
Recent Advances in the Biological Investigation of Organometallic Platinum-Group Metal (Ir, Ru, Rh, Os, Pd, Pt) Complexes as Antimalarial Agents




Abstract
1. Introduction
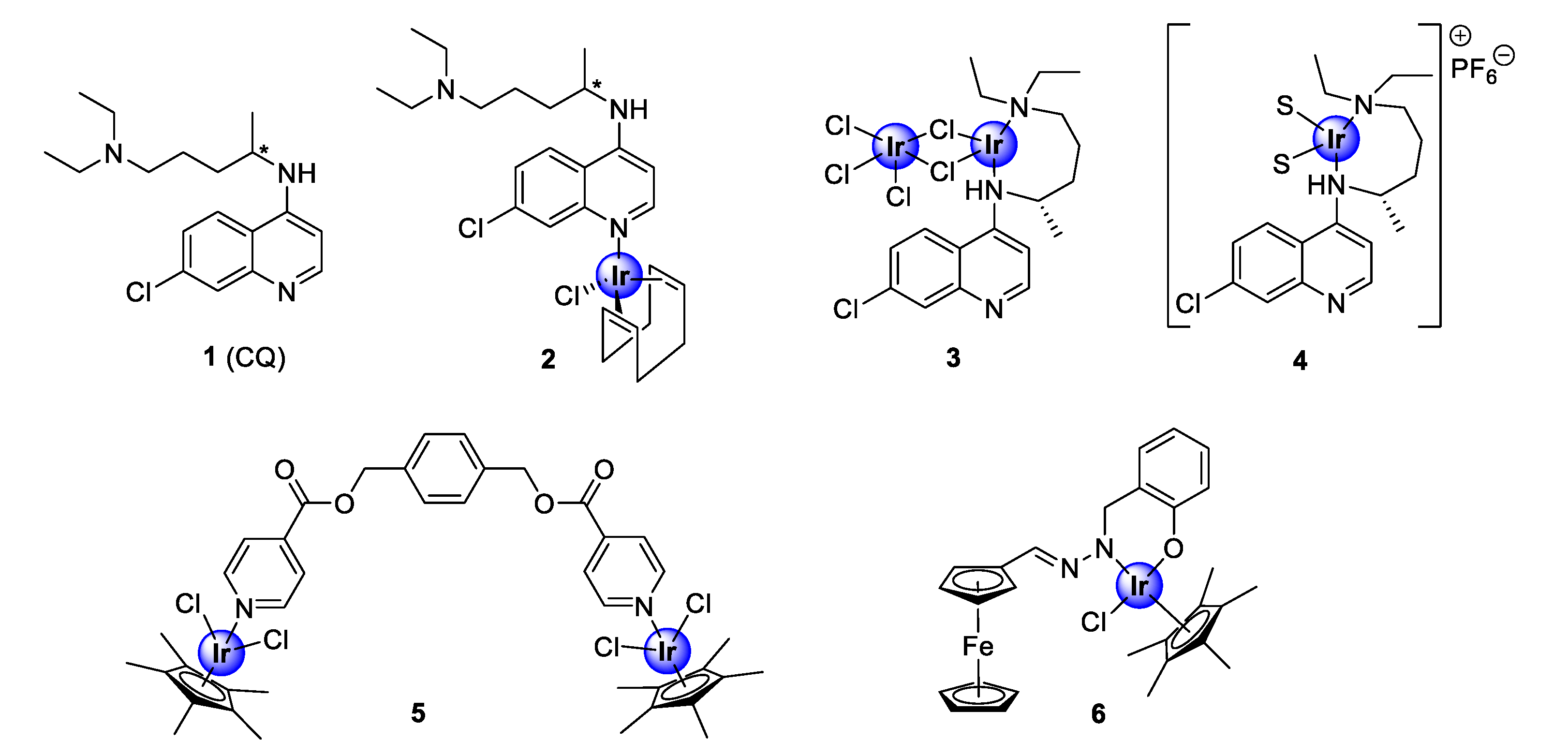
2. Iridium Complexes
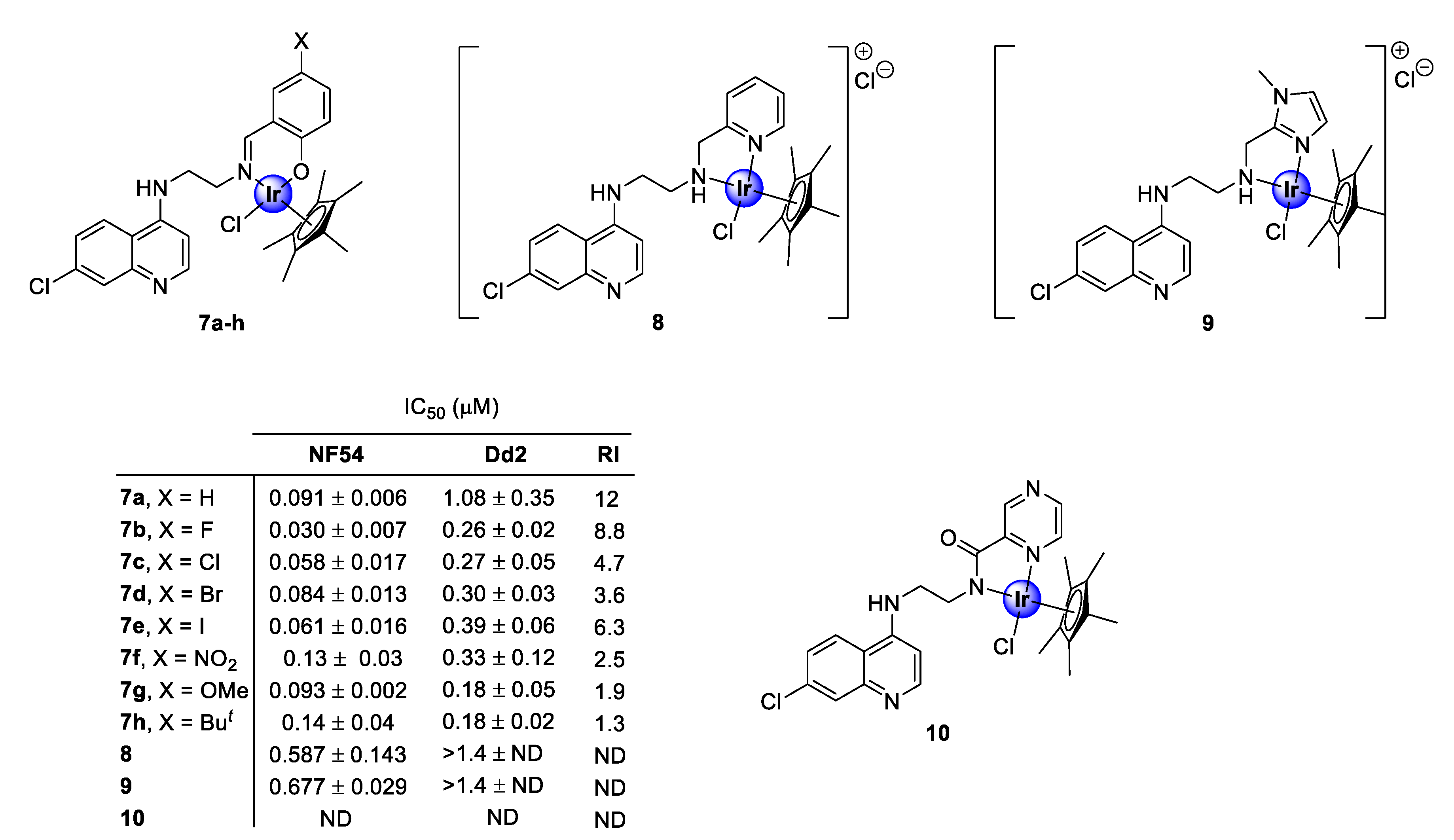
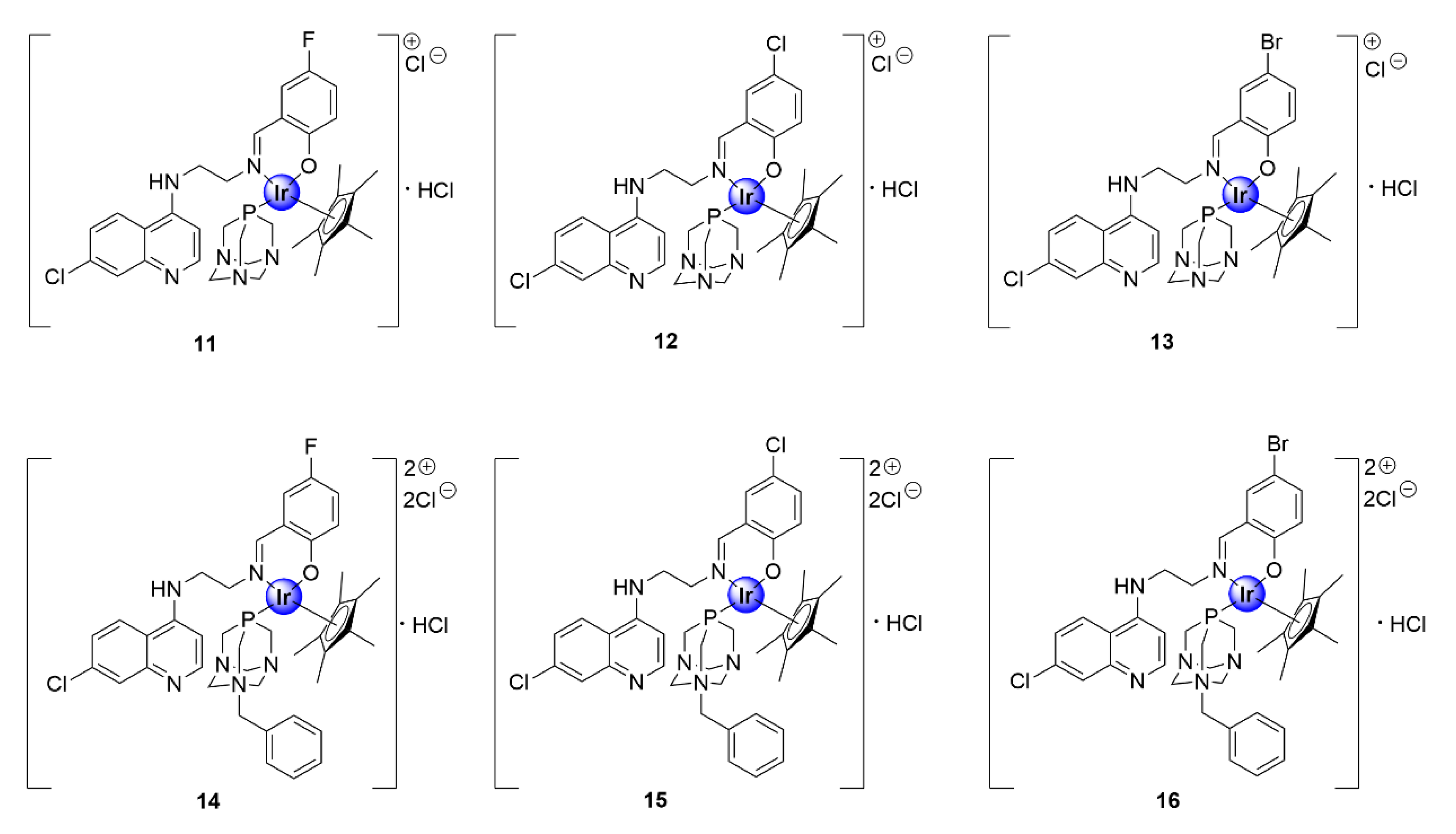
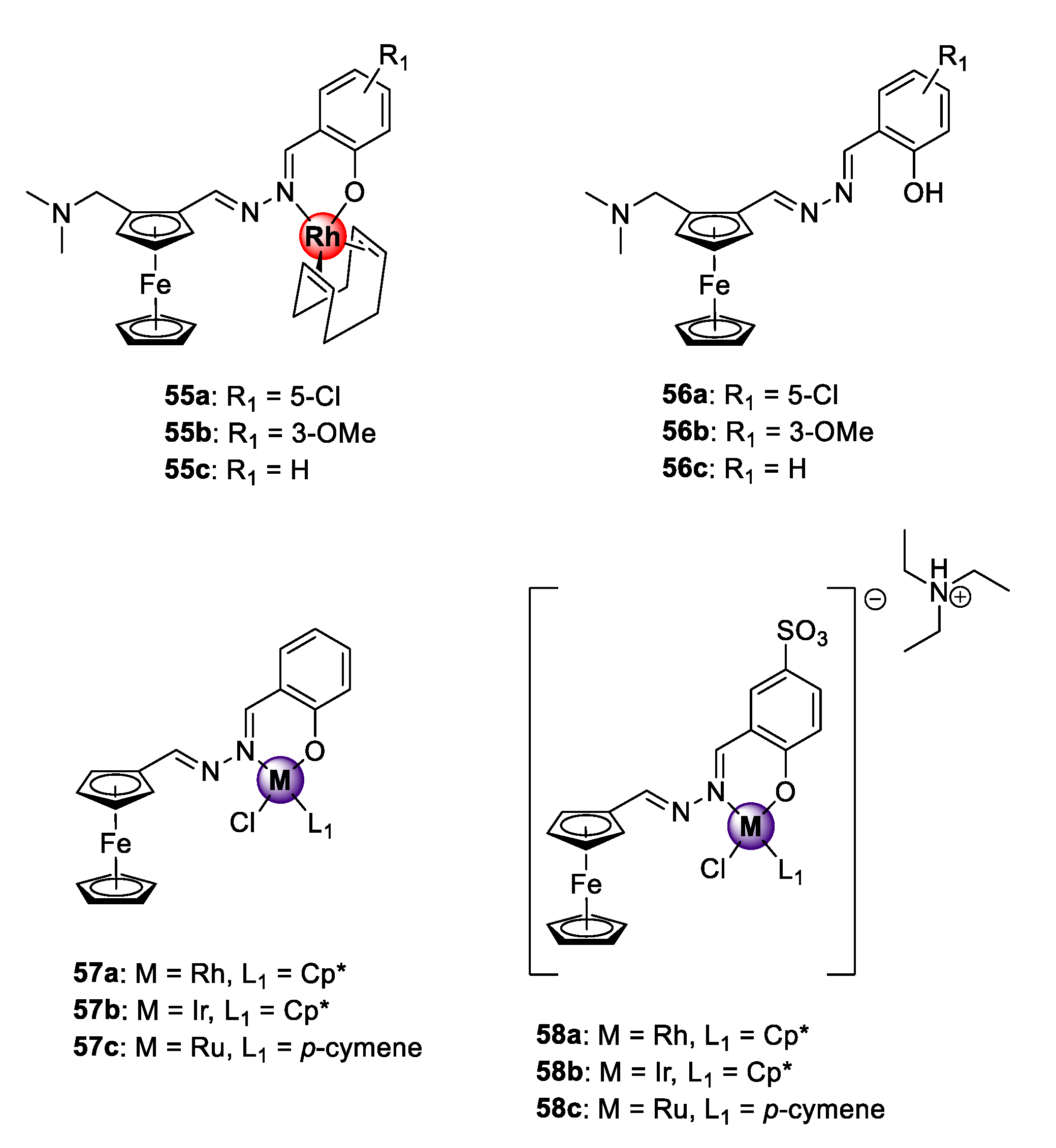
2.1. Quinoline-Salicylaldiminato/Picolamine/Imidazole Ligands
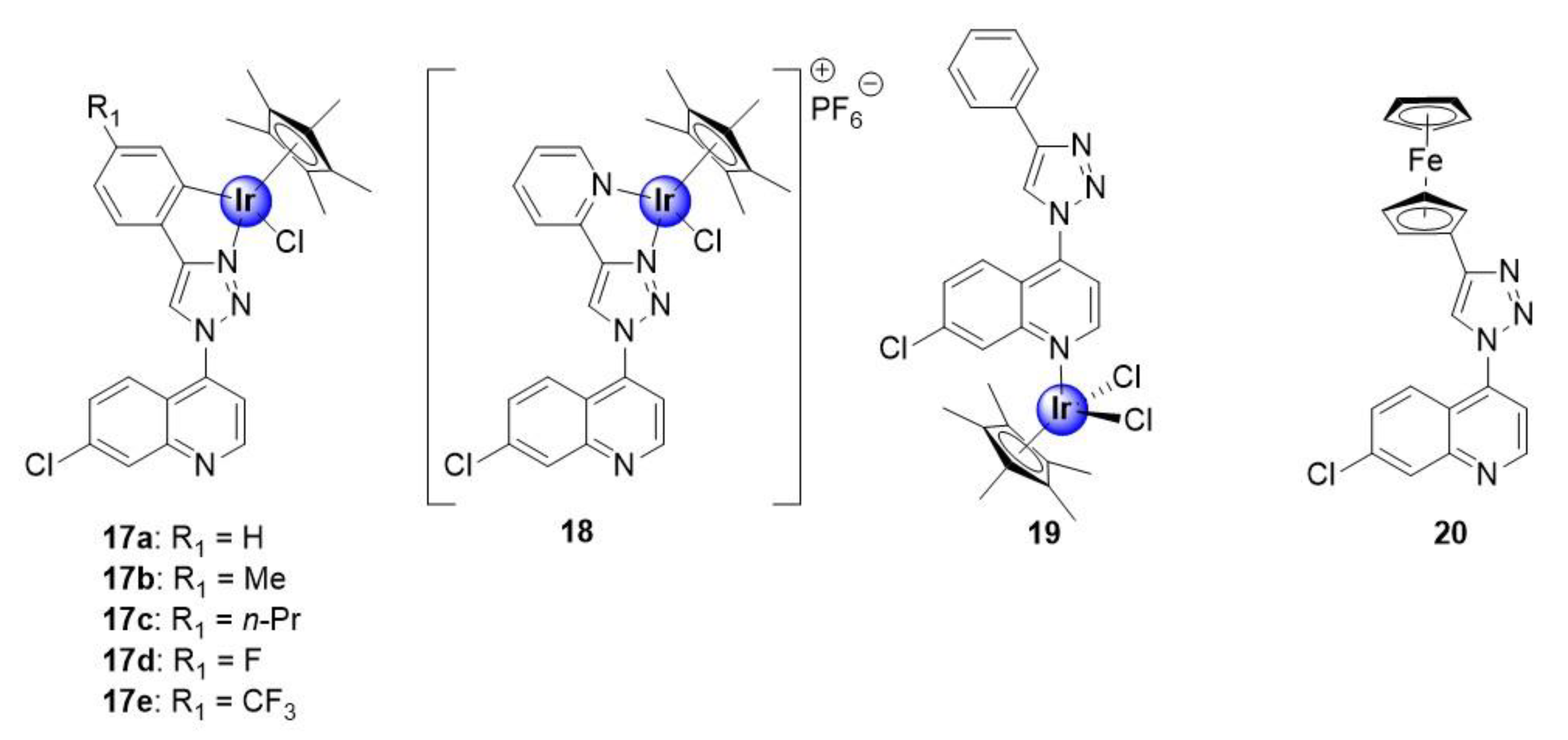
2.2. Quinoline-Triazole Ligands
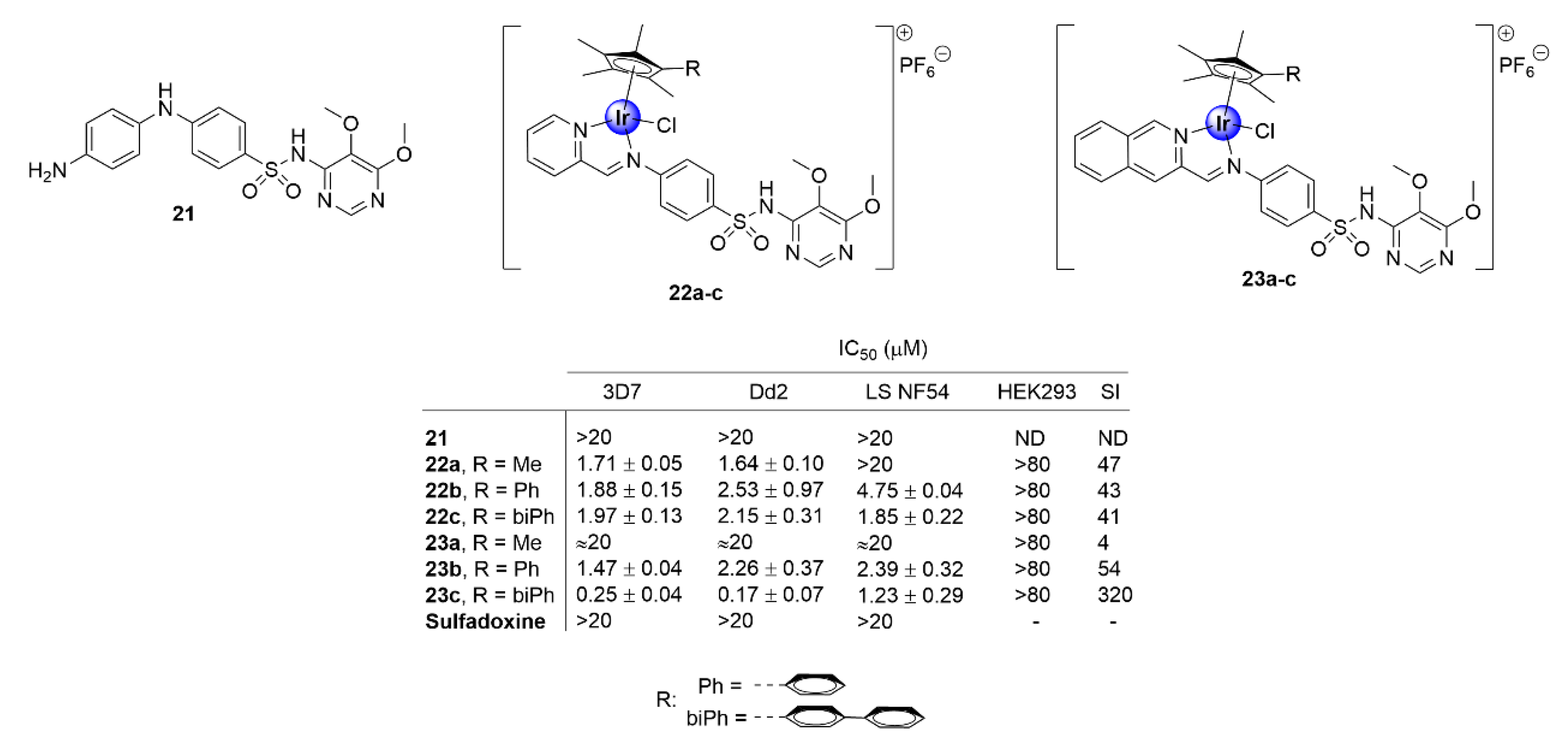
2.3. Sulfadoxine Ligands
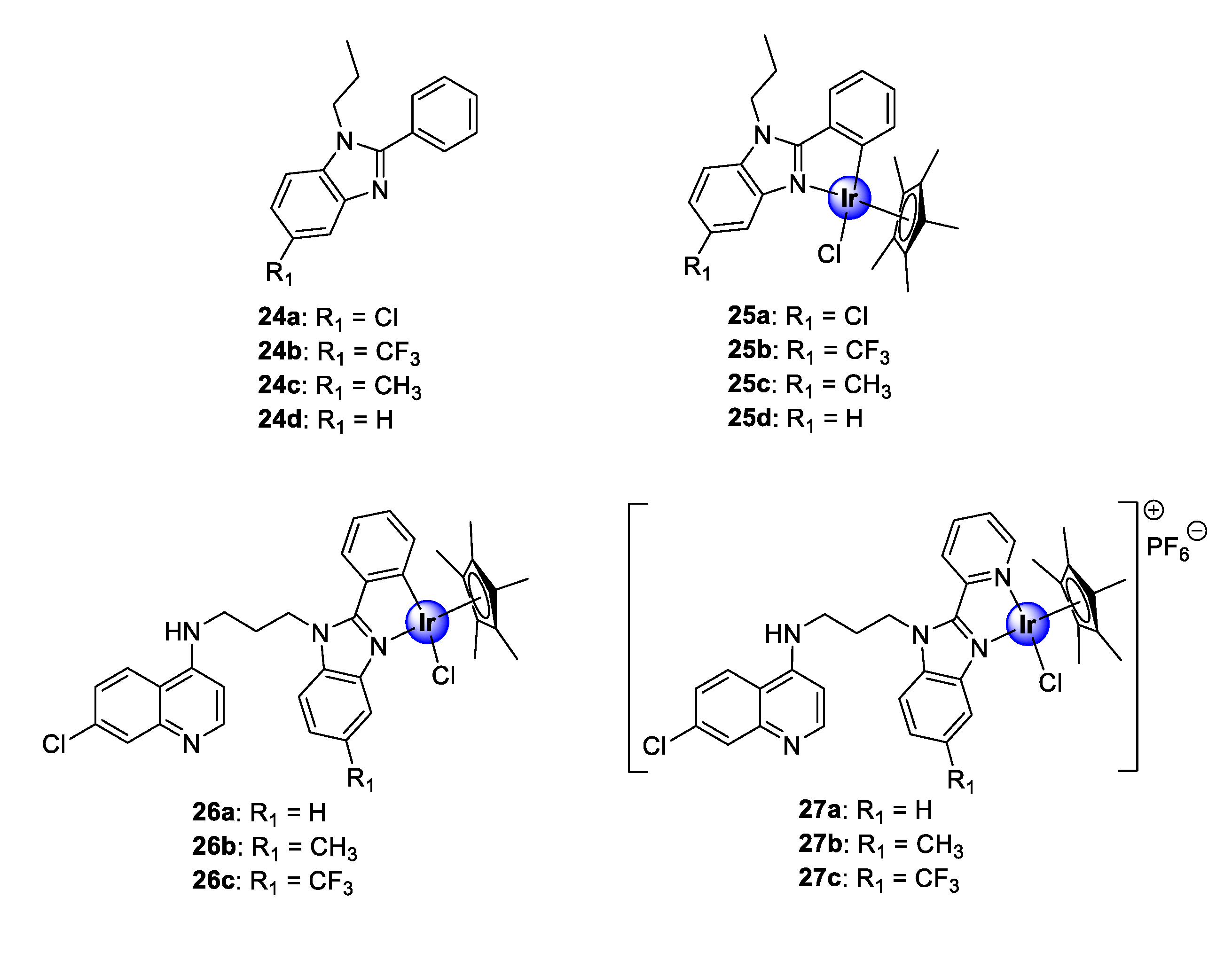
2.4. Benzimidazole (Hybrid) Ligands
3. Ruthenium Complexes
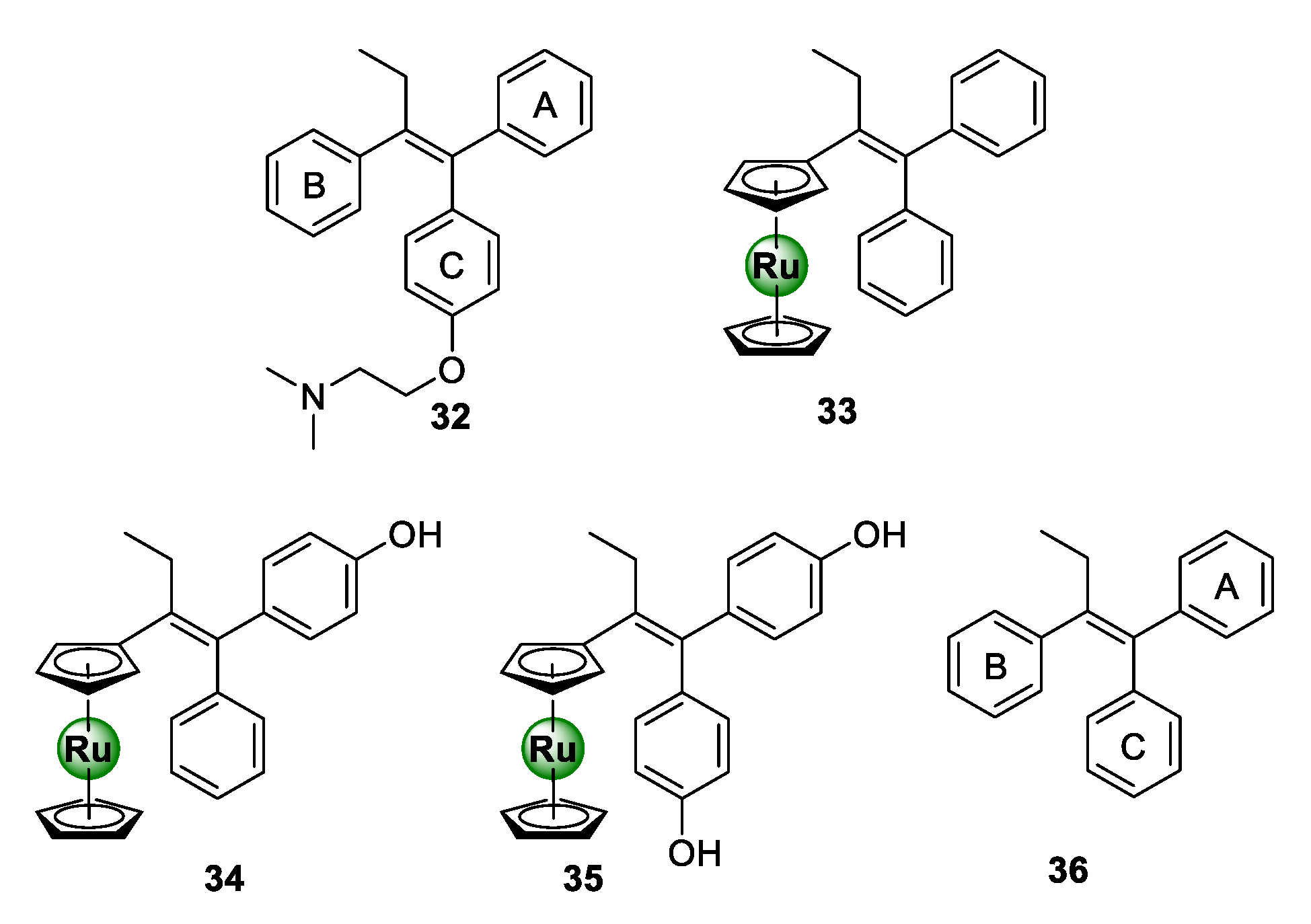
3.1. Tamoxifen Derivatives
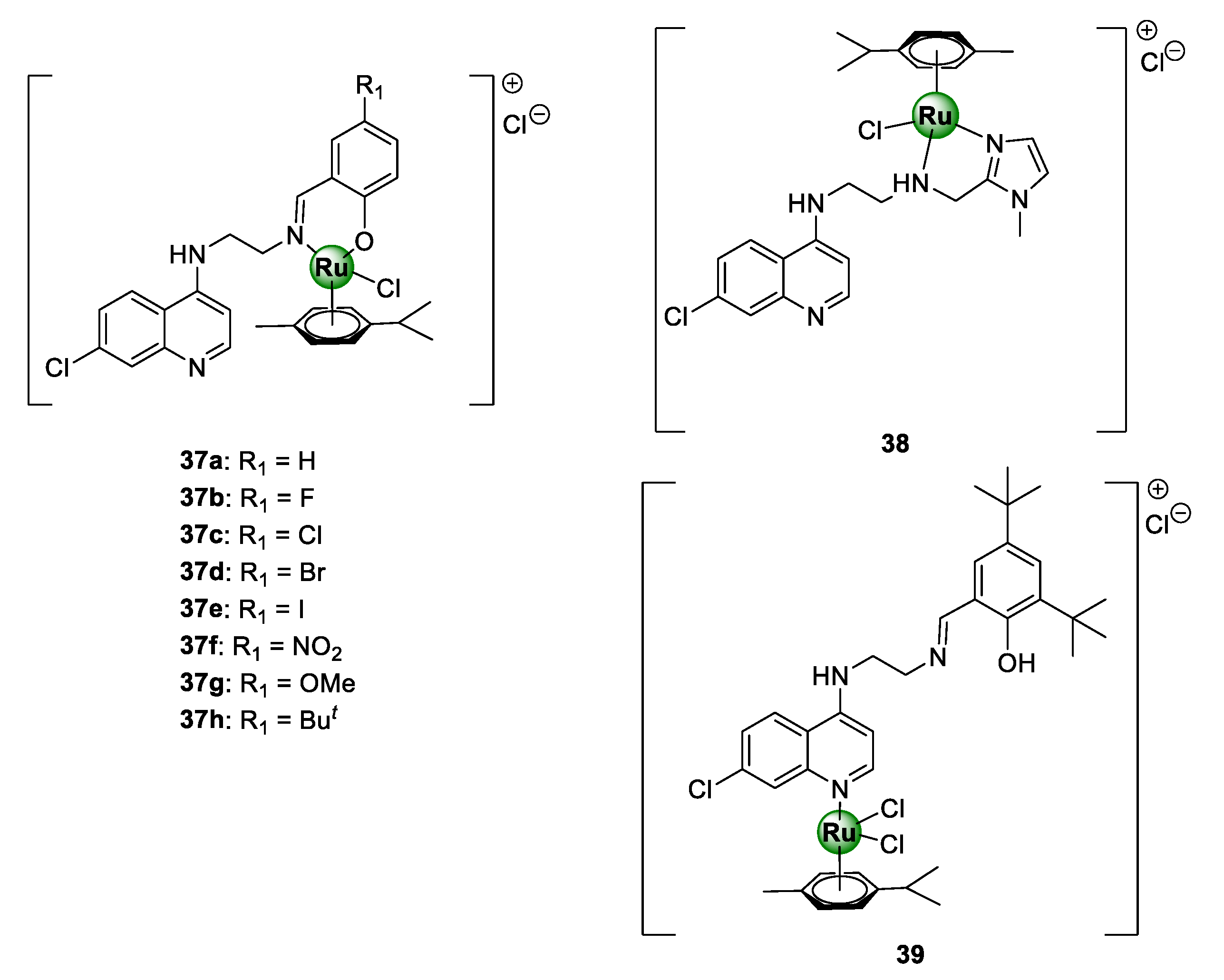
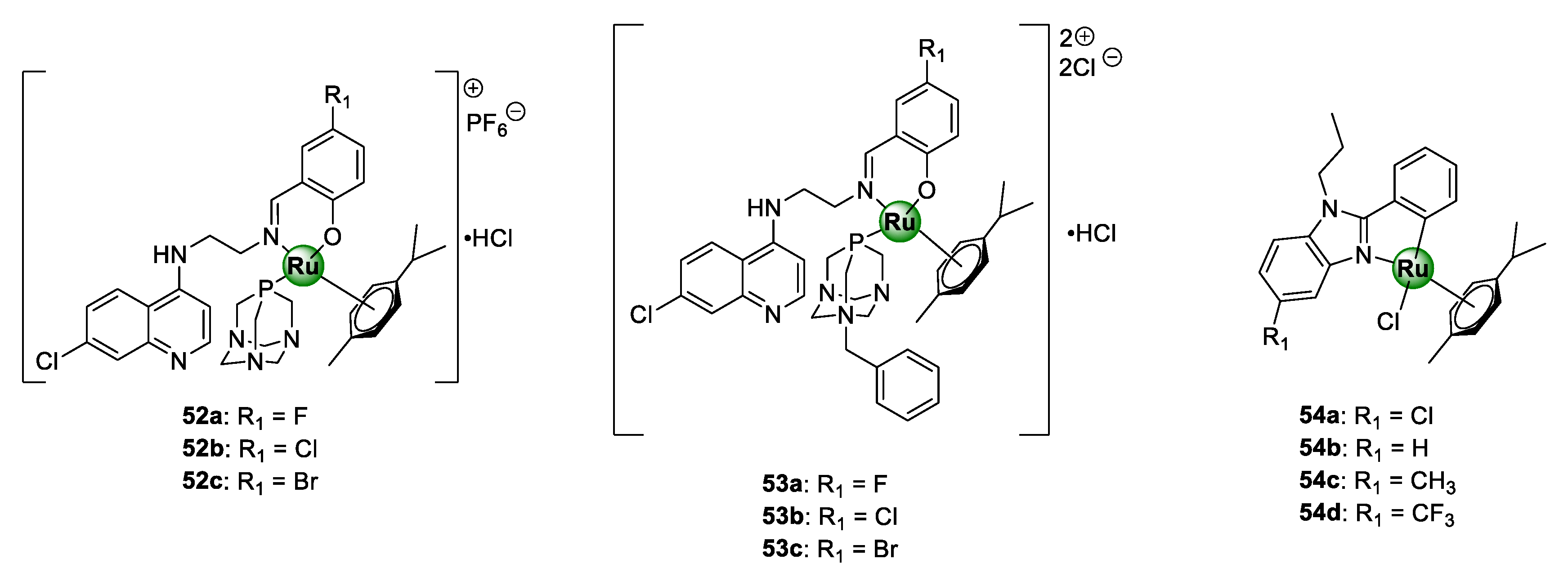
3.2. Quinoline-Salicylaldiminato/Imidazole Ligands
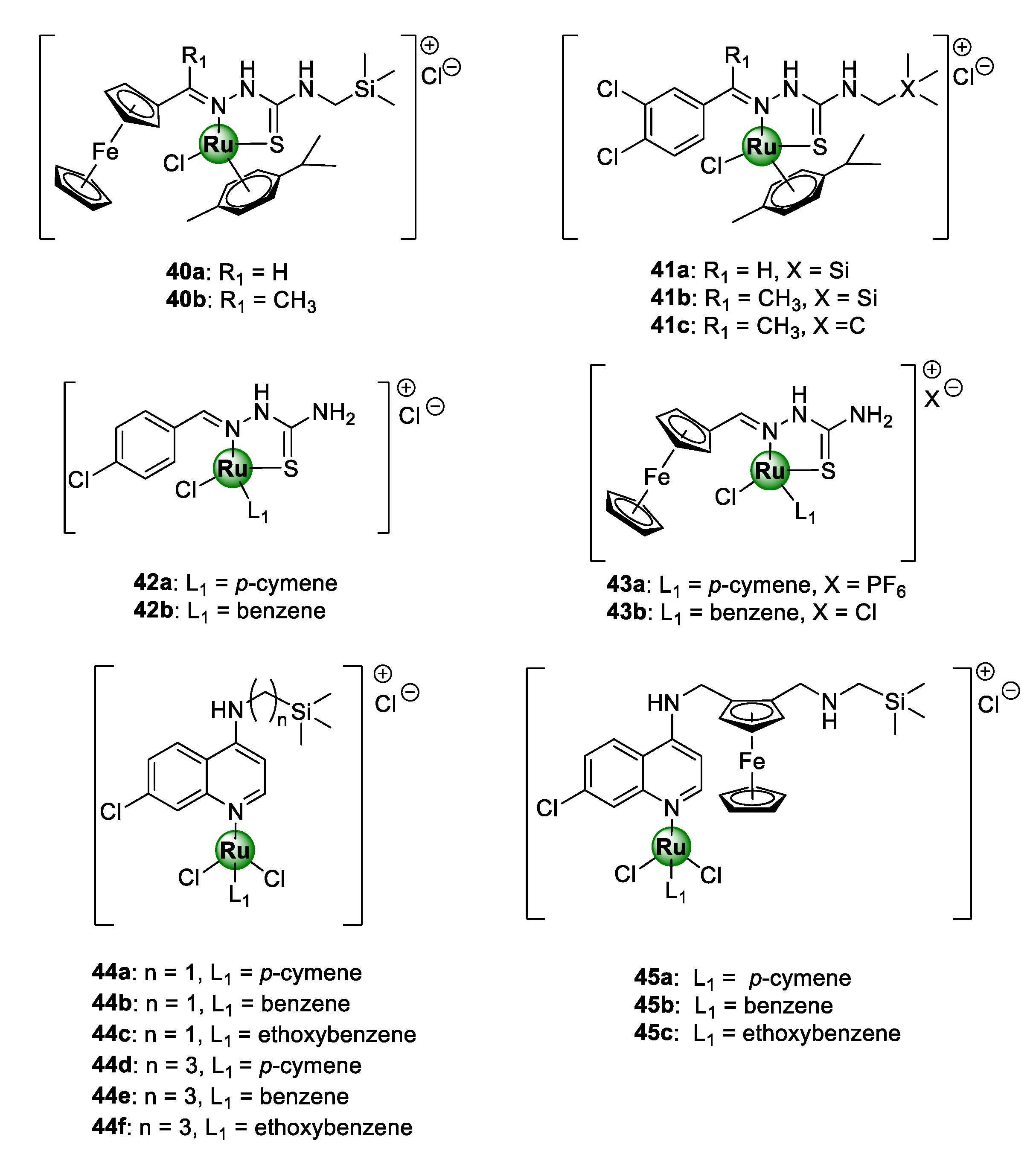
3.3. Thiosemicarbazone and Organosilane Derivatives
3.4. Quinoline-Trioxane Ligands
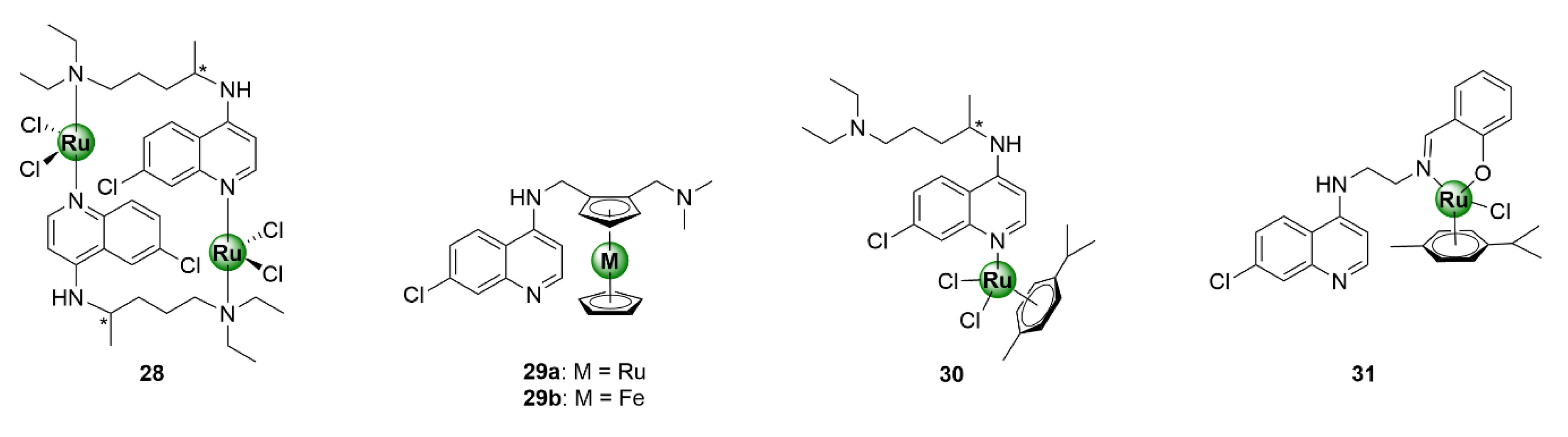
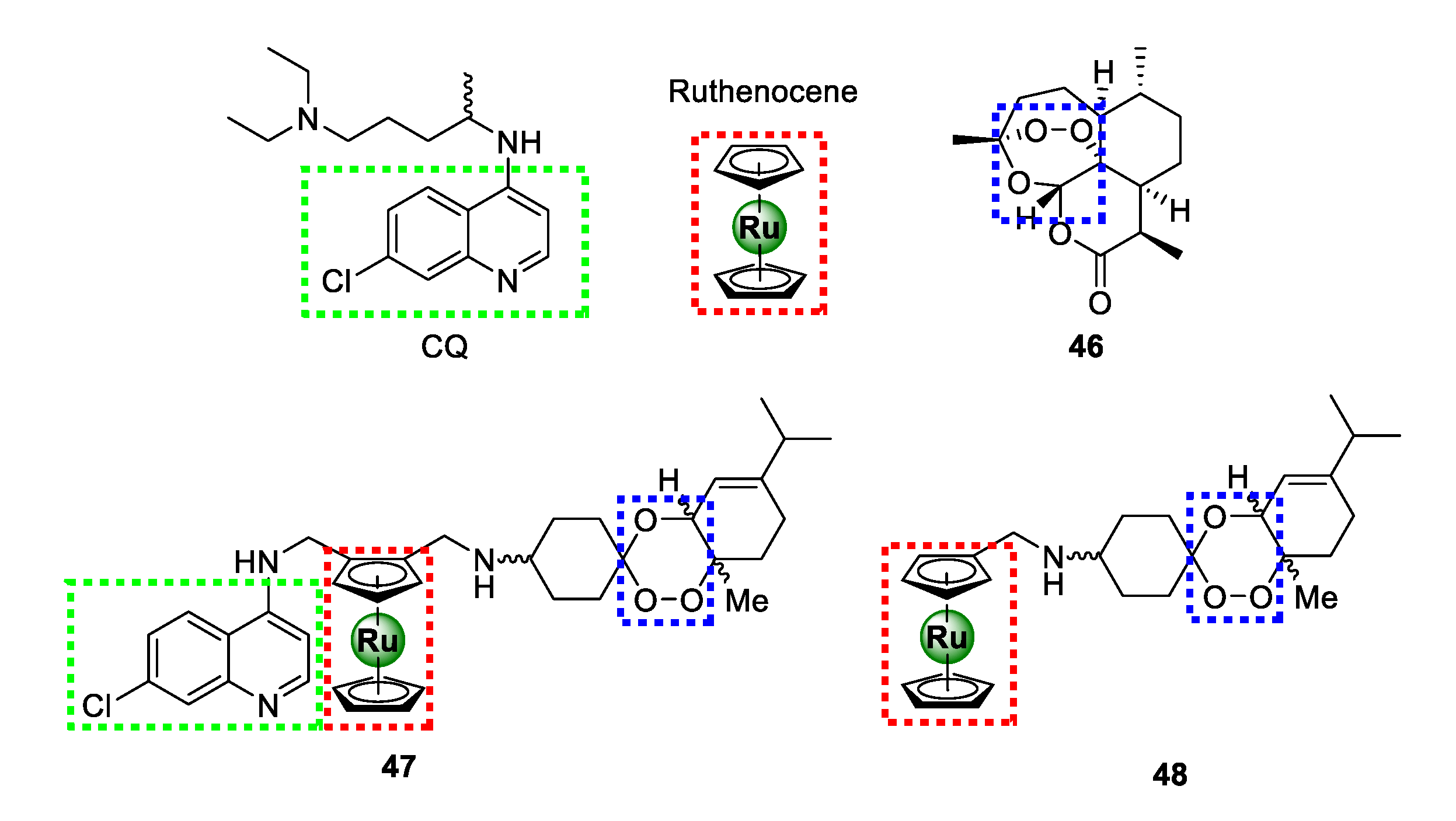
3.5. Heteroaromatic Ligands
3.6. Quinoline/Benzimidazole Ligands
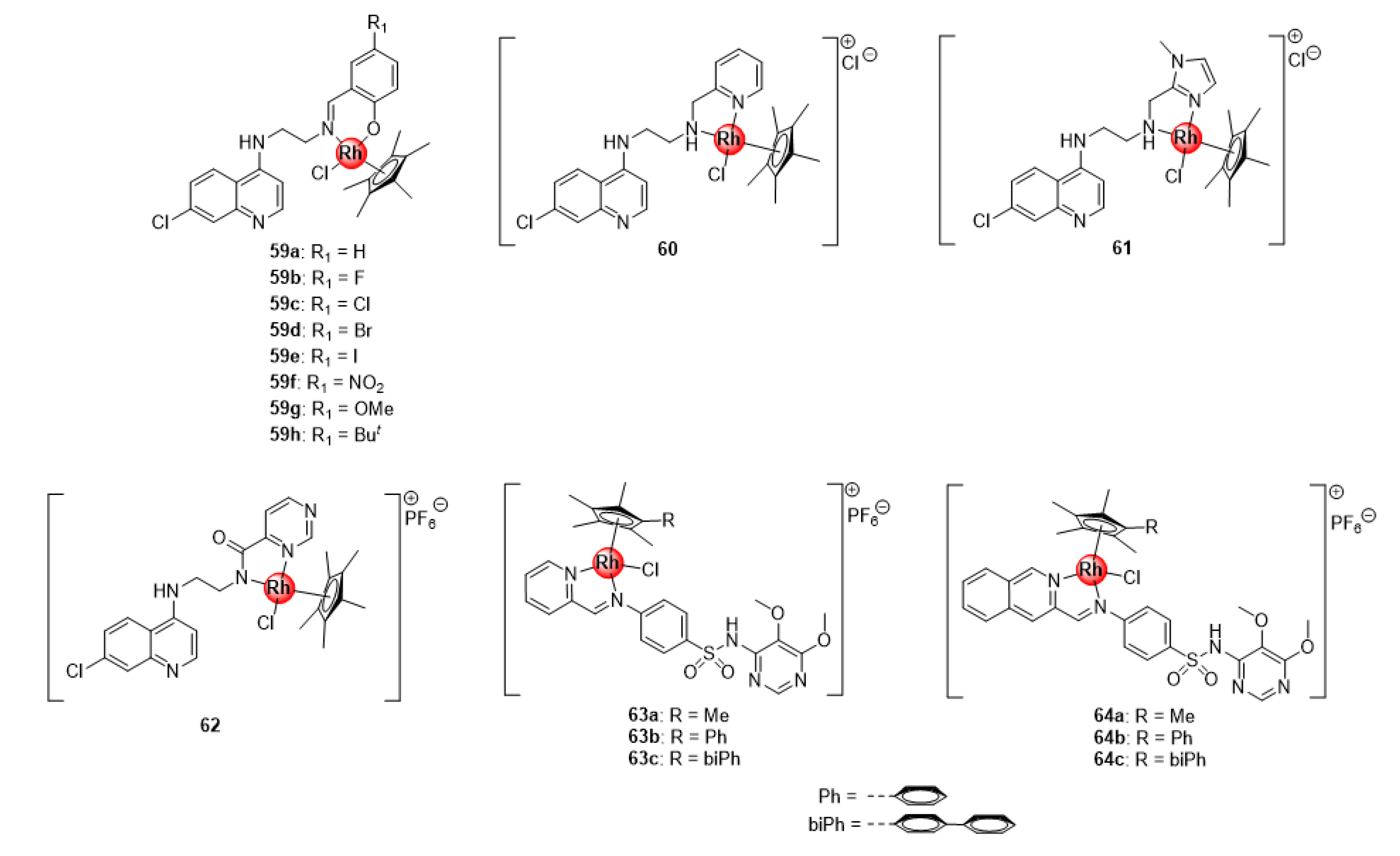
4. Rhodium Complexes
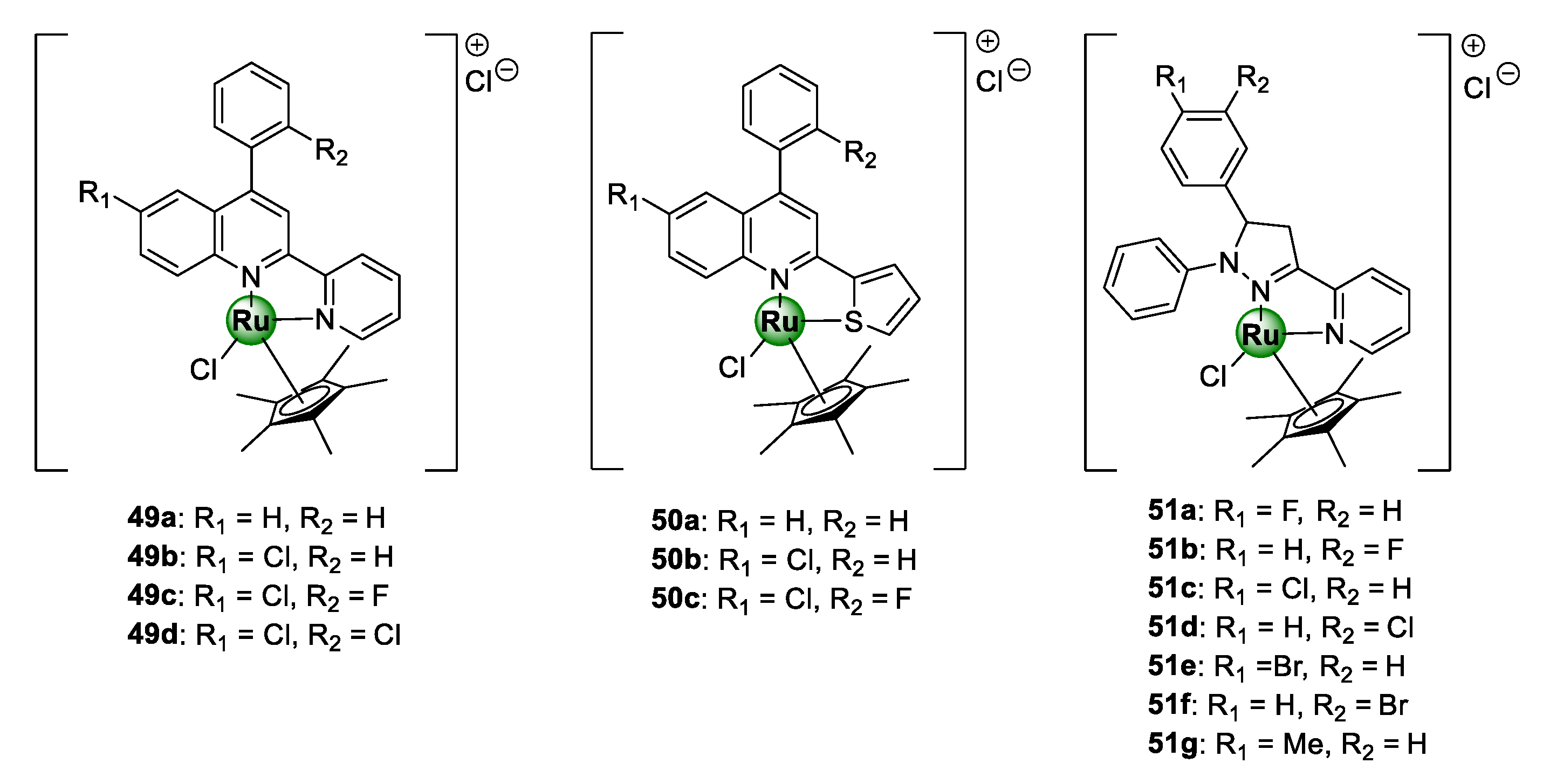
4.1. Salicylaldiminato Ligands
4.2. Quinoline-Salicylaldiminato and Sulfadoxine Ligands
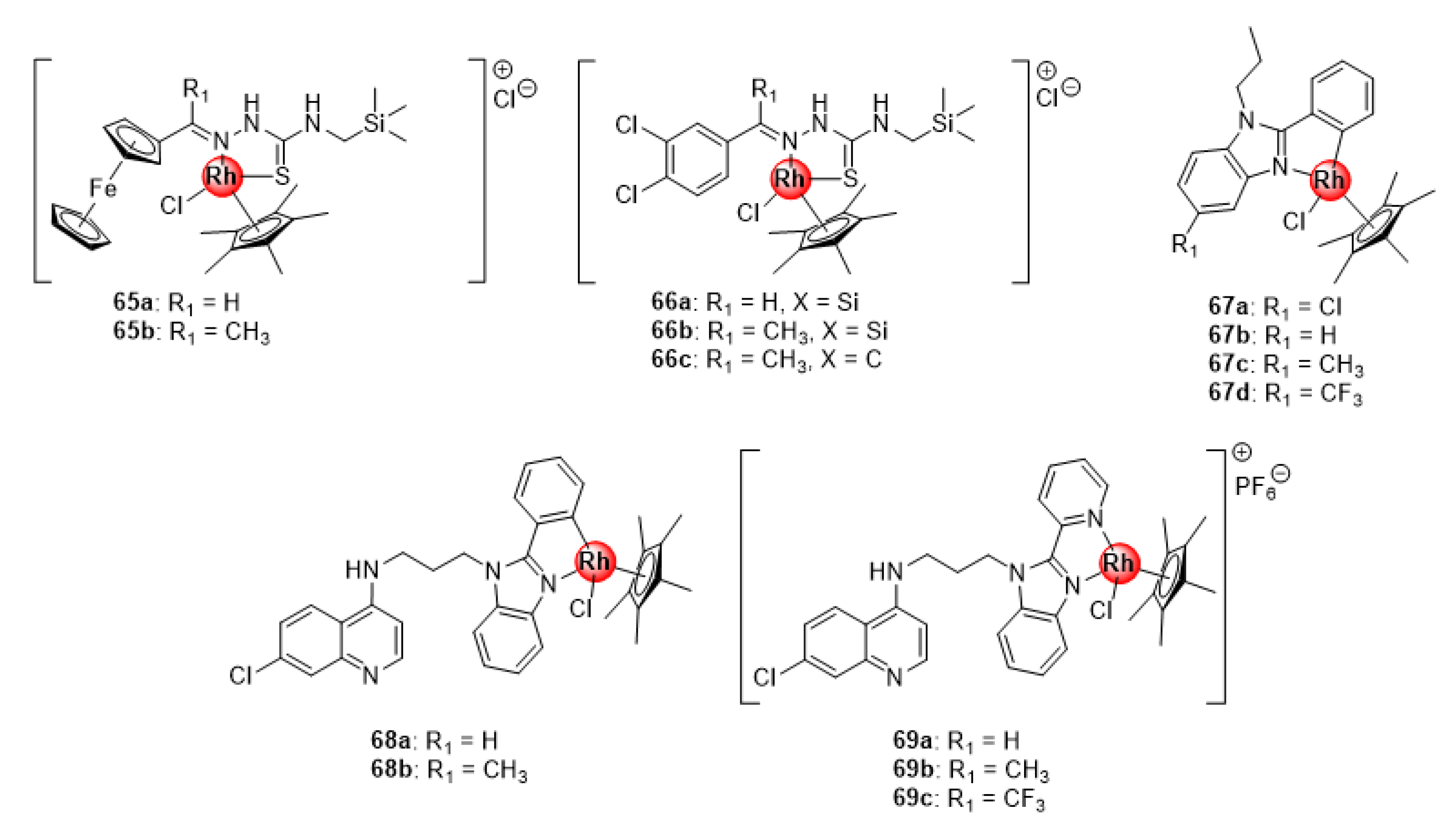
4.3. Thiosemicarbazone and Benzimidazole (Hybrid) Ligands
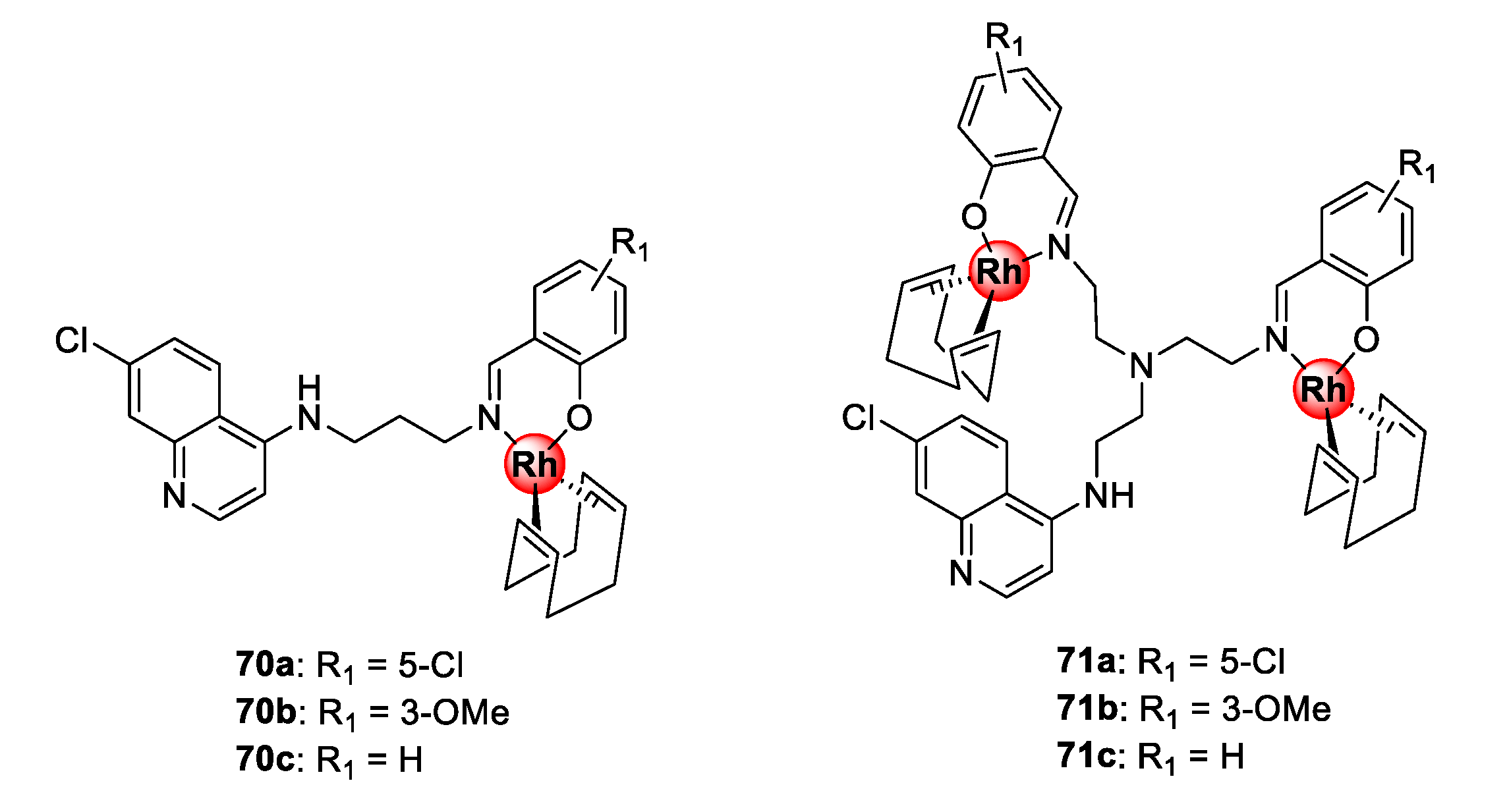
4.4. Quinoline-Polyamine Scaffolds
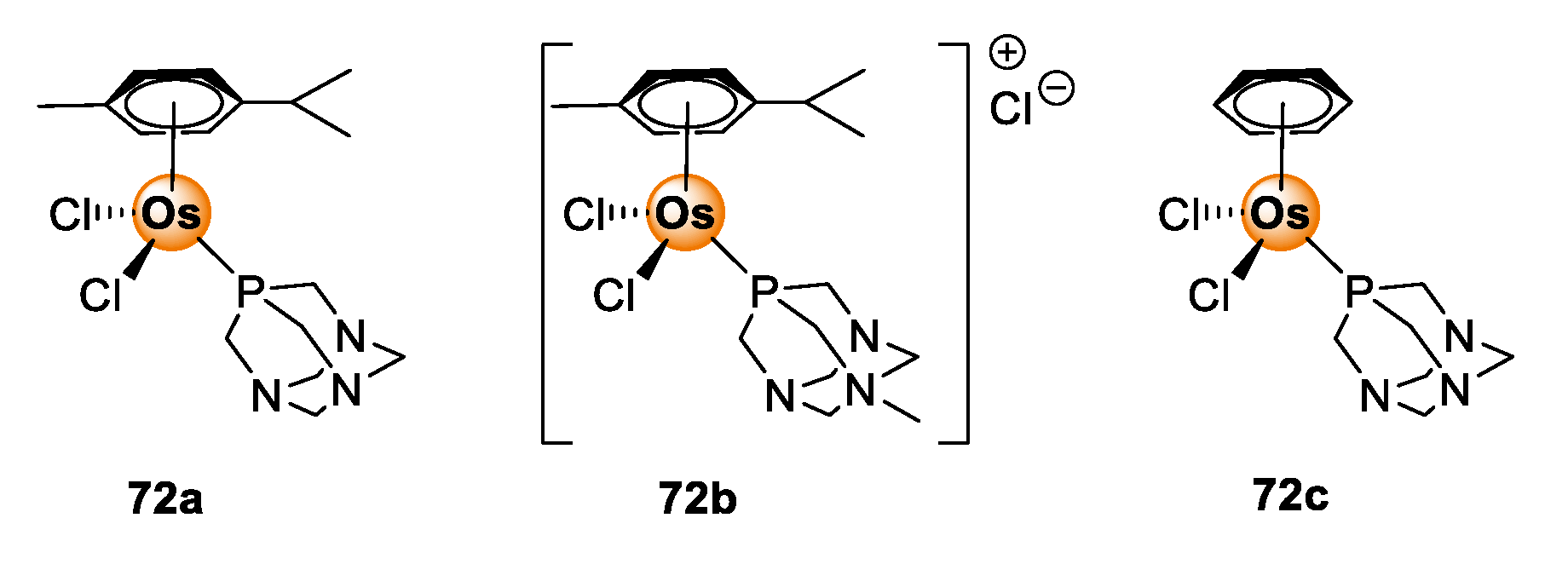
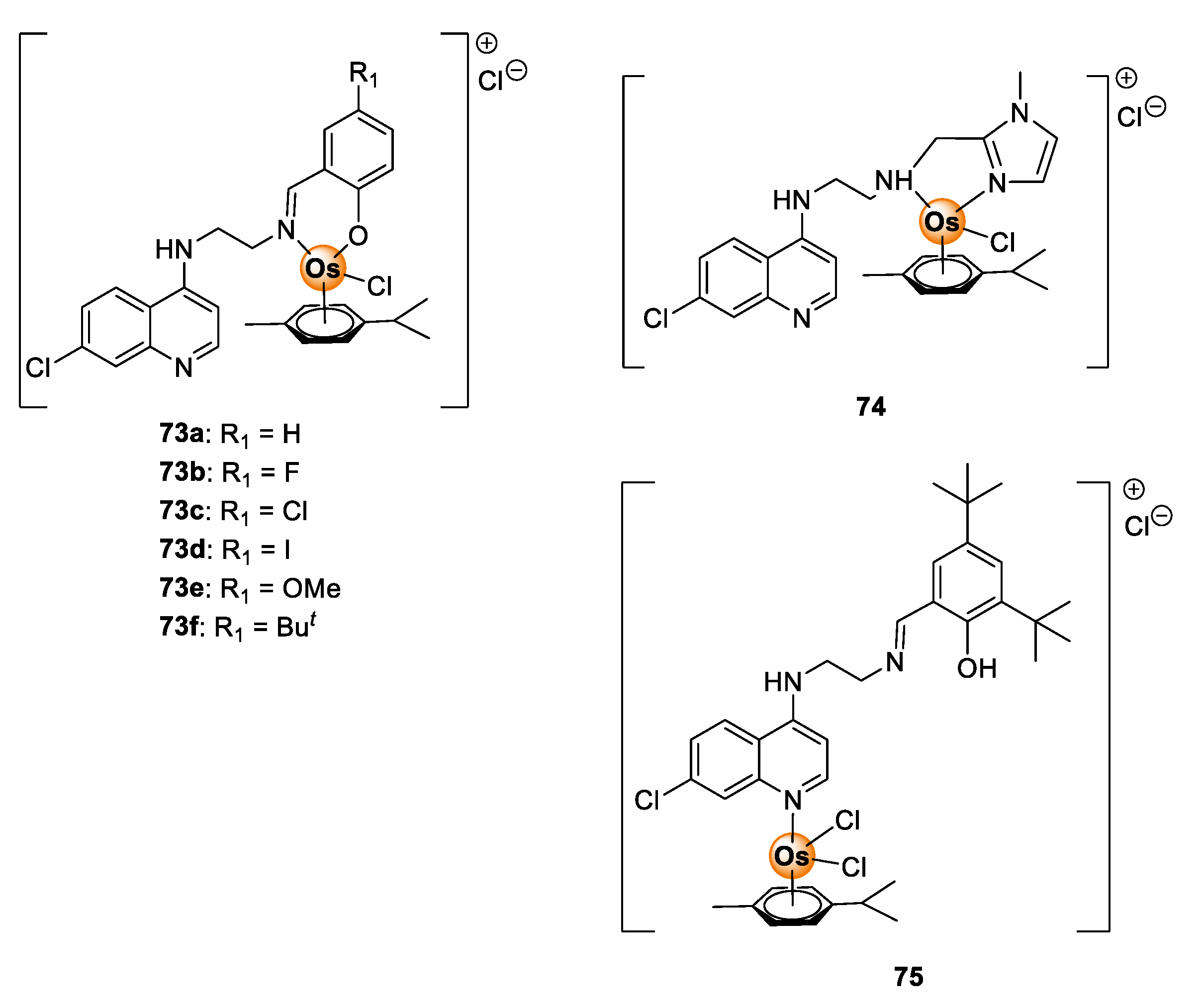
5. Osmium Complexes
Quinoline-Salicylaldiminato/Imidazole Ligands
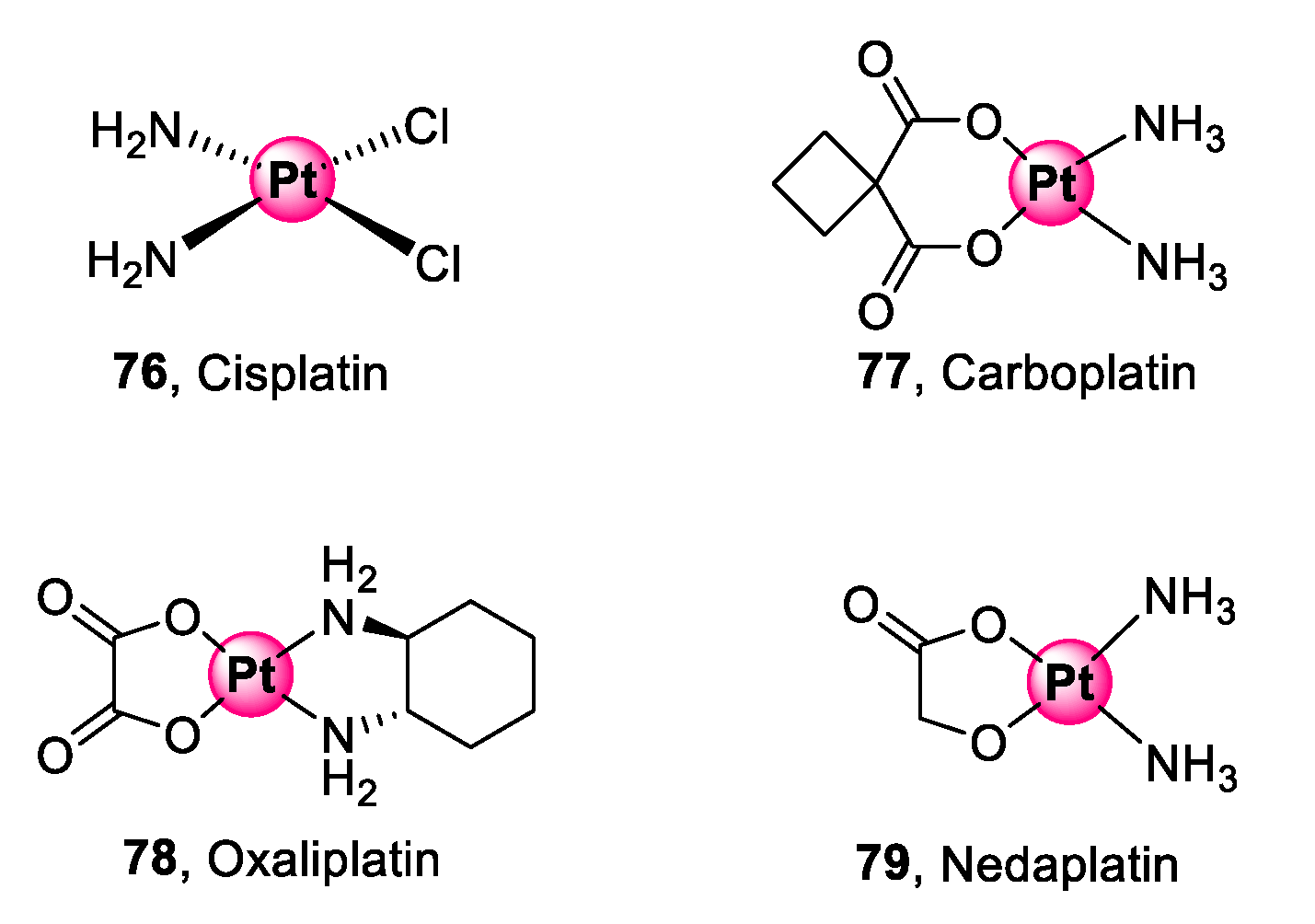
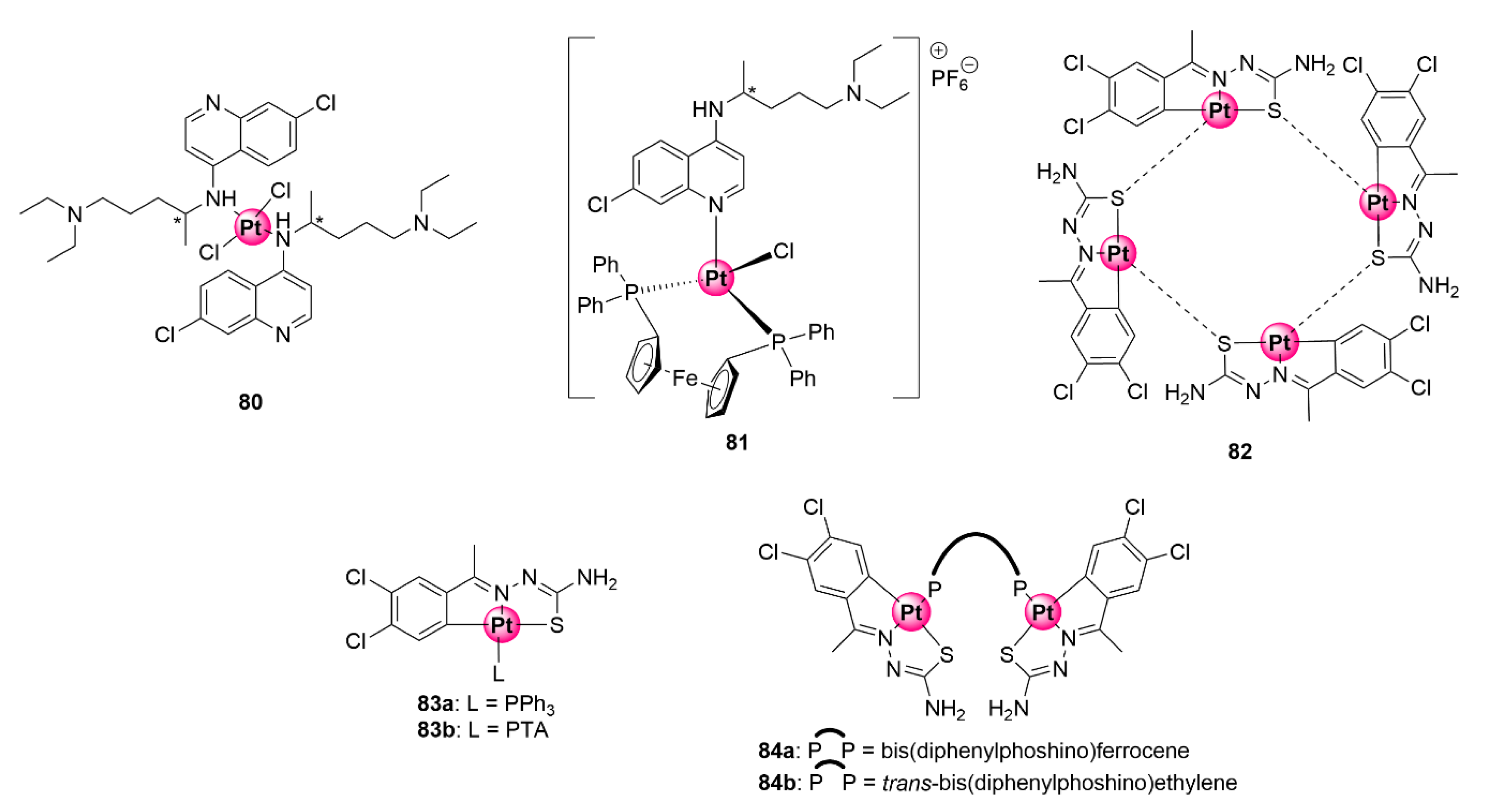
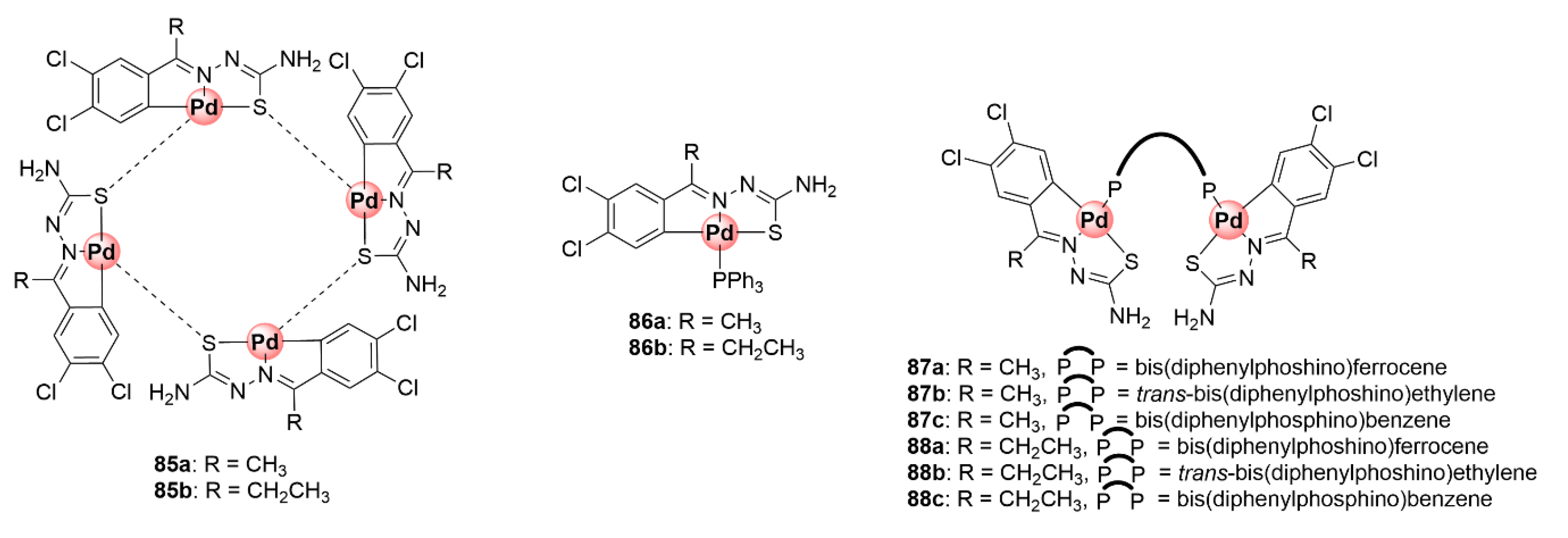
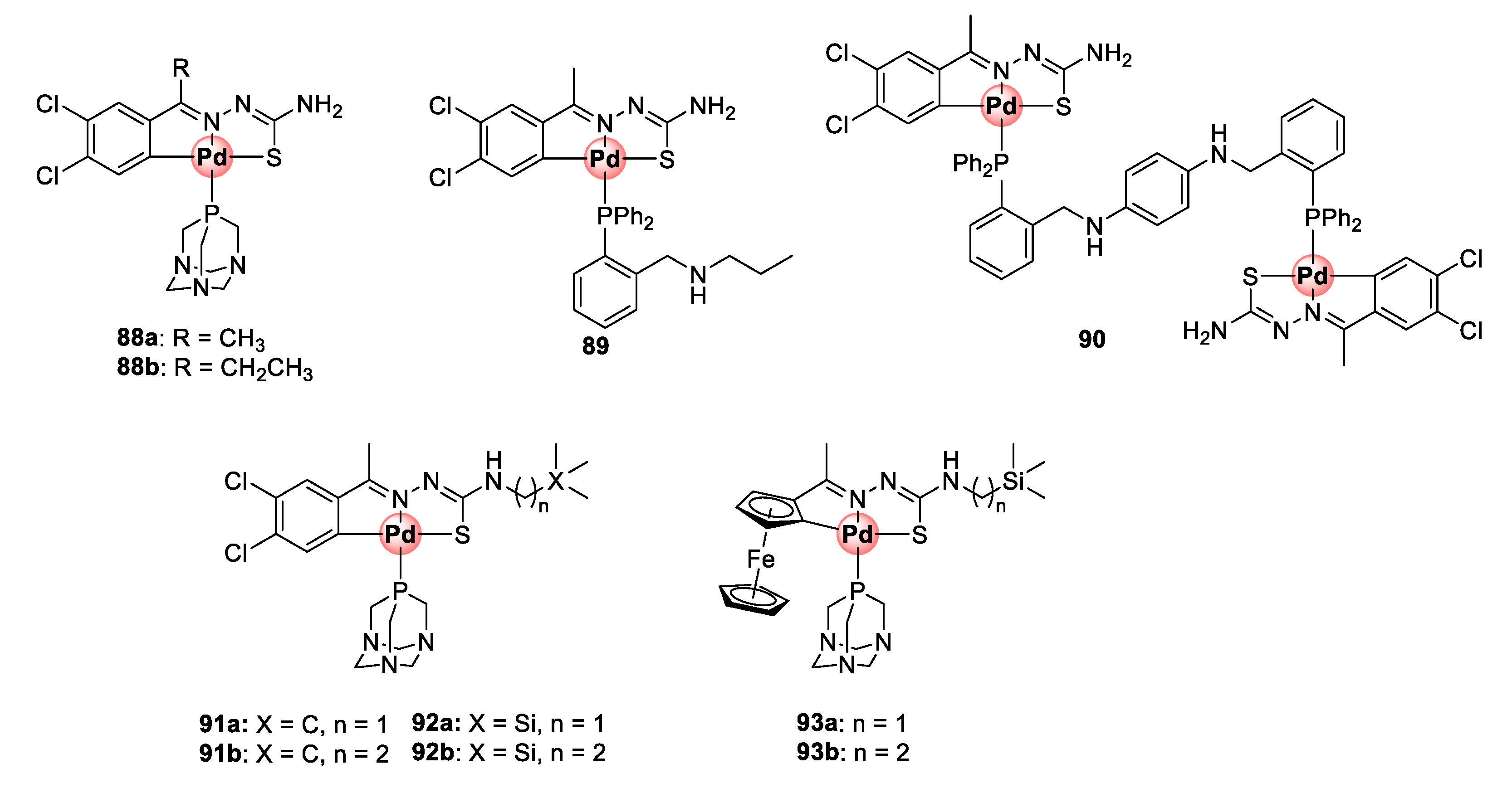
6. Platinum and Palladium Complexes
Thiosemicarbazone Ligands
7. Insights into Antiplasmodial Mechanisms of Action of Organometallic PGM Complexes
7.1. Blocking the Plasmodial Heme Biocrystallisation Pathway
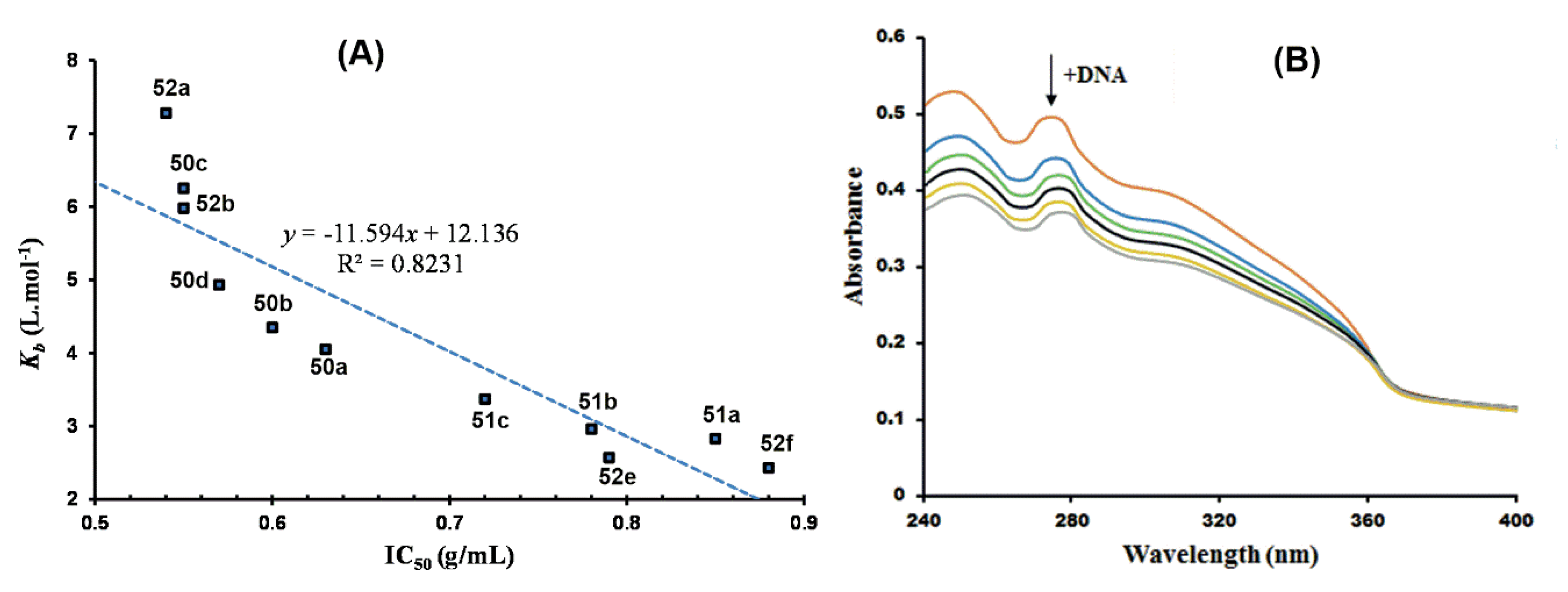
7.2. Targeting DNA Interaction for Antiplasmodial Activity
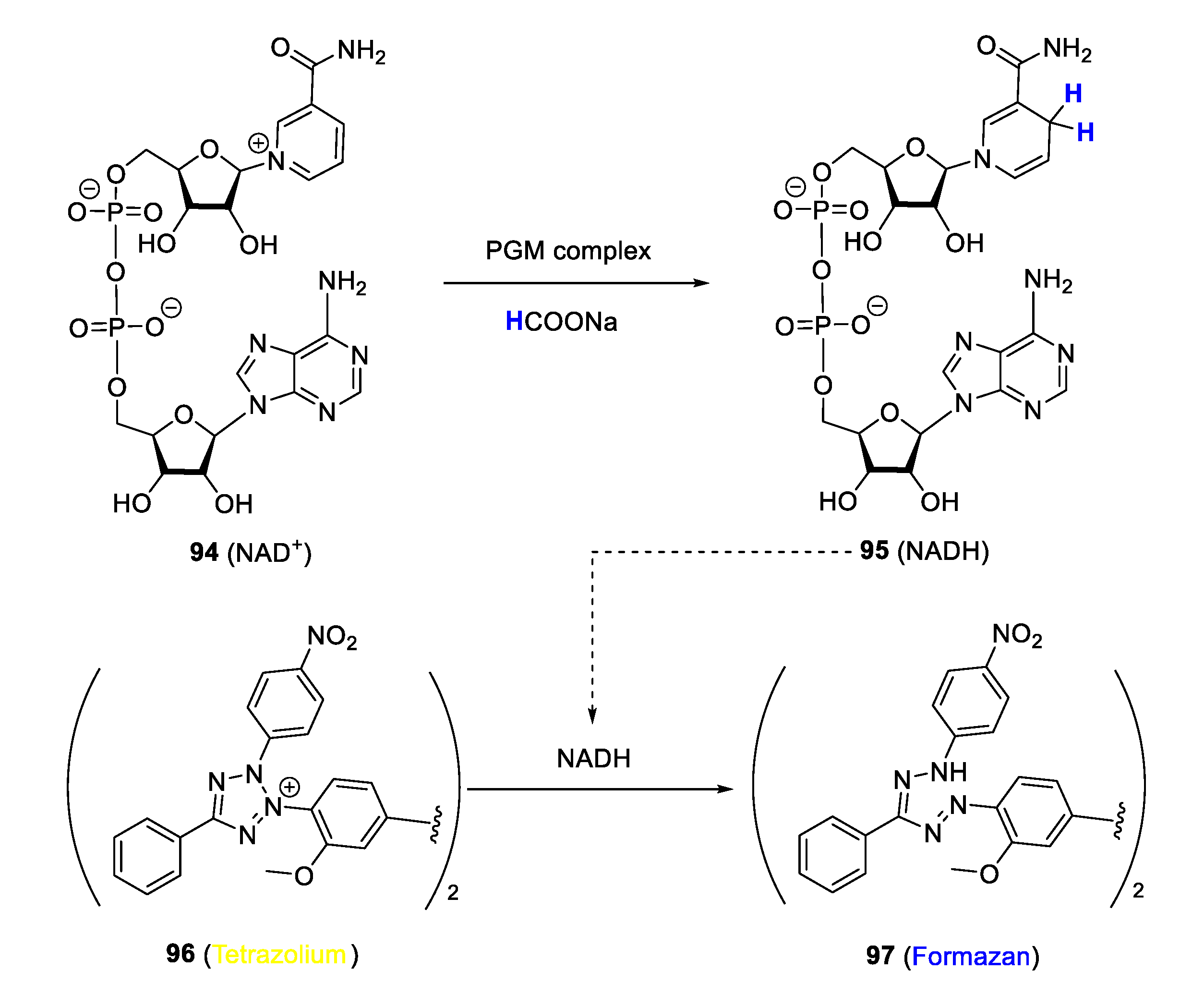
7.3. Disruption of Plasmodial Biochemical Processes by Intracellular Catalysis
8. Conclusions and Future Outlook
Author Contributions
Funding
Conflicts of Interest
References
- Global Polio Eradication Initiative Applauds WHO African Region for Wild Polio-Free Certification. Available online: https://www.who.int/news/item/25-08-2020-global-polio-eradication-initiative-applauds-who-african-region-for-wild-polio-free-certification (accessed on 15 October 2020).
- Malaria. Available online: https://www.who.int/news-room/fact-sheets/detail/malaria (accessed on 15 October 2020).
- Ashley, E.A.; White, N.J. Artemisinin-based combinations. Curr. Opin. Infect. Dis. 2005, 18, 531–536. [Google Scholar] [CrossRef]
- World Health Organization. World Malaria Report 2018; World Health Organization: Geneva, Switzerland, 2019. [Google Scholar]
- World Health Organization. Status Report on Artemisinin Resistance; World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Gasser, G.; Metzler-Nolte, N. The potential of organometallic complexes in medicinal chemistry. Curr. Opin. Chem. Biol. 2012, 16, 84–91. [Google Scholar] [CrossRef]
- Ghosh, S. Cisplatin: The first metal based anticancer drug. Bioorg. Chem. 2019, 88, 102925. [Google Scholar] [CrossRef]
- Hanif, M.; Hartinger, C.G. Anticancer metallodrugs: Where is the next cisplatin? Future Med. Chem. 2018, 10, 615–617. [Google Scholar] [CrossRef]
- Hartley, F.R. Chemistry of the Platinum Group Metals: Recent Developments; Elsevier Science Publishers B.V.: Amsterdam, The Netherlands, 1991; p. 639. [Google Scholar]
- Murray, B.S.; Babak, M.V.; Hartinger, C.G.; Dyson, P.J. The development of RAPTA compounds for the treatment of tumors. Coord. Chem. Rev. 2016, 306, 86–114. [Google Scholar] [CrossRef]
- Thota, S.; Rodrigues, D.A.; Crans, D.C.; Barreiro, E.J. Ru (II) compounds: Next-generation anticancer metallotherapeutics? J. Med. Chem. 2018, 61, 5805–5821. [Google Scholar] [CrossRef] [PubMed]
