1. Introduction
Many tyrosine kinase receptors are located upstream of mitogen-activated protein kinase (MAPK) pathways in colorectal cancer (CRC) cells, and the MAPK signaling pathway was found to be related to oncogenic processes [
1]. The function of recombinant activated factor (RAF) family kinases in CRC cells is known to play a role in proliferation, differentiation, and survival [
2]. Sorafenib is an oral kinase inhibitor that has activity in patients with advanced solid tumors, and it is the most successful multi-kinase inhibitor targeting the RAF/mitogen-activated protein kinase (MEK)/extracellular signal-regulated kinase (ERK) pathway [
1,
3]. Therefore, sorafenib can also be expected to be effective for the treatment of patients with advanced CRC. The excellence of sorafenib as a targeted therapy for CRC was verified through non-clinical and clinical studies [
4,
5,
6,
7].
As a single agent, sorafenib can decrease the viability of human CRC cells or reduce carcinoembryonic antigens. The antitumor effect of sorafenib in combination with erlotinib or cetuximab on human CRC cells was also investigated [
4,
5]. In patients with metastatic CRC, sorafenib has been found to increase the median progression-free survival and overall survival when single-treated or combined with an anti-angiogenic agent, such as bevacizumab [
6,
7]. However, sorafenib may cause resistance, which has become an obstacle in its continuous use in cancer patients. As primary resistance, the abnormal activation of epithelial growth factor receptor (EGFR) and overexpression of its ligands due to the genetic heterogeneity of cancer was reported to lower the antitumor effect of sorafenib [
8]. The phosphoinositide 3-kinases (PI3K)/Akt pathway regulates many molecules involved in most aspects of cancer growth and cross-talks with the RAF/MEK/ERK pathway [
9]. Therefore, in acquired resistance, the expression of the phosphorylated PI3K/Akt pathway is maintained by sorafenib, and downstream factors, such as mammalian target of rapamycin (mTOR), are activated to act as a potential compensation mechanism [
3,
9].
Focal adhesion kinase (FAK) is a functioning upstream activator of PI3K and stimulates cancer cell proliferation and migration via the Akt-dependent pathway [
10]. FAK promotes phosphorylation of the p85 subunit of PI3K and then indirectly interacts with a collaborative companion protein that can bind to Akt to form a complex [
10,
11]. It is well established that CRC cells increase FAK expression in the early stages of carcinogenesis [
11]. The upregulation of FAK promotes the adhesive properties, survival, and local invasion of CRC cells [
11]. Therefore, targeting FAK would be one way to overcome the acquired resistance caused by sorafenib, and an enhanced therapeutic effect on CRC can be expected simultaneously. Our recent study demonstrated that a sustained calcium influx can destroy the constituents of the focal contacts of FAK subject to downregulation of the steroid receptor coactivator (Src), which could involve the inhibition of epithelial-mesenchymal transition signaling, leading to significant suppression of CRC cell invasion and motility [
12,
13].
In the present study, we investigated whether calcium-mediated deactivation of FAK led to the inhibition of Akt signaling, bypassing the inhibitory pathway of RAF family kinases by sorafenib, and resulting in an enhanced antitumor effect on CRC cells.
