Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment



Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Nucleic Acid Biophysical Properties and Therapeutic Uses
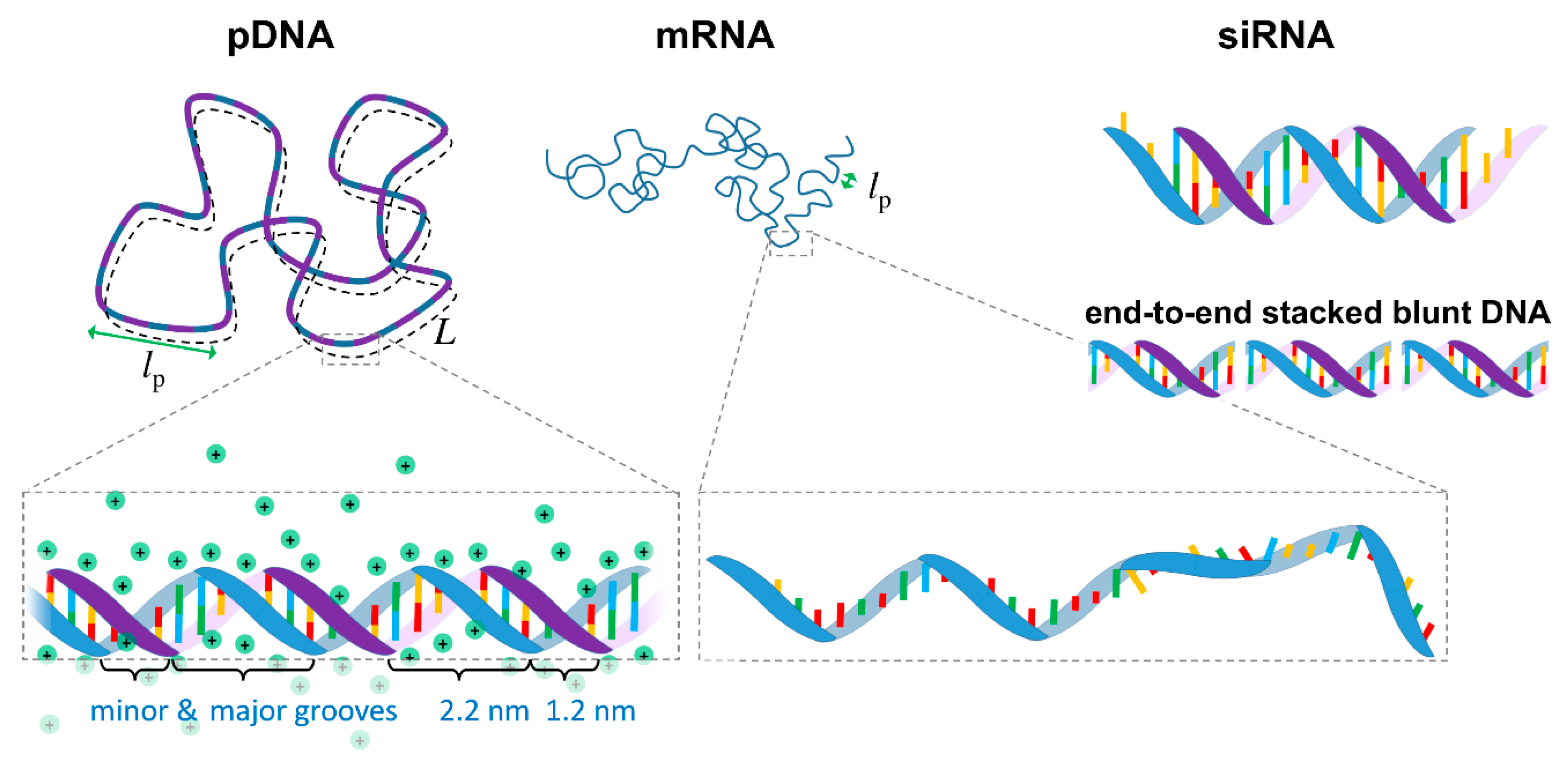
2.1. DNA
2.2. siRNA
2.3. mRNA
2.4. miRNA