- Gichumbi, J.M.; Friedrich, H.B. Half-sandwich complexes of platinum group metals (Ir, Rh, Ru and Os) and some recent biological and catalytic applications. J. Organomet. Chem. 2018, 866, 123–143. [Google Scholar] [CrossRef]
- Deo, K.M.; Ang, D.L.; McGhie, B.; Rajamanickam, A.; Dhiman, A.; Khoury, A.; Holland, J.; Bjelosevic, A.; Pages, B.; Gordon, C. Platinum coordination compounds with potent anticancer activity. Coord. Chem. Rev. 2018, 375, 148–163. [Google Scholar] [CrossRef]
- Odularu, A.T.; Ajibade, P.A.; Mbese, J.Z.; Oyedeji, O.O. Developments in Platinum-Group Metals as Dual Antibacterial and Anticancer Agents. J. Chem. 2019, 2019, 5459461. [Google Scholar] [CrossRef]
- Karaküçük-İyidoğan, A.; Taşdemir, D.; Oruç-Emre, E.E.; Balzarini, J. Novel platinum (II) and palladium (II) complexes of thiosemicarbazones derived from 5-substitutedthiophene-2-carboxaldehydes and their antiviral and cytotoxic activities. Eur. J. Med. Chem. 2011, 46, 5616–5624. [Google Scholar] [CrossRef]
- Imran, M.; Ayub, W.; Butler, I.S. Photoactivated platinum-based anticancer drugs. Coord. Chem. Rev. 2018, 376, 405–429. [Google Scholar] [CrossRef]
- McFarland, S.A.; Mandel, A.; Dumoulin-White, R.; Gasser, G. Metal-based photosensitizers for photodynamic therapy: The future of multimodal oncology? Curr. Opin. Chem. Biol. 2020, 56, 23–27. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Delgado, R.A.; Anzellotti, A. Metal complexes as chemotherapeutic agents against tropical diseases: Trypanosomiasis, malaria and leishmaniasis. Mini Rev. Med. Chem. 2004, 4, 23–30. [Google Scholar]
- Biot, C.; Castro, W.; Botté, C.Y.; Navarro, M. The therapeutic potential of metal-based antimalarial agents: Implications for the mechanism of action. Dalton Trans. 2012, 41, 6335–6349. [Google Scholar] [CrossRef]
- Macedo, T.S.; Villarreal, W.; Couto, C.C.; Moreira, D.R.; Navarro, M.; Machado, M.; Prudêncio, M.; Batista, A.A.; Soares, M.B. Platinum (ii)–chloroquine complexes are antimalarial agents against blood and liver stages by impairing mitochondrial function. Metallomics 2017, 9, 1548–1561. [Google Scholar] [CrossRef]
- Navarro, M.; Pekerar, S.; Pérez, H.A. Synthesis, characterization and antimalarial activity of new iridium–chloroquine complexes. Polyhedron 2007, 26, 2420–2424. [Google Scholar] [CrossRef]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Taylor, D.; Smith, P.J.; Riedel, T.; Dyson, P.J.; Chibale, K. Synthesis and evaluation of new polynuclear organometallic Ru (II), Rh (III) and Ir (III) pyridyl ester complexes as in vitro antiparasitic and antitumor agents. Dalton Trans. 2014, 43, 513–526. [Google Scholar] [CrossRef]
- Nkoana, W.; Nyoni, D.; Chellan, P.; Stringer, T.; Taylor, D.; Smith, P.J.; Hutton, A.T.; Smith, G.S. Heterometallic half-sandwich complexes containing a ferrocenyl motif: Synthesis, molecular structure, electrochemistry and antiplasmodial evaluation. J. Organomet. Chem. 2014, 752, 67–75. [Google Scholar] [CrossRef]
- Ekengard, E.; Kumar, K.; Fogeron, T.; de Kock, C.; Smith, P.J.; Haukka, M.; Monari, M.; Nordlander, E. Pentamethylcyclopentadienyl-rhodium and iridium complexes containing (N^ N and N^ O) bound chloroquine analogue ligands: Synthesis, characterization and antimalarial properties. Dalton Trans. 2016, 45, 3905–3917. [Google Scholar] [CrossRef] [PubMed]
- Ekengard, E.; Bergare, I.; Hansson, J.; Doverbratt, I.; Monari, M.; Gordhan, B.; Kana, B.; Kock, C.D.; Smith, P.J.; Nordlander, E. A pyrazine amide-4-aminoquinoline hybrid and its rhodium and iridium pentamethylcyclopentadienyl complexes; evaluation of anti-mycobacterial and anti-plasmodial activities. J. Mex. Chem. 2017, 61, 158–166. [Google Scholar] [CrossRef]
- Stringer, T.; Quintero, M.A.S.; Wiesner, L.; Smith, G.S.; Nordlander, E. Evaluation of PTA-derived ruthenium (II) and iridium (III) quinoline complexes against chloroquine-sensitive and resistant strains of the Plasmodium falciparum malaria parasite. J. Inorg. Biochem. 2019, 191, 164–173. [Google Scholar] [CrossRef] [PubMed]
- Su, W.; Li, Y.; Li, P. Design of Ru-arene complexes for antitumor drugs. Mini Rev. Med. Chem. 2018, 18, 184–193. [Google Scholar] [CrossRef] [PubMed]