3. Discussion
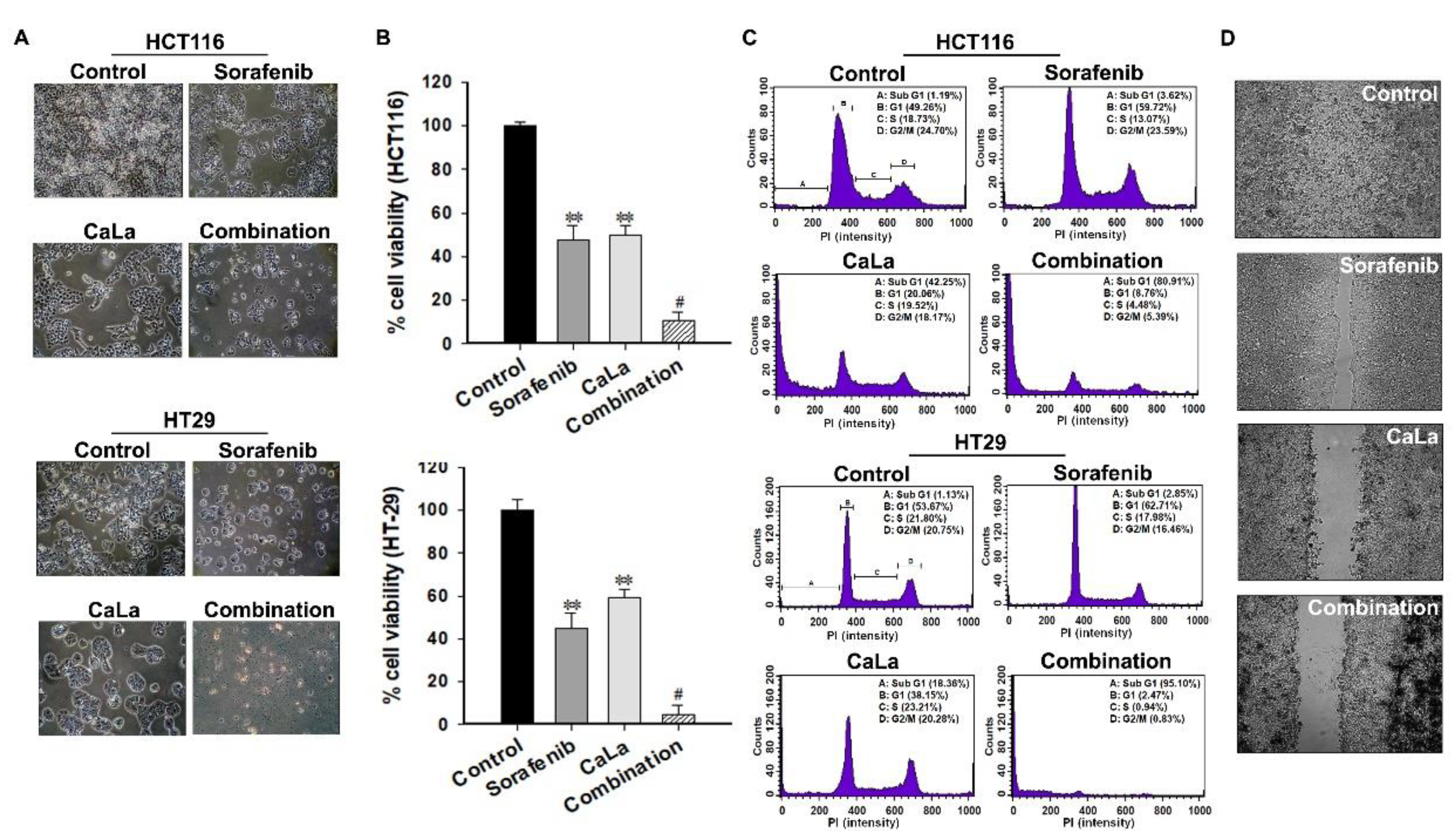
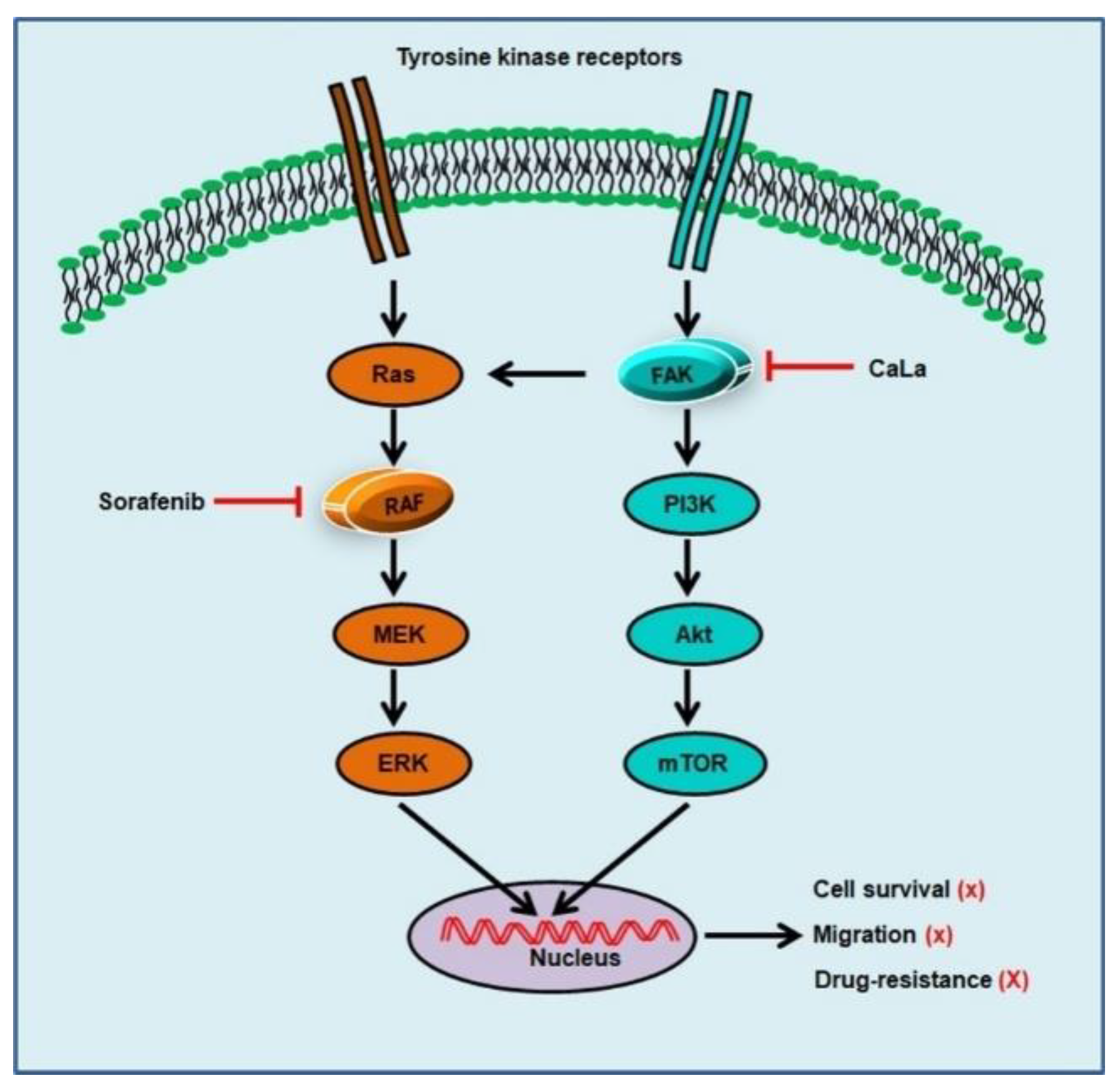
In this study, we investigated whether sorafenib could show an enhanced anticancer effect against CRC cells following calcium-mediated inhibition of the bypassable survival signaling pathways of Src/FAK/Akt/mTOR. The anticancer efficacy of sorafenib was related to the inhibition of CRC cell growth through the inhibition of MAPK signal transduction via RAF downregulation, and enhanced antitumor effect of sorafenib was possible following the inhibition of sub-signals from FAK downregulation mediated by calcium influx (
Figure 5).
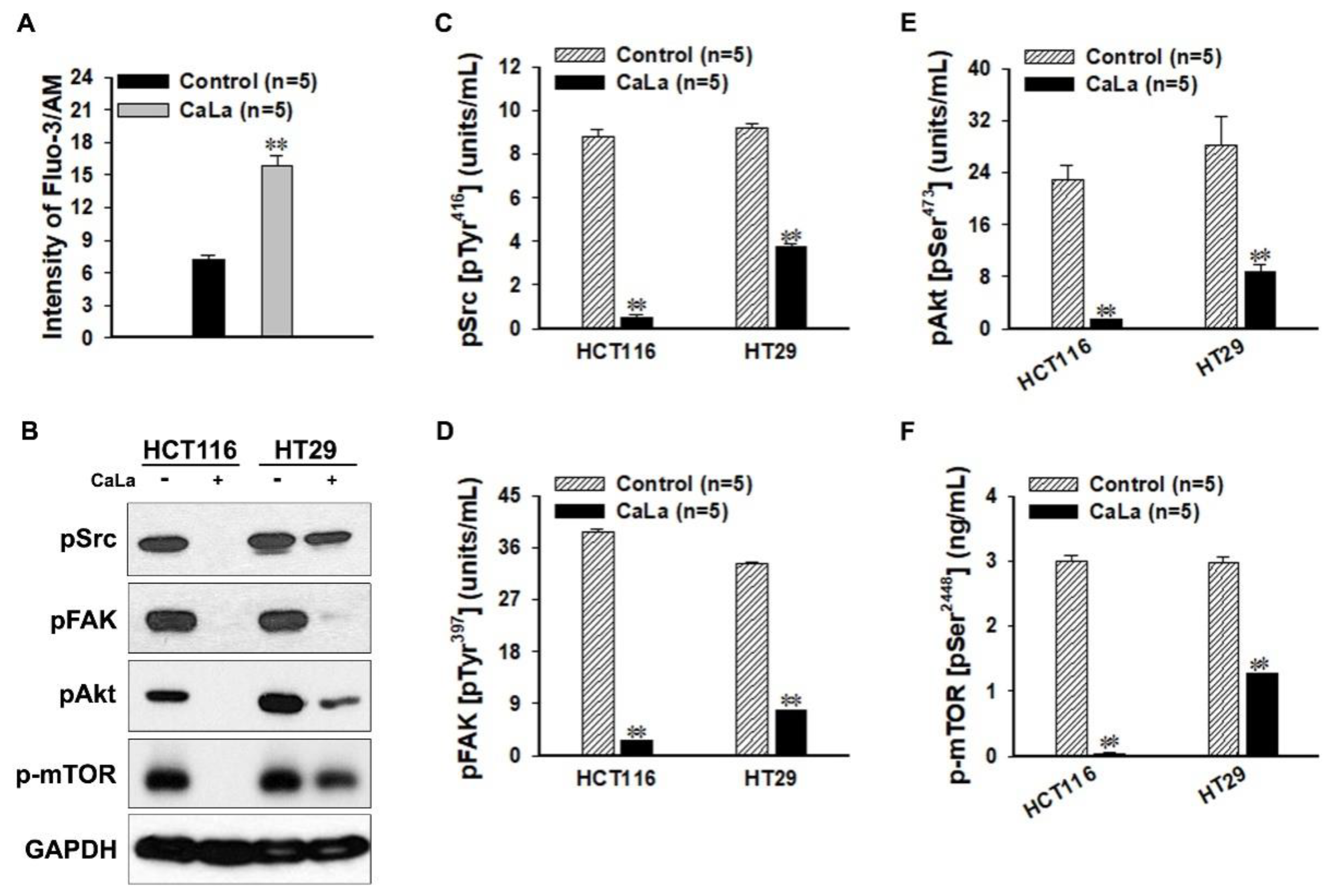
CaLa is a crystalline salt formed by a reaction between lactic acid and calcium carbonate, and its neutral nature allows it to diffuse easily by penetrating the cell membrane; therefore, it was utilized for sustained calcium supply to CRC cells [