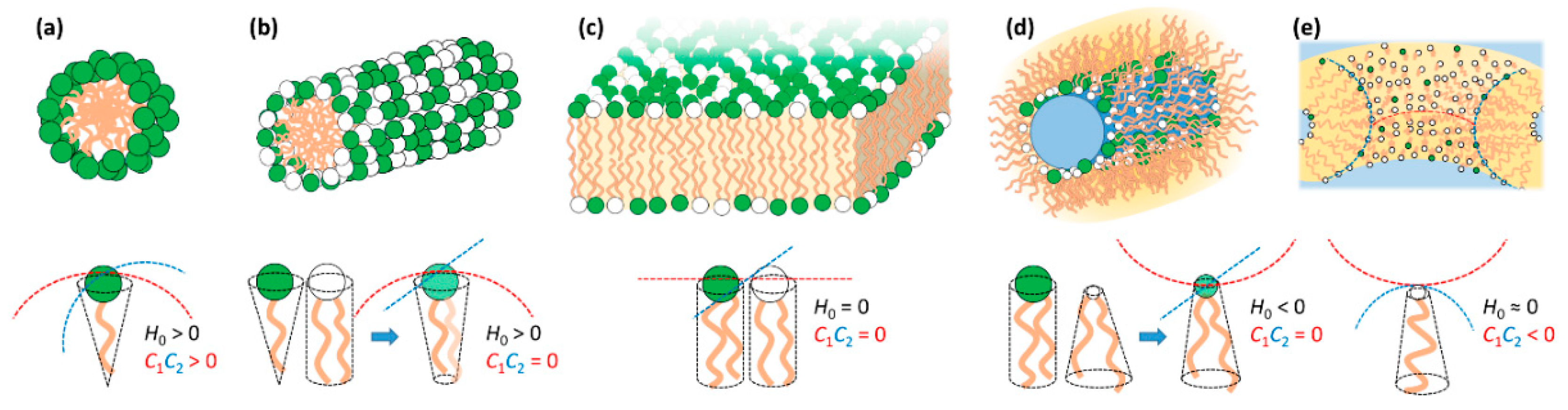
3. Lipid Self-Assembly and Lipid Film Elastic Energy
3.1. Lipid Film Elastic Energy
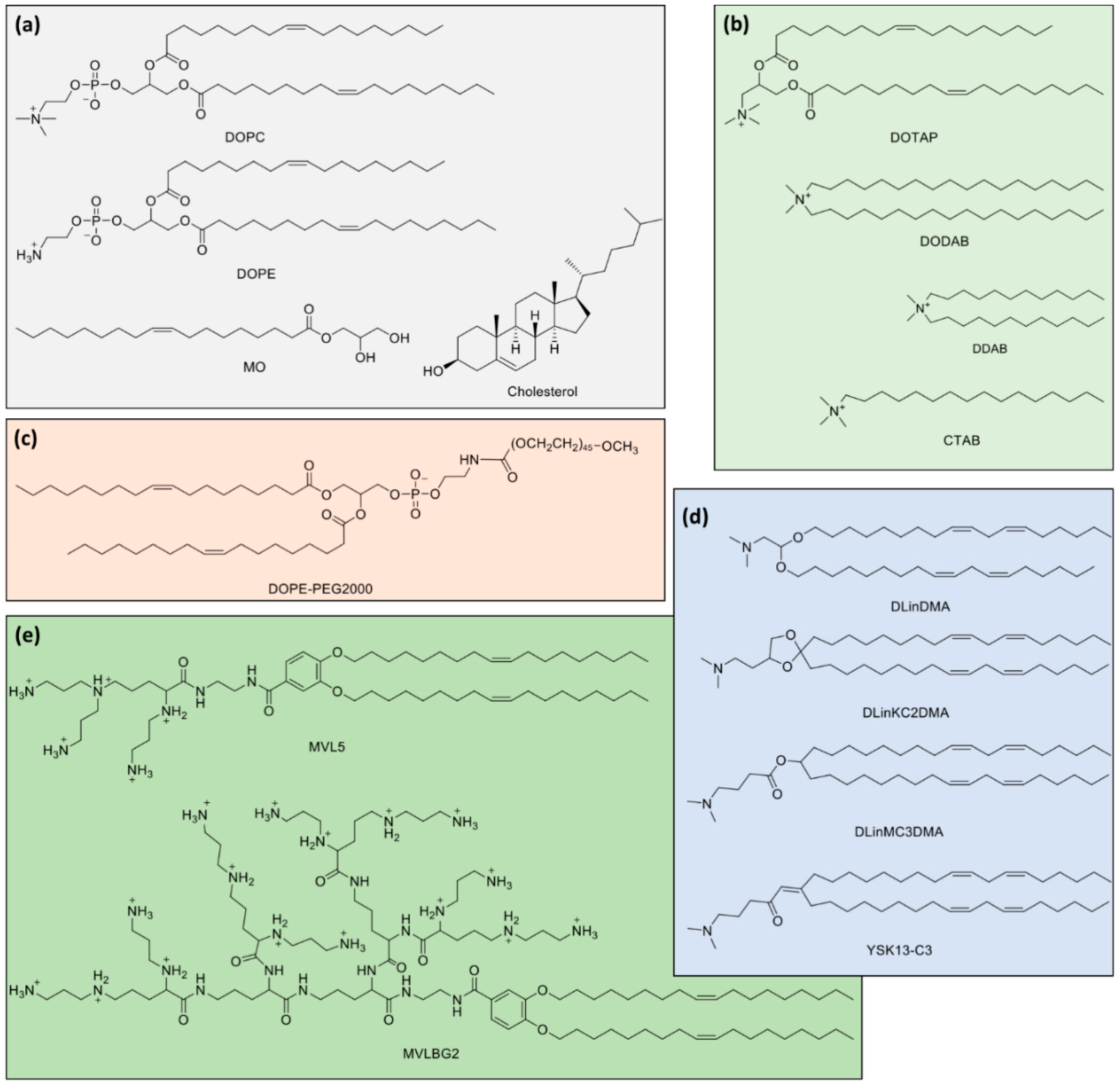
3.2. Lipid Mixtures
4. Lipid-NA Nanoparticle Formation, Structure and Stability
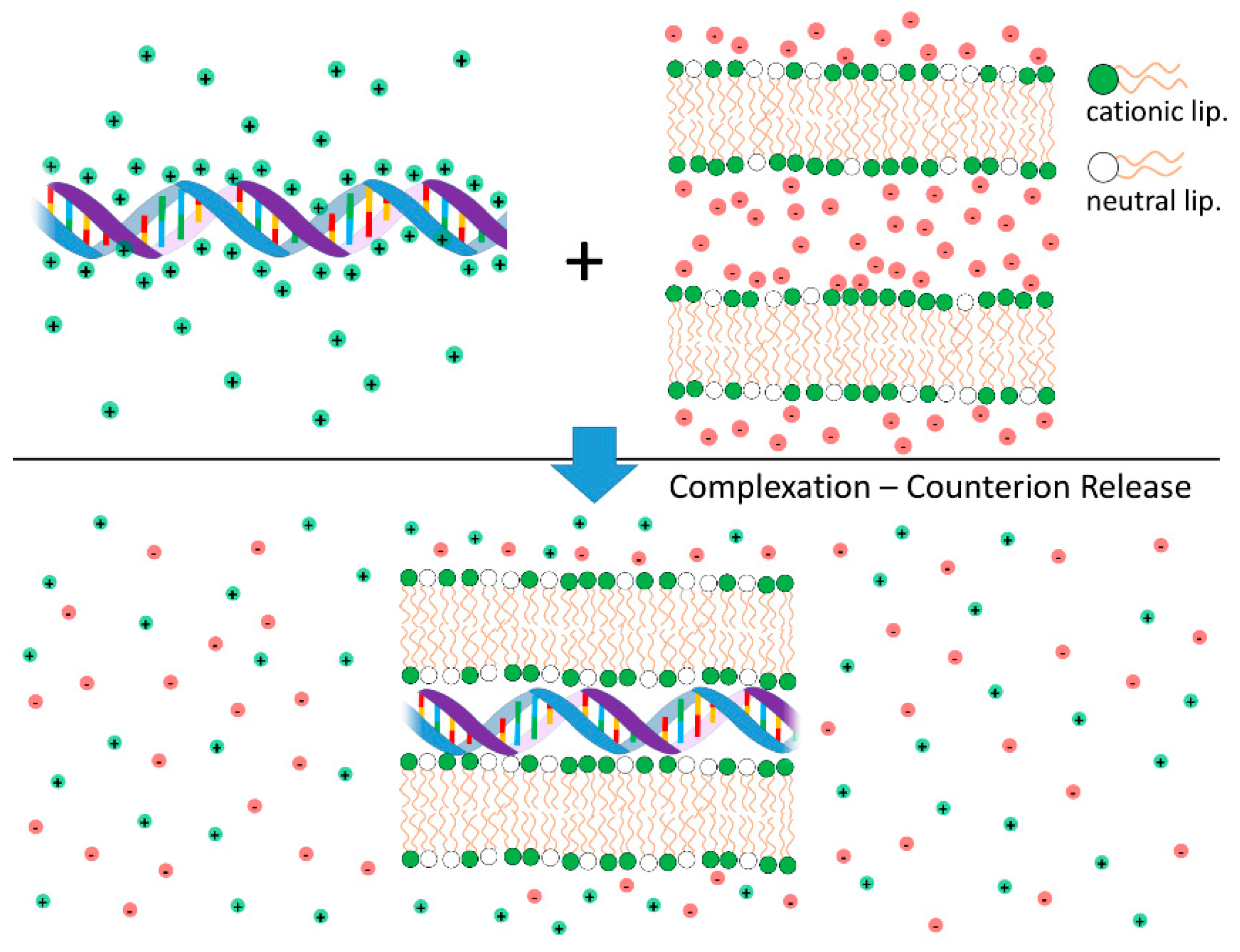