- Lehane, A.M.; Kirk, K. Efflux of a range of antimalarial drugs and ‘chloroquine resistance reversers’ from the digestive vacuole in malaria parasites with mutant PfCRT. Mol. Microbiol. 2010, 77, 1039–1051. [Google Scholar] [CrossRef] [PubMed]
- Melis, D.R.; Barnett, C.B.; Wiesner, L.; Nordlander, E.; Smith, G.S. Quinoline-triazole half-sandwich iridium (III) complexes: Synthesis, antiplasmodial activity and preliminary transfer hydrogenation studies. Dalton Trans. 2020, 49, 11543–11555. [Google Scholar] [CrossRef] [PubMed]
- Chellan, P.; Avery, V.M.; Duffy, S.; Triccas, J.A.; Nagalingam, G.; Tam, C.; Cheng, L.W.; Liu, J.; Land, K.M.; Clarkson, G.J. Organometallic conjugates of the drug sulfadoxine for combatting antimicrobial resistance. Chem. Eur. J. 2018, 24, 10078–10090. [Google Scholar] [CrossRef] [PubMed]
- Rylands, L.-I.; Welsh, A.; Maepa, K.; Stringer, T.; Taylor, D.; Chibale, K.; Smith, G.S. Structure-activity relationship studies of antiplasmodial cyclometallated ruthenium (II), rhodium (III) and iridium (III) complexes of 2-phenylbenzimidazoles. Eur. J. Med. Chem. 2019, 161, 11–21. [Google Scholar] [CrossRef]
- Baartzes, N.; Jordaan, A.; Warner, D.F.; Combrinck, J.; Taylor, D.; Chibale, K.; Smith, G.S. Antimicrobial evaluation of neutral and cationic iridium (III) and rhodium (III) aminoquinoline-benzimidazole hybrid complexes. Eur. J. Med. Chem. 2020, 206, 112694. [Google Scholar] [CrossRef]
- Grubbs, R.H. Olefin-metathesis catalysts for the preparation of molecules and materials (Nobel lecture). Angew. Chem. Int. Ed. 2006, 45, 3760–3765. [Google Scholar] [CrossRef]
- Chakrabarti, M.H.; Roberts, E.P.L.; Bae, C.; Saleem, M. Ruthenium based redox flow battery for solar energy storage. Energy Convers. Manag. 2011, 52, 2501–2508. [Google Scholar] [CrossRef]
- Gill, M.R.; Thomas, J.A. Ruthenium (II) polypyridyl complexes and DNA—From structural probes to cellular imaging and therapeutics. Chem. Soc. Rev. 2012, 41, 3179–3192. [Google Scholar] [CrossRef]
- Levina, A.; Mitra, A.; Lay, P.A. Recent developments in ruthenium anticancer drugs. Metallomics 2009, 1, 458–470. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Lai, H.; Xiong, Z.; Chen, B.; Chen, T. Functionalization and cancer-targeting design of ruthenium complexes for precise cancer therapy. Chem. Commun. 2019, 55, 9904–9914. [Google Scholar] [CrossRef]
- Sánchez-Delgado, R.A.; Navarro, M.; Pérez, H.; Urbina, J.A. Toward a novel metal-based chemotherapy against tropical diseases. 2. Synthesis and antimalarial activity in vitro and in vivo of new ruthenium− and rhodium− chloroquine complexes. J. Med. Chem. 1996, 39, 1095–1099. [Google Scholar] [CrossRef] [PubMed]
- Beagley, P.; Blackie, M.A.; Chibale, K.; Clarkson, C.; Moss, J.R.; Smith, P.J. Synthesis and antimalarial activity in vitro of new ruthenocene–chloroquine analogues. J. Chem. Soc. Dalton Trans. 2002, 4426–4433. [Google Scholar] [CrossRef]
- Martínez, A.; Rajapakse, C.S.; Jalloh, D.; Dautriche, C.; Sánchez-Delgado, R.A. The antimalarial activity of Ru–chloroquine complexes against resistant Plasmodium falciparum is related to lipophilicity, basicity, and heme aggregation inhibition ability near water/n-octanol interfaces. J. Biol. Inorg. Chem. 2009, 14, 863–871. [Google Scholar] [CrossRef] [PubMed]
- Rajapakse, C.S.; Martinez, A.; Naoulou, B.; Jarzecki, A.A.; Suarez, L.; Deregnaucourt, C.; Sinou, V.; Schrevel, J.; Musi, E.; Ambrosini, G. Synthesis, characterization, and in vitro antimalarial and antitumor activity of new ruthenium (II) complexes of chloroquine. Inorg. Chem. 2009, 48, 1122–1131. [Google Scholar] [CrossRef]
- Glans, L.; Ehnbom, A.; De Kock, C.; Martínez, A.; Estrada, J.; Smith, P.J.; Haukka, M.; Sánchez-Delgado, R.A.; Nordlander, E. Ruthenium (II) arene complexes with chelating chloroquine analogue ligands: Synthesis, characterization and in vitro antimalarial activity. Dalton Trans. 2012, 41, 2764–2773. [Google Scholar] [CrossRef]
- Biot, C.; Dubar, F.; Khalife, J.; Slomianny, C. Opening up the advantages of the ruthenocenic bioprobes of ferroquine: Distribution and localization in Plasmodium falciparum-infected erythrocytes. Metallomics 2012, 4, 780–783. [Google Scholar] [CrossRef]