12]. To observe the inhibitory effect of the FAK/Akt/mTOR pathways following calcium influx, the sensitivity of the phosphorylated form in signaling pathways was determined at the protein level. The Src family of protein tyrosine kinases is important in the regulation of growth and differentiation of cancer cells. Src activity is upregulated upon phosphorylation at Tyr
416 in the kinase domain [
14]. FAK is a non-receptor protein tyrosine kinase that acts as a substrate for Src and is a key element of integrin sub-signaling. FAK plays an important role in the cell proliferation, migration, cell death, and acceleration of the cell cycle in the G1 to S phase transition [
15]. Tyr
397 is the autophosphorylation site of FAK, and this phosphorylated site binds the SH2 domains of Src.
Akt, which is known as protein kinase B, is a ubiquitous kinase that plays an important role in diverse biological responses, such as cell survival and growth, by phosphorylating multiple proteins [
16]. Akt promotes cell survival by inhibiting apoptosis, and the activation of Akt is dependent on a regulatory mechanism that requires its phosphorylation on Ser
473 [
17]. The active form of mTOR is determined by its phosphorylation at Ser
2448 via the PI3K/Akt signaling pathway and it plays a key role in cell growth [
18].
There are two potential reasons for the downregulation of these signaling molecules at the protein level. One is the downregulation of signaling cascades by enzymatic activation. The other is the collective enhancement of dephosphorylation at the active site belonging to signaling molecules. The continuous influx of calcium is the fundamental reason that makes the above two hypotheses possible because the existing studies have well established the role of various proteolytic and catabolic breakdown enzymes in the action of calcium, such as the activation of proteases and phosphatases [
19,
20]. However, proof of the concept related to enzymatic activation was not planned in the present study. Therefore, further studies will be carried out to examine a variety of possibilities related to the activity of calcium-based enzymes to provide a clear basis for calcium-dependent downregulation of the FAK signaling pathway.