4.1. Lipid-NA Complexation: Counterion Release as the Main Driving Force for Lipid-NA Association
4.2. Cationic-to-Anionic Charge Ratio, Overcharging and Colloidal Stability of Lipid-NA Complexes
4.3. Lipid-NA Structure
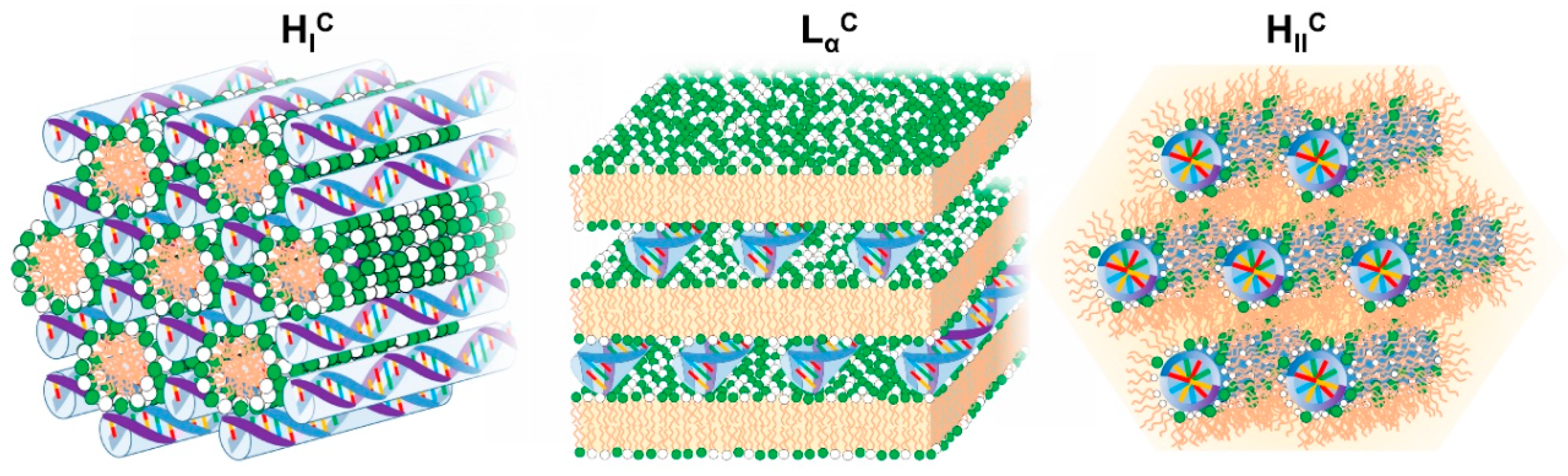
4.3.1. Lamellar Complexes—LαC
4.3.2. Inverted Hexagonal Complexes—HIIC
4.3.3. Inverse Bicontinuous Cubic Complexes—QIIC
4.3.4. Normal Bicontinuous Cubic Complexes—QIC
4.3.5. Normal Columnar Complexes—HIC and SIC
4.3.6. Other Normal Phases