- Souza, N.B.D.; Aguiar, A.C.C.; Oliveira, A.C.D.; Top, S.; Pigeon, P.; Jaouen, G.; Goulart, M.O.F.; Krettli, A.U. Antiplasmodial activity of iron (II) and ruthenium (II) organometallic complexes against Plasmodium falciparum blood parasites. Mem. Inst. Oswaldo Cruz 2015, 110, 981–988. [Google Scholar] [CrossRef]
- Ekengard, E.; Glans, L.; Cassells, I.; Fogeron, T.; Govender, P.; Stringer, T.; Chellan, P.; Lisensky, G.C.; Hersh, W.H.; Doverbratt, I. Antimalarial activity of ruthenium (II) and osmium (II) arene complexes with mono-and bidentate chloroquine analogue ligands. Dalton Trans. 2015, 44, 19314–19329. [Google Scholar] [CrossRef]
- Adams, M.; de Kock, C.; Smith, P.J.; Land, K.M.; Liu, N.; Hopper, M.; Hsiao, A.; Burgoyne, A.R.; Stringer, T.; Meyer, M. Improved antiparasitic activity by incorporation of organosilane entities into half-sandwich ruthenium (II) and rhodium (III) thiosemicarbazone complexes. Dalton Trans. 2015, 44, 2456–2468. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.; Li, Y.; Khot, H.; De Kock, C.; Smith, P.J.; Land, K.; Chibale, K.; Smith, G.S. The synthesis and antiparasitic activity of aryl-and ferrocenyl-derived thiosemicarbazone ruthenium (II)–arene complexes. Dalton Trans. 2013, 42, 4677–4685. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; de Kock, C.; Smith, P.J.; Guzgay, H.; Hendricks, D.T.; Naran, K.; Mizrahi, V.; Warner, D.F.; Chibale, K.; Smith, G.S. Synthesis, characterization, and pharmacological evaluation of silicon-containing aminoquinoline organometallic complexes as antiplasmodial, antitumor, and antimycobacterial agents. Organometallics 2013, 32, 141–150. [Google Scholar] [CrossRef]
- Li, Y.; de Kock, C.; Smith, P.J.; Chibale, K.; Smith, G.S. Synthesis and evaluation of a carbosilane congener of ferroquine and its corresponding half-sandwich ruthenium and rhodium complexes for antiplasmodial and β-hematin inhibition activity. Organometallics 2014, 33, 4345–4348. [Google Scholar] [CrossRef]
- Mills, J.S.; Showell, G.A. Exploitation of silicon medicinal chemistry in drug discovery. Expert Opin. Investig. Drugs 2004, 13, 1149–1157. [Google Scholar] [CrossRef] [PubMed]
- Englebienne, P.; Hoonacker, A.V.; Herst, C.V. The place of the bioisosteric sila-substitution in drug design. Drug Des. Rev. Online 2005, 2, 467–483. [Google Scholar] [CrossRef]
- Tacke, R.; Heinrich, T.; Bertermann, R.; Burschka, C.; Hamacher, A.; Kassack, M.U. Sila-haloperidol: A silicon analogue of the dopamine (D2) receptor antagonist haloperidol. Organometallics 2004, 23, 4468–4477. [Google Scholar] [CrossRef]
- Tacke, R.; Schmid, T.; Penka, M.; Burschka, C.; Bains, W.; Warneck, J. Syntheses and pharmacological properties of the histaminic H1 antagonists sila-terfenadine-A, sila-terfenadine-B, disila-terfenadine, and sila-fexofenadine: A study on C/Si bioisosterism. Organometallics 2004, 23, 4915–4923. [Google Scholar] [CrossRef]
- Martínez, A.; Deregnaucourt, C.; Sinou, V.; Latour, C.; Roy, D.; Schrével, J.; Sánchez-Delgado, R.A. Synthesis of an organo-ruthenium aminoquinoline-trioxane hybrid and evaluation of its activity against Plasmodium falciparum and its toxicity toward normal mammalian cells. Med. Chem. Res. 2017, 26, 473–483. [Google Scholar] [CrossRef]
- Mehta, J.V.; Gajera, S.B.; Raval, D.B.; Thakkar, V.R.; Patel, M.N. Biological assessment of substituted quinoline based heteroleptic organometallic compounds. MedChemComm 2016, 7, 1617–1627. [Google Scholar] [CrossRef]
- Mehta, J.V.; Gajera, S.B.; Patel, M.N. Biological applications of pyrazoline-based half-sandwich ruthenium (III) coordination compounds. J. Biomol. Struct. Dyn. 2017, 35, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Brabec, V.; Nováková, O. DNA binding mode of ruthenium complexes and relationship to tumor cell toxicity. Drug Resist. Updat. 2006, 9, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Moucheron, C. From cisplatin to photoreactive Ru complexes: Targeting DNA for biomedical applications. New J. Chem. 2009, 33, 235–245. [Google Scholar] [CrossRef]
- Ma, D.-L.; Wang, M.; Mao, Z.; Yang, C.; Ng, C.-T.; Leung, C.-H. Rhodium complexes as therapeutic agents. Dalton Trans. 2016, 45, 2762–2771. [Google Scholar] [CrossRef] [PubMed]