Cell cycle arrest is the point of disruption in the cell cycle, where the cells are no longer involved in the processes surrounding division, and it is possible to observe signs of growth inhibition or cell death [
21]. The G1 phase of the cell cycle is an intermediate phase occupying the time between the end of cell division in mitosis and the beginning of DNA replication [
21,
22]. Since the cells grow in preparation for DNA replication in this phase, cell growth could be suppressed under proliferation-quiescence through G1 arrest [
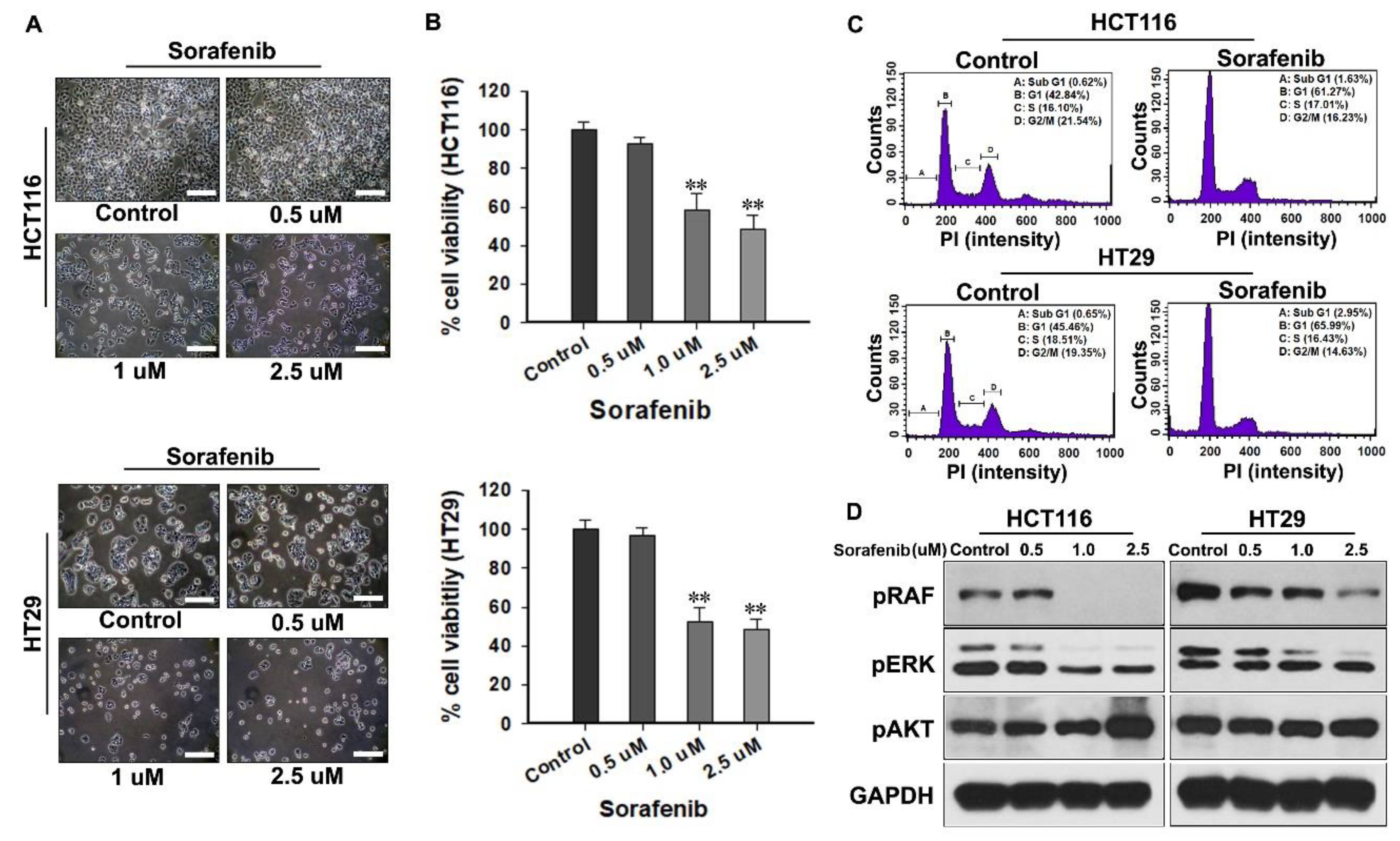
22]. When CRC cells were treated with sorafenib, floating or lysed cells could not be observed in morphological analysis; therefore, cell cycle analysis was a good basis for supporting the mechanism of growth inhibition by sorafenib. Of course, a high concentration of sorafenib could induce other cell arrests; however, only G1 arrest was observed at low concentrations of sorafenib.
