanson, R.V. N-linked glycosylation of dipeptidyl peptidase IV (CD26): Effects on enzyme activity, homodimer formation, and adenosine deaminase binding. Protein Sci. 2004, 13, 145–154. [Google Scholar] [CrossRef]
- Bassendine, M.F.; Bridge, S.H.; McCaughan, G.W.; Gorrell, M.D. COVID-19 and comorbidities: A role for dipeptidyl peptidase 4 (DPP4) in disease severity? J. Diabetes Res. 2020, 12, 649–658. [Google Scholar] [CrossRef]
- Du, H.; Wang, D.W.; Chen, C. The potential effects of DPP-4 inhibitors on cardiovascular system in COVID-19 patients. J. Cell. Mol. Med. 2020, 24, 10274–10278. [Google Scholar] [CrossRef] [PubMed]
- Varin, E.M.; Mulvihill, E.E.; Beaudry, J.L.; Pujadas, G.; Fuchs, S.; Tanti, J.F.; Drucker, D.J. Circulating Levels of Soluble Dipeptidyl Peptidase-4 Are Dissociated from Inflammation and Induced by Enzymatic DPP4 Inhibition. Cell Metab. 2018, 29, 320–334.e5. [Google Scholar] [CrossRef] [PubMed]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Tan, W. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef]
- Verity, R.; Okell, L.C.; Dorigatti, I.; Winskill, P.; Whittaker, C.; Imai, N.; Ferguson, N.M. Estimates of the severity of coronavirus disease 2019: A model-based analysis. Lancet Infect. Dis. 2020, 20, 669–677. [Google Scholar] [CrossRef]
- Lu, G.; Hu, Y.; Wang, Q.; Qi, J.; Gao, F.; Li, Y.; Gao, G.F. Molecular basis of binding between novel human coronavirus MERS-CoV and its receptor CD26. Nature 2013, 500, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Wang, Y.; Wang, N.; Wang, D.; Guo, J.; Fu, L.; Shi, X. Identification of residues on human receptor DPP4 critical for MERS-CoV binding and entry. Virology 2014, 471–473, 49–53. [Google Scholar] [CrossRef]
- Vankadari, N.; Wilce, J.A. Emerging WuHan (COVID-19) coronavirus: Glycan shield and structure prediction of spike glycoprotein and its interaction with human CD26. Emerg. Microbes Infect. 2020, 9, 601–604. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Pohlmann, S. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280 e8. [Google Scholar] [CrossRef]
- Shang, J.; Ye, G.; Shi, K.; Wan, Y.; Luo, C.; Aihara, H.; Li, F. Structural basis of receptor recognition by SARS-CoV-2. Nature 2020, 581, 221–224. [Google Scholar] [CrossRef]
- Shang, J.; Wan, Y.; Luo, C.; Ye, G.; Geng, Q.; Auerbach, A.; Li, F. Cell entry mechanisms of SARS-CoV-2. Proc. Natl. Acad. Sci. USA 2020, 117, 11727–11734. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Shi, Z.L. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270–273. [Google Scholar] [CrossRef] [PubMed]
- Letko, M.; Marzi, A.; Munster, V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat. Rev. Microbiol. 2020, 5, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Dobers, J.; Zimmermann-Kordmann, M.; Leddermann, M.; Schewe, T.; Reutter, W.; Fan, H. Expression, purification, and characterization of human dipeptidyl peptidase IV/CD26 in Sf9 insect cells. Protein Expr. Purif. 2020, 25, 527–532. [Google Scholar] [CrossRef]
- Park, J.; Knott, H.M.; Nadvi, N.A.; Collyer, C.A.; Wang, X.M.; Church, W.B.; Gorrell, M.D. Reversible inactivation of human dipeptidyl peptidases 8 and 9 by oxidation. Open Enzym. Inhib. J. 2008, 1, 52–61. [Google Scholar] [CrossRef]
- Thoma, R.; Loffler, B.; Stihle, M.; Huber, W.; Ruf, A.; Hennig, M. Structural basis of proline-specific exopeptidase activity as observed in human dipeptidyl peptidase-IV. Structure 2003, 11, 947–959. [Google Scholar] [CrossRef]
- Jarvis, D.L. Baculovirus-insect cell expression systems. Meth. Enzymol. 2009, 463, 191–222. [Google Scholar] [CrossRef]
- Mannix, C.; Jarman, R.F. A Guide to Successful Scale-up of the Baculovirus Expression System. In Cell Engineering; Al-Rubeai, M., Ed.; Springer: Dordrecht, The Netherlands, 2020. [Google Scholar] [CrossRef]