- Stringer, T.; Guzgay, H.; Combrinck, J.M.; Hopper, M.; Hendricks, D.T.; Smith, P.J.; Land, K.M.; Egan, T.J.; Smith, G.S. Synthesis, characterization and pharmacological evaluation of ferrocenyl azines and their rhodium (I) complexes. J. Organomet. Chem. 2015, 788, 1–8. [Google Scholar] [CrossRef]
- Baartzes, N.; Stringer, T.; Chellan, P.; Combrinck, J.M.; Smith, P.J.; Hutton, A.T.; Smith, G.S. Synthesis, characterization, antiplasmodial evaluation and electrochemical studies of water-soluble heterobimetallic ferrocenyl complexes. Inorg. Chim. Acta 2016, 446, 111–115. [Google Scholar] [CrossRef]
- Dubar, F.; Slomianny, C.; Khalife, J.; Dive, D.; Kalamou, H.; Guerardel, Y.; Grellier, P.; Biot, C. The ferroquine antimalarial conundrum: Redox activation and reinvasion inhibition. Angew. Chem. 2013, 125, 7844–7847. [Google Scholar] [CrossRef]
- Patra, M.; Gasser, G. The medicinal chemistry of ferrocene and its derivatives. Nat. Rev. Chem. 2017, 1, 1–12. [Google Scholar] [CrossRef]
- Stringer, T.; Taylor, D.; Guzgay, H.; Shokar, A.; Au, A.; Smith, P.J.; Hendricks, D.T.; Land, K.M.; Egan, T.J.; Smith, G.S. Polyamine quinoline rhodium complexes: Synthesis and pharmacological evaluation as antiparasitic agents against Plasmodium falciparum and Trichomonas vaginalis. Dalton Trans. 2015, 44, 14906–14917. [Google Scholar] [CrossRef]
- Le Bozec, H.; Touchard, D.; Dixneuf, P.H. Organometallic chemistry of arene ruthenium and osmium complexes. In Advances in Organometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 1989; Volume 29, pp. 163–247. [Google Scholar]
- Zhang, P.; Huang, H. Future potential of osmium complexes as anticancer drug candidates, photosensitizers and organelle-targeted probes. Dalton Trans. 2018, 47, 14841–14854. [Google Scholar] [CrossRef]
- Kumaresan, D.; Shankar, K.; Vaidya, S.; Schmehl, R.H. Photochemistry and photophysics of coordination compounds: Osmium. In Photochemistry and Photophysics of Coordination Compounds II; Springer: Berlin/Heidelberg, Germany, 2007; pp. 101–142. [Google Scholar]
- Lazic, S.; Kaspler, P.; Shi, G.; Monro, S.; Sainuddin, T.; Forward, S.; Kasimova, K.; Hennigar, R.; Mandel, A.; McFarland, S. Novel Osmium-based Coordination Complexes as Photosensitizers for Panchromatic Photodynamic Therapy. Photochem. Photobiol. 2017, 93, 1248–1258. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wang, Y.; Qiu, K.; Zhao, Z.; Hu, R.; He, C.; Zhang, Q.; Chao, H. A NIR phosphorescent osmium (II) complex as a lysosome tracking reagent and photodynamic therapeutic agent. Chem. Commun. 2017, 53, 12341–12344. [Google Scholar] [CrossRef] [PubMed]
- Hanif, M.; Babak, M.V.; Hartinger, C.G. Development of anticancer agents: Wizardry with osmium. Drug Discov. Today 2014, 19, 1640–1648. [Google Scholar] [CrossRef] [PubMed]
- Meier-Menches, S.M.; Gerner, C.; Berger, W.; Hartinger, C.G.; Keppler, B.K. Structure–activity relationships for ruthenium and osmium anticancer agents–towards clinical development. Chem. Soc. Rev. 2018, 47, 909–928. [Google Scholar] [CrossRef]
- Nabiyeva, T.; Marschner, C.; Blom, B. Synthesis, structure and anti-cancer activity of osmium complexes bearing π-bound arene substituents and phosphane Co-Ligands: A review. Eur. J. Med. Chem. 2020, 201, 112483. [Google Scholar] [CrossRef]
- Rosenberg, B.; Vancamp, L.; Trosko, J.E.; Mansour, V.H. Platinum compounds: A new class of potent antitumour agents. Nature 1969, 222, 385–386. [Google Scholar] [CrossRef]
- Johnstone, T.C.; Suntharalingam, K.; Lippard, S.J. The next generation of platinum drugs: Targeted Pt (II) agents, nanoparticle delivery, and Pt (IV) prodrugs. Chem. Rev. 2016, 116, 3436–3486. [Google Scholar] [CrossRef]
- Dilruba, S.; Kalayda, G.V. Platinum-based drugs: Past, present and future. Cancer Chemother. Pharmacol. 2016, 77, 1103–1124. [Google Scholar] [CrossRef]
- Baartzes, N.; Stringer, T.; Smith, G.S. Targeting Sensitive-Strain and Resistant-Strain Malaria Parasites through a Metal-Based Approach. In Advances in Bioorganometallic Chemistry; Elsevier: Amsterdam, The Netherlands, 2019; pp. 193–213. [Google Scholar]
- Sekhon, B.S.; Bimal, N. Transition metal-based anti-malarial. J. Pharm. Educ. Res. 2012, 20112, 52–63. [Google Scholar]