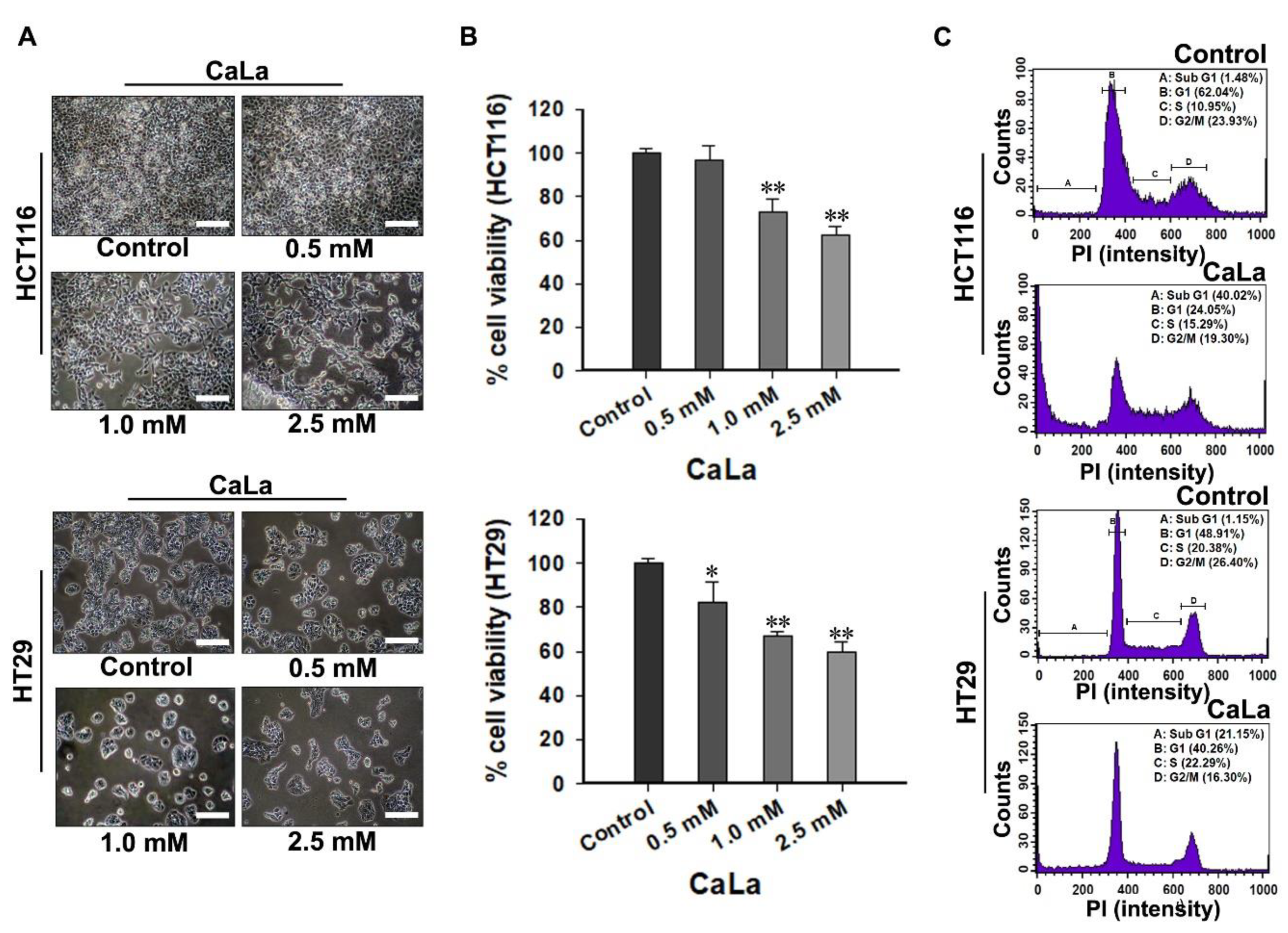
The increase in the sub-G1 accumulation of CRC cells was confirmed following treatment with CaLa. During apoptosis, genomic DNA is cleaved into smaller fragments [
21]. This is a specific marker of apoptosis, for which propidium iodide-stained cells show a peak in the sub-G1 phase [
21,
22]. The appropriate CaLa concentration was applied for the combination study by fulfilling an equal screening method with sorafenib, and partial cell death was observed at selected concentrations. At this point, it must not be concluded that RAF is only responsible for cell growth and that FAK is only associated with cell survival. As it is impossible to determine the predominant signaling pathway between RAF and FAK in relation to the proliferation and survival of CRC cells, and RAF or FAK signaling pathways can complement each other for cancer cell proliferation and survival, the binary distinction must not be acceptable.