- Ajami, K.; Pitman, M.R.; Wilson, C.H.; Park, J.; Menz, R.I.; Starr, A.E.; Cox, J.H.; Abbott, C.A.; Overall, C.M.; Gorrell, M.D. Stromal cell-derived factors 1 alpha and 1 beta, inflammatory protein-10 and interferon-inducible T cell chemo-attractant are novel substrates of dipeptidyl peptidase 8. FEBS Lett. 2008, 582, 819–825. [Google Scholar] [CrossRef]
- Ajami, K.; Abbott, C.A.; Obradovic, M.; Gysbers, V.; Kähne, T.; McCaughan, G.W.; Gorrell, M.D. Structural requirements for catalysis, expression and dimerisation in the CD26/DPIV gene family. Biochemistry 2003, 42, 694–701. [Google Scholar] [CrossRef]
- Oravecz, T.; Pall, M.; Roderiquez, G.; Gorrell, M.D.; Ditto, M.; Nguyen, N.Y.; Norcross, M.A. Regulation of the receptor specificity and function of the chemokine RANTES (regulated on activation normal T cell expressed and activated) by dipeptidyl peptidase IV (CD26)-mediated cleavage. J. Exp. Med. 1997, 186, 1865–1872. [Google Scholar] [CrossRef]
- Osborne, B.; Yao, T.-W.; Wang, X.M.; Chen, Y.; Kotan, L.D.; Nadvi, N.A.; Gorrell, M.D. A rare variant in human fibroblast activation protein associated with ER stress, loss of function and loss of cell surface localisation. Biochim. Biophys. Acta 2014, 1844, 1248–1259. [Google Scholar] [CrossRef] [PubMed]
- Buckley, S.J.; Collins, P.J.; O’Connor, B.F. The purification and characterisation of novel dipeptidyl peptidase IV-like activity from bovine serum. Int. J. Biochem. Cell Biol. 2014, 36, 1281–1296. [Google Scholar] [CrossRef] [PubMed]
- Leatherbarrow, R.J.; Dean, P.D. Studies on the mechanism of binding of serum albumins to immobilised cibacron blue F3G A. Biochem. J. 1980, 189, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, N.; Shi, X.; Jiang, L.; Zhang, S.; Wang, D.; Tong, P.; Wang, X. Structure of MERS-CoV spike receptor-binding domain complexed with human receptor DPP4. Cell Res. 2013, 23, 986–993. [Google Scholar] [CrossRef]
- Weihofen, W.A.; Liu, J.; Reutter, W.; Saenger, W.; Fan, H. Crystal structures of HIV-1 Tat-derived nonapeptides Tat-(1–9) and Trp2-Tat-(1–9) bound to the active site of dipeptidyl-peptidase IV (CD26). J. Biol. Chem. 2015, 280, 14911–14917. [Google Scholar] [CrossRef]
- Watanabe, Y.; Allen, J.D.; Wrapp, D.; McLellan, J.S.; Crispin, M. Site-specific analysis of the SARS-CoV-2 glycan shield. Science 2020, 369, 330–333. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Wang, X. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Daddona, P.E.; Kelley, W.N. Human adenosine deaminase binding protein. Assay, purification, and properties. J. Biol. Chem. 1978, 253, 4617–4623. [Google Scholar]
- Sinnathurai, P.; Lau, W.; Vieira de Ribeiro, A.J.; Bachovchin, W.W.; Englert, H.; Howe, G.; Gorrell, M.D. Circulating fibroblast activation protein and dipeptidyl peptidase 4 in rheumatoid arthritis and systemic sclerosis. Int. J. Rheum. Dis. 2018, 21, 1915–1923. [Google Scholar] [CrossRef]
- Teo, A.S.; Ramos, J.D.; Lee, B.W.; Cheong, N.; Chua, K.Y. Expression of the Blomia tropicalis paramyosin Blo t 11 and its immunodominant peptide in insect cells. Appl. Biochem. Biotechnol. 2006, 45, 13–21. [Google Scholar] [CrossRef]
- Summers, M.D.; Smith, G.D. A manual of Methods for Baculovirus Vectors and Insect Cell Culture Procedures; Texas Agriculture Experiment Station Bulletin: College Station, TX, USA, 1978. [Google Scholar]
- Abbott, C.A.; Yu, D.M.T.; Woollatt, E.; Sutherland, G.R.; McCaughan, G.W.; Gorrell, M.D. Cloning, expression and chromosomal localization of a novel human dipeptidyl peptidase (DPP) IV homolog, DPP8. Chem. Eur. J. 2020, 267, 6140–6150. [Google Scholar] [CrossRef]
- Amanat, F.; Stadlbauer, D.; Strohmeier, S.; Nguyen, T.H.O.; Chromikova, V.; McMahon, M.; Krammer, F. A serological assay to detect SARS-CoV-2 seroconversion in humans. Nat. Med. 2020, 26, 1033–1036. [Google Scholar] [CrossRef] [PubMed]