- Marcelino, P.R.F.; Moreira, M.B.; Lacerda, T.M.; da Silva, S.S. Metal-Based Drugs for Treatment of Malaria. In Biomedical Applications of Metals; Rai, M., Ingle, A.P., Medici, S., Eds.; Springer International Publishing: Cham, Switzerland, 2018; pp. 167–193. [Google Scholar]
- Navarro, M.; Castro, W.; Madamet, M.; Amalvict, R.; Benoit, N.; Pradines, B. Metal-chloroquine derivatives as possible anti-malarial drugs: Evaluation of anti-malarial activity and mode of action. Malar. J. 2014, 13, 471. [Google Scholar] [CrossRef]
- Chellan, P.; Land, K.M.; Shokar, A.; Au, A.; An, S.H.; Clavel, C.M.; Dyson, P.J.; Kock, C.D.; Smith, P.J.; Chibale, K. Exploring the versatility of cycloplatinated thiosemicarbazones as antitumor and antiparasitic agents. Organometallics 2012, 31, 5791–5799. [Google Scholar] [CrossRef]
- Livingstone, S.E. 7—PLATINUM. In The Chemistry of Ruthenium, Rhodium, Palladium, Osmium, Iridium and Platinum; Pergamon: Bergama, Turkey, 1973; Volume 25, pp. 1330–1370. [Google Scholar]
- Livingstone, S.E. 6—PALLADIUM. In The Chemistry of Ruthenium, Rhodium, Palladium, Osmium, Iridium and Platinum; Pergamon: Bergama, Turkey, 1973; Volume 25, pp. 1274–1330. [Google Scholar]
- Kapdi, A.R.; Fairlamb, I.J. Anti-cancer palladium complexes: A focus on PdX 2 L 2, palladacycles and related complexes. Chem. Soc. Rev. 2014, 43, 4751–4777. [Google Scholar] [CrossRef] [PubMed]
- Lazarević, T.; Rilak, A.; Bugarčić, Ž.D. Platinum, palladium, gold and ruthenium complexes as anticancer agents: Current clinical uses, cytotoxicity studies and future perspectives. Eur. J. Med. Chem. 2017, 142, 8–31. [Google Scholar] [CrossRef] [PubMed]
- Graham, R.D.; Williams, D.R. The synthesis and screening for anti-bacterial,-cancer,-fungicidal and-viral activities of some complexes of palladium and nickel. J. Inorg. Nucl. Chem. 1979, 41, 1245–1249. [Google Scholar] [CrossRef]
- Chellan, P.; Shunmoogam-Gounden, N.; Hendricks, D.T.; Gut, J.; Rosenthal, P.J.; Lategan, C.; Smith, P.J.; Chibale, K.; Smith, G.S. Synthesis, structure and in vitro biological screening of palladium (II) complexes of functionalised salicylaldimine thiosemicarbazones as antimalarial and anticancer agents. Eur. J. Inorg. Chem. 2010, 2010, 3520–3528. [Google Scholar] [CrossRef]
- Quirante, J.; Ruiz, D.; Gonzalez, A.; López, C.; Cascante, M.; Cortés, R.; Messeguer, R.; Calvis, C.; Baldomà, L.; Pascual, A. Platinum (II) and palladium (II) complexes with (N, N′) and (C, N, N′)− ligands derived from pyrazole as anticancer and antimalarial agents: Synthesis, characterization and in vitro activities. J. Inorg. Biochem. 2011, 105, 1720–1728. [Google Scholar] [CrossRef]
- Chellan, P.; Nasser, S.; Vivas, L.; Chibale, K.; Smith, G.S. Cyclopalladated complexes containing tridentate thiosemicarbazone ligands of biological significance: Synthesis, structure and antimalarial activity. J. Organomet. Chem. 2010, 695, 2225–2232. [Google Scholar] [CrossRef]
- Adams, M.; de Kock, C.; Smith, P.J.; Chibale, K.; Smith, G.S. Synthesis, characterization and antiplasmodial evaluation of cyclopalladated thiosemicarbazone complexes. J. Organomet. Chem. 2013, 736, 19–26. [Google Scholar] [CrossRef]
- Adams, M.; Barnard, L.; de Kock, C.; Smith, P.J.; Wiesner, L.; Chibale, K.; Smith, G.S. Cyclopalladated organosilane–tethered thiosemicarbazones: Novel strategies for improving antiplasmodial activity. Dalton Trans. 2016, 45, 5514–5520. [Google Scholar] [CrossRef]
- Begum, K.; Kim, H.-S.; Kumar, V.; Stojiljkovic, I.; Wataya, Y. In vitro antimalarial activity of metalloporphyrins against Plasmodium falciparum. Parasitol. Res. 2003, 90, 221–224. [Google Scholar] [CrossRef]
- Aggarwal, S. A histochemical approach to the mechanism of action of cisplatin and its analogues. J. Histochem. Cytochem. 1993, 41, 1053–1073. [Google Scholar] [CrossRef] [PubMed]
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Hempelmann, E. Hemozoin biocrystallization in Plasmodium falciparum and the antimalarial activity of crystallization inhibitors. Parasitol. Res. 2007, 100, 671–676. [Google Scholar] [CrossRef] [PubMed]
- Foley, M.; Tilley, L. Quinoline antimalarials: Mechanisms of action and resistance and prospects for new agents. Pharmacol. Ther. 1998, 79, 55–87. [Google Scholar] [CrossRef]