FAK is not only a critical modulator of signaling pathways mediated by tyrosine kinase receptors but also responds to stimuli from cell–cell adhesion proteins to control a variety of oncogenic cellular responses [
11,
16]. Src is a common intracellular point of convergence in the signaling initiated by integrin-extracellular matrix interactions [
14]. The tyrosine kinase Src phosphorylates FAK, leading to increased FAK activity [
11]. Further, the interaction between FAK and Src plays an important role in facilitating the intracellular signaling pathways for migration and invasion by inducing epithelial-mesenchymal transition [
11,
12,
14].
FAK deactivation is considered to play a huge role in the calcium-mediated inhibition of migration, and combination with sorafenib could be a potential therapeutic strategy for the advanced stage of CRC. However, the significance of the calcium supply in the prevention or treatment of CRC is still controversial [
23,
24]. Therefore, it is necessary to establish a clear mode of action and reproducibility of the effect for clinical applications, and there is a need to additionally secure the applicable dosage and non-clinical toxicity.
In conclusion, our results indicated that calcium influx induced the deactivation of Src/FAK/Akt/mTOR, which is a bypassable signaling pathway of sorafenib. The combination of sorafenib with CaLa enhanced apoptosis and inhibited the metastatic features of CRC cells even when a lower dose of sorafenib was used. A clearer mode of action should be investigated in a future study with CRC cells that are completely unresponsive to sorafenib. Although the immediate clinical application may be premature, our findings indicate that the anticancer effect of sorafenib on patients with advanced CRC could be enhanced in combination with CaLa, if an appropriate usage/dosage is provided.
4. Materials and Methods
4.1. Cell Culture and Reagents
Human CRC cell lines (HCT-116 and HT-29) were purchased from American Type Culture Collection (Manassa, VA, USA). The cells were maintained in RPMI1640 medium (Welgene, Kyung-San, Korea) supplemented with 10% fetal bovine serum (Welgene), 10,000 units/mL penicillin, and 10,000 μg/mL streptomycin (Welgene) in a humidified atmosphere at 37 °C containing 5% CO2. Sorafenib [N-(3-trifluoromethyl-4-chlorophenyl)-N′-(4-(2-methyl carbamoyl pyridin-4-yl)oxyphenyl)urea] was synthesized at Bayer Corporation (West Haven, CT, USA), and CaLa was purchased from Sigma-Aldrich (St. Louis, MO, USA).
4.2. Cell Viability Assay
CRC cells were seeded at a density of 5000 cells/well in a 96-well plate and treated with the indicated concentrations of sorafenib and CaLa, alone and in combination. After 72 h, the medium was removed and 100 µL of 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT, 5 mg/mL; Sigma-Aldrich) was added to each well, and the cells were incubated for 2 h at 37 °C in a humidified environment at 5% atmospheric CO2. The cells were then lysed, the formazan crystals formed were dissolved in 100 μL of dimethyl sulfoxide (DMSO, Sigma-Aldrich), and the absorbance was measured at 570 nm using an Epoch Micro-Volume spectrophotometer system (Bio-Tek, Winooski, VT, USA).
4.3. Cell Cycle Analysis
For cell cycle distribution analysis, 3 × 105 CRC cells were seeded in 60 mm dishes and treated with CaLa and sorafenib, alone and in combination, for 72 h. After the treatment, the cells were collected through trypsinization (Sigma-Aldrich), centrifuged at 3000 rpm for 3 min at 4 °C, washed twice with ice-cold phosphate buffered saline (PBS, Welgene), and fixed with 70% cold ethanol (Sigma-Aldrich). The cells were then treated with 5 μL of a 10 mg/mL solution of ribonuclease and were stained with 10 μL of a 1 mg/mL solution of propidium iodide for 30 min at 37 °C in the dark (BD bioscience, Franklin Lakes, NJ, USA). The DNA content of the cells was measured using flow cytometry (BD FACSCalibur™, BD bioscience, NJ, USA), and the percentage of cells in each cell cycle phase was evaluated using BD FACS CompTM software (BD bioscience).