- Stadlbauer, D.; Amanat, F.; Chromikova, V.; Jiang, K.; Strohmeier, S.; Arunkumar, G.A.; Krammer, F. SARS-CoV-2 Seroconversion in Humans: A Detailed Protocol for a Serological Assay, Antigen Production, and Test Setup. Curr. Protoc. Microbiol. 2020, 57, e100. [Google Scholar] [CrossRef] [PubMed]
- Keane, F.M.; Yao, T.-W.; Seelk, S.; Gall, M.G.; Chowdhury, S.; Poplawski, S.E.; Gorrell, M.D. Quantitation of fibroblast activation protein (FAP)-specific protease activity in mouse, baboon and human fluids and organs. FEBS Open Bio 2014, 4, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Frey, G.; Chen, J.; Rits-Volloch, S.; Freeman, M.M.; Zolla-Pazner, S.; Chen, B. Distinct conformational states of HIV-1 gp41 are recognized by neutralizing and non-neutralizing antibodies. Nat. Struct. Mol. Biol. 2010, 17, 1486–1491. [Google Scholar] [CrossRef] [PubMed]
- Grehan, K.; Ferrara, F.; Temperton, N. An optimised method for the production of MERS-CoV spike expressing viral pseudotypes. MethodsX 2015, 2, 379–384. [Google Scholar] [CrossRef]
- Ying, T.; Du, L.; Ju, T.W.; Prabakaran, P.; Lau, C.C.; Lu, L.; Dimitrov, D.S. Exceptionally potent neutralization of Middle East respiratory syndrome coronavirus by human monoclonal antibodies. J. Virol. 2014, 88, 7796–7805. [Google Scholar] [CrossRef]
Sample Availability: Samples of novel materials are available from the authors. |






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Volume (mL) | Protein (mg/mL) | Total Protein (mg) | Total Activity (U) | Specific Activity (U/mg) | Fold-Purification * | Yield (%) ** | Step-Wise DPP4 Loss (%) # | Step-Wise Total Protein Loss (%) ## | |
|---|---|---|---|---|---|---|---|---|---|
| Cell culture supernatant | 1000 | 7.3 | 7300 | 144 | 0.020 | 1 | 100 | 0 | 0 |
| 35% AS | 1046 | 3.9 | 4079 | 109 | 0.027 | 1.4 | 76 | 24 | 44 |
| 80% AS | 82 | 7.2 | 589 | 85 | 0.14 | 7.0 | 59 | 22 | 86 |
| Phenyl Sepharose | 93 | 4.1 | 381 | 77 | 0.20 | 10 | 53 | 9.4 | 35 |
| Ni Sepharose | 41 | 0.034 | 1.4 | 64 | 46 | 2300 | 44 | 16 | 99.6 |
| DEAE Sepharose | 4.4 | 0.23 | 1.0 | 42 | 42 | 2100 | 29 | 35 | 29 |
| Purification Replicate | Culture Supernatant (L) | Total Protein (mg) | Enzyme Activity (U/mL) | Specific Activity (U/mg) | Final Yield (mg/L Culture) |
|---|---|---|---|---|---|
| 1 | 1 | 1.2 | 45 | 37 | 1.2 |
| 2 | 1.1 | 1.2 | 52 | 43 | 1.1 |
| 3 | 0.98 | 1.5 | 44 | 29 | 1.6 |
| 4 | 0.98 | 1.8 | 61 | 34 | 1.9 |
| 5 | 0.99 | 1.0 | 42 | 40 | 1.0 |
| Mean ± SD | 1.0 ± 0.1 | 1.3 ± 0.3 | 49 ± 7.8 | 36 ± 5.4 | 1.4 ± 0.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xi, C.R.; Di Fazio, A.; Nadvi, N.A.; Patel, K.; Xiang, M.S.W.; Zhang, H.E.; Deshpande, C.; Low, J.K.K.; Wang, X.T.; Chen, Y.; et al. A Novel Purification Procedure for Active Recombinant Human DPP4 and the Inability of DPP4 to Bind SARS-CoV-2. Molecules 2020, 25, 5392. https://doi.org/10.3390/molecules25225392
Xi CR, Di Fazio A, Nadvi NA, Patel K, Xiang MSW, Zhang HE, Deshpande C, Low JKK, Wang XT, Chen Y, et al. A Novel Purification Procedure for Active Recombinant Human DPP4 and the Inability of DPP4 to Bind SARS-CoV-2. Molecules. 2020; 25(22):5392. https://doi.org/10.3390/molecules25225392
Chicago/Turabian StyleXi, Cecy R, Arianna Di Fazio, Naveed Ahmed Nadvi, Karishma Patel, Michelle Sui Wen Xiang, Hui Emma Zhang, Chandrika Deshpande, Jason K K Low, Xiaonan Trixie Wang, Yiqian Chen, and et al. 2020. "A Novel Purification Procedure for Active Recombinant Human DPP4 and the Inability of DPP4 to Bind SARS-CoV-2" Molecules 25, no. 22: 5392. https://doi.org/10.3390/molecules25225392
APA StyleXi, C. R., Di Fazio, A., Nadvi, N. A., Patel, K., Xiang, M. S. W., Zhang, H. E., Deshpande, C., Low, J. K. K., Wang, X. T., Chen, Y., McMillan, C. L. D., Isaacs, A., Osborne, B., Vieira de Ribeiro, A. J., McCaughan, G. W., Mackay, J. P., Church, W. B., & Gorrell, M. D. (2020). A Novel Purification Procedure for Active Recombinant Human DPP4 and the Inability of DPP4 to Bind SARS-CoV-2. Molecules, 25(22), 5392. https://doi.org/10.3390/molecules25225392