- Egan, T.J. Quinoline antimalarials. Opin. Ther. Pat. 2001, 11, 185–209. [Google Scholar] [CrossRef]
- Dubar, F.; Khalife, J.; Brocard, J.; Dive, D.; Biot, C. Ferroquine, an ingenious antimalarial drug–thoughts on the mechanism of action. Molecules 2008, 13, 2900–2907. [Google Scholar] [CrossRef]
- Navarro, M.; Castro, W.; Biot, C. Bioorganometallic compounds with antimalarial targets: Inhibiting hemozoin formation. Organometallics 2012, 31, 5715–5727. [Google Scholar] [CrossRef]
- Dubar, F.; Egan, T.J.; Pradines, B.; Kuter, D.; Ncokazi, K.K.; Forge, D.; Paul, J.-F.; Pierrot, C.; Kalamou, H.; Khalife, J. The antimalarial ferroquine: Role of the metal and intramolecular hydrogen bond in activity and resistance. ACS Chem. Biol. 2011, 6, 275–287. [Google Scholar] [CrossRef]
- Ncokazi, K.K.; Egan, T.J. A colorimetric high-throughput β-hematin inhibition screening assay for use in the search for antimalarial compounds. Anal. Biochem. 2005, 338, 306–319. [Google Scholar] [CrossRef]
- Aravind, L.; Iyer, L.M.; Wellems, T.E.; Miller, L.H. Plasmodium biology: Genomic gleanings. Cell 2003, 115, 771–785. [Google Scholar] [CrossRef]
- Woynarowski, J.M.; Krugliak, M.; Ginsburg, H. Pharmacogenomic analyses of targeting the AT-rich malaria parasite genome with AT-specific alkylating drugs. Mol. Biochem. Parasitol. 2007, 154, 70–81. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, N.I.; Chavain, N.; Wang, Y.; Friebolin, W.; Maes, L.; Pradines, B.; Lanzer, M.; Yardley, V.; Brun, R.; Herold-Mende, C. Antimalarial versus cytotoxic properties of dual drugs derived from 4-aminoquinolines and Mannich bases: Interaction with DNA. J. Med. Chem. 2010, 53, 3214–3226. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Kaur, H.; Smith, P.; de Kock, C.; Chibale, K.; Balzarini, J. Quinoline–pyrimidine hybrids: Synthesis, antiplasmodial activity, SAR, and mode of action studies. J. Med. Chem. 2014, 57, 435–448. [Google Scholar] [CrossRef] [PubMed]
- Ali, I.; Saleem, K.; Wesselinova, D.; Haque, A. Synthesis, DNA binding, hemolytic, and anti-cancer assays of curcumin I-based ligands and their ruthenium (III) complexes. Med. Chem. Res. 2013, 22, 1386–1398. [Google Scholar] [CrossRef]
- Ali, I.; A Wani, W.; Saleem, K.; Wesselinova, D. Syntheses, DNA binding and anticancer profiles of L-glutamic acid ligand and its copper (II) and ruthenium (III) complexes. Med. Chem. 2013, 9, 11–21. [Google Scholar] [CrossRef]
- Prakash, G.; Manikandan, R.; Viswanathamurthi, P.; Velmurugan, K.; Nandhakumar, R. Ruthenium (III) S-methylisothiosemicarbazone Schiff base complexes bearing PPh3/AsPh3 coligand: Synthesis, structure and biological investigations, including antioxidant, DNA and protein interaction, and in vitro anticancer activities. J. Photochem. Photobiol. B 2014, 138, 63–74. [Google Scholar] [CrossRef]
- Aziz, A.A.A.; Elbadawy, H.A. Spectral, electrochemical, thermal, DNA binding ability, antioxidant and antibacterial studies of novel Ru (III) Schiff base complexes. Spectrochim. Acta A 2014, 124, 404–415. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Romero-Canelón, I.; Habtemariam, A.; Sadler, P.J. Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design. Nat. Commun. 2015, 6, 1–9. [Google Scholar] [CrossRef]
- Bose, S.; Ngo, A.H.; Do, L.H. Intracellular transfer hydrogenation mediated by unprotected organoiridium catalysts. J. Am. Chem. Soc. 2017, 139, 8792–8795. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Metzler-Nolte, N. Intracellular catalysis with selected metal complexes and metallic nanoparticles: Advances toward the development of catalytic metallodrugs. Chem. Rev. 2019, 119, 829–869. [Google Scholar] [CrossRef]
- Coverdale, J.P.; Romero-Canelón, I.; Sanchez-Cano, C.; Clarkson, G.J.; Habtemariam, A.; Wills, M.; Sadler, P.J. Asymmetric transfer hydrogenation by synthetic catalysts in cancer cells. Nat. Chem. 2018, 10, 347. [Google Scholar] [CrossRef] [PubMed]
- Biancalana, L.; Abdalghani, I.; Chiellini, F.; Zacchini, S.; Pampaloni, G.; Crucianelli, M.; Marchetti, F. Ruthenium Arene Complexes with α-Aminoacidato Ligands: New Insights into Transfer Hydrogenation Reactions and Cytotoxic Behaviour. Eur. J. Inorg. Chem. 2018, 2018, 3041–3057. [Google Scholar] [CrossRef]