4.4. Western Blot Analysis
CRC cells were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer (1% NP-40, 0.25% sodium deoxycholate, 50 mM Tris-HCl pH 7.4, 150 mM NaCl, 1 mM ethylenediaminetetraacetic acid (EDTA), 1% Triton X-100, 10% glycerol, 100 mM NaF, 1 mM NA3VO4, 10 mM NaPP, and a protease inhibitor cocktail (Roche, Basel, Switzerland)). The lysates were quantitated using a bicinchoninic acid assay kit (Thermo Scientific, Waltham, MA, USA) and were transferred to a polyvinylidene fluoride membrane (EMD Millipore, Billerica, MA, USA). After blocking in tris-buffered saline (TBS) with tween 20 (100 mM Tris-HCl, 1.5 M NaCl, 0.5% Tween-20, pH 7.5; Sigma-Aldrich) containing 5% non-fat milk (Biorad, CA, USA) for 1 h, the membranes were incubated overnight in a 4 °C cold room with a primary antibody at an appropriate dilution in TBS-T containing 5% BSA and 0.1% sodium azide (Sigma-Aldrich). The primary antibodies used in the study were: GAPDH (1:10,000, Cell Signaling, Danvers, MA, USA), pRAF (1:1000, Cell Signaling, Danvers, MA, USA), pERK (1:1000, Cell Signaling, Danvers, MA, USA), pSrc (1:1000, Cell Signaling, Danvers, MA, USA), pFAK (1:1000, Cell Signaling, Danvers, MA, USA), pAkt (1:1000, Cell Signaling, Danvers, MA, USA), and p-mTOR (1:1000, Cell Signaling, Danvers, MA, USA). After incubation, the membranes were incubated for 2 h with an anti-rabbit secondary antibody (1:5000, Cell Signaling, Danvers, MA, USA). The immunoblots were developed using western blotting detection reagents (Abclone, Seoul, Korea) and were exposed to an X-ray film (Agfa, Leverkusen, Germany) according to the manufacturer’s recommended protocol.
4.5. Intracellular Calcium Concentration
The cytosolic free calcium ion concentration was measured using a confocal laser scanning microscope (Leica, Heidelberg, Germany). Cultured HCT116 cells were loaded with 10 μM Fluo-3/AM and 1 μL of 25% Pluronic F-127 in dimethyl sulfoxide (Thermo Scientific, Waltham, MA, USA) and were incubated for 30 min at 37 °C. After loading the fluorescence probes, 2.5 mM CaLa was added and the intensity of green fluorescence was measured using Image J (
imagej.nih.gov/ij).
4.6. Enzyme-Linked Immunosorbent Assay (ELISA)
The lysates from cultured CRC cells were concentrated using VIVASPIN® (Sartorius Stedim Biotech, Goettingen, Germany). Quantitative analysis was performed using a human pSrc ELISA kit (Cell Signaling, Danvers, MA, USA) and human pFAK, pAkt, and p-mTOR ELISA kits (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions.
4.7. Wound Healing Assay
For the wound healing assay, a culture insert consisting of two reservoirs separated by a 500 µm thick wall was used (ibidi, München, Germany). The CRC cells were seeded into a two-chamber (100 µL of 4 × 105 cells/mL) culture insert (ibidi, München, Germany). After 24 h of incubation, the chamber was gently removed, creating a gap of ~500 µm. The cells were then allowed to migrate into the bare areas for 12 h. The cell imaging was performed using the JuLi Br Live-cell analyzer (NanoEnTek Inc., Guro-Dong, Korea).
4.8. Statistical Analysis
Data are presented as the mean ± standard deviation (SD). Statistical significance was analyzed using Student’s t-test or the Mann–Whitney rank-sum test depending on the normality of the data. A difference of p < 0.05 was considered statistically significant. All statistical analyses were performed using Sigma Stat ver. 3.5 (Systat Software Inc., San Jose, CA, USA).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