- Chen, F.; Romero-Canelón, I.; Soldevila-Barreda, J.J.; Song, J.-I.; Coverdale, J.P.; Clarkson, G.J.; Kasparkova, J.; Habtemariam, A.; Wills, M.; Brabec, V. Transfer hydrogenation and antiproliferative activity of tethered half-sandwich organoruthenium catalysts. Organometallics 2018, 37, 1555–1566. [Google Scholar] [CrossRef] [PubMed]
- Soldevila-Barreda, J.J.; Habtemariam, A.; Romero-Canelón, I.; Sadler, P.J. Half-sandwich rhodium (III) transfer hydrogenation catalysts: Reduction of NAD+ and pyruvate, and antiproliferative activity. J. Inorg. Biochem. 2015, 153, 322–333. [Google Scholar] [CrossRef]
- Stringer, T.; Melis, D.R.; Smith, G.S. N, O-Chelating quinoline-based half-sandwich organorhodium and-iridium complexes: Synthesis, antiplasmodial activity and preliminary evaluation as transfer hydrogenation catalysts for the reduction of NAD+. Dalton Trans. 2019, 48, 13143–13148. [Google Scholar] [CrossRef]
- Makler, M.T.; Hinrichs, D.J. Measurement of the lactate dehydrogenase activity of Plasmodium falciparum as an assessment of parasitemia. Am. J. Trop. Med. Hyg. 1993, 48, 205–210. [Google Scholar] [CrossRef]
- Lo, H.C.; Buriez, O.; Kerr, J.B.; Fish, R.H. Regioselective Reduction of NAD+ Models with [Cp* Rh (bpy) H]+: Structure–Activity Relationships and Mechanistic Aspects in the Formation of the 1, 4-NADH Derivatives. Angew. Chem. Int. Ed. 1999, 38, 1429–1432. [Google Scholar] [CrossRef]
- Lo, H.C.; Leiva, C.; Buriez, O.; Kerr, J.B.; Olmstead, M.M.; Fish, R.H. Bioorganometallic Chemistry. 13. Regioselective Reduction of NAD+ Models, 1-Benzylnicotinamde Triflate and β-Nicotinamide Ribose-5′-methyl Phosphate, with in Situ Generated [Cp* Rh (Bpy) H]+: Structure− Activity Relationships, Kinetics, and Mechanistic Aspects in the Formation of the 1, 4-NADH Derivatives. Inorg. Chem. 2001, 40, 6705–6716. [Google Scholar]


























{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (μM) a | RI b | SI c | ||
|---|---|---|---|---|---|
| NF54 | Dd2 | CHO | |||
| 40b | 7.81 ± 0.56 | – d | – | – | – |
| 41a | 2.92 ± 0.33 | 4.28 ± 0.33 | 71.8 ± 8.11 | 1.47 | 24.6 |
| 41b | 4.19 ± 0.12 | 6.66 ± 2.58 | 21.5 ± 0.80 | 1.59 | 5.14 |
| 41c | 2.57 ± 0.99 | 2.29 ± 0.25 | 3.65 ± 0.630 | 0.89 | 1.42 |
| 42a | 6.20 ± 1.00 | – | – | – | – |
| 42b | 2.90 ± 0.90 | 3.8 ± 0.2 | – | – | – |
| 43a | 16.5 ± 1.00 | 14.1 ± 0.90 | – | – | – |
| 43b | 8.60 ± 0.60 | 6.60 ± 0.30 | – | – | – |
| 44a | 276.8 ± 31.0 | 526.56 ± 70.85 | – | 1.90 | – |
| 44b | 61.4 ± 10.3 | >1749 | – | >28.5 | – |
| 44c | 270.2 ± 35.9 | 835.09 ± 190.03 | – | 3.09 | – |
| 44d | 81.6 ± 7.40 | 228.96 ± 4.06 | – | 2.81 | – |
| 44e | 71.7 ± 4.80 | 211.77 ± 22.68 | – | 2.95 | – |
| 44f | 151.9 ± 14.1 | 385.61 ± 8.48 | – | 2.54 | – |
| 45a | 8.27 ± 0.38 | 42.73 ± 12.78 | – | 5.17 | – |
| 45b | 30.7 ± 2.70 | 42.99 ± 1.08 | – | 1.40 | – |
| 45c | 4.96 ± 0.76 | 36.64 ± 4.33 | – | 7.39 | – |
| CQ | 5.43 ± 2.13 | 108.36 ± 1.10 | – | 19.95 | – |
| FQ | 42.6 ± 9.91 | 27.67 ± 6.46 | – | 0.65 | – |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mbaba, M.; Golding, T.M.; Smith, G.S. Recent Advances in the Biological Investigation of Organometallic Platinum-Group Metal (Ir, Ru, Rh, Os, Pd, Pt) Complexes as Antimalarial Agents. Molecules 2020, 25, 5276. https://doi.org/10.3390/molecules25225276
Mbaba M, Golding TM, Smith GS. Recent Advances in the Biological Investigation of Organometallic Platinum-Group Metal (Ir, Ru, Rh, Os, Pd, Pt) Complexes as Antimalarial Agents. Molecules. 2020; 25(22):5276. https://doi.org/10.3390/molecules25225276
Chicago/Turabian StyleMbaba, Mziyanda, Taryn M. Golding, and Gregory S. Smith. 2020. "Recent Advances in the Biological Investigation of Organometallic Platinum-Group Metal (Ir, Ru, Rh, Os, Pd, Pt) Complexes as Antimalarial Agents" Molecules 25, no. 22: 5276. https://doi.org/10.3390/molecules25225276
APA StyleMbaba, M., Golding, T. M., & Smith, G. S. (2020). Recent Advances in the Biological Investigation of Organometallic Platinum-Group Metal (Ir, Ru, Rh, Os, Pd, Pt) Complexes as Antimalarial Agents. Molecules, 25(22), 5276. https://doi.org/10.3390/molecules25225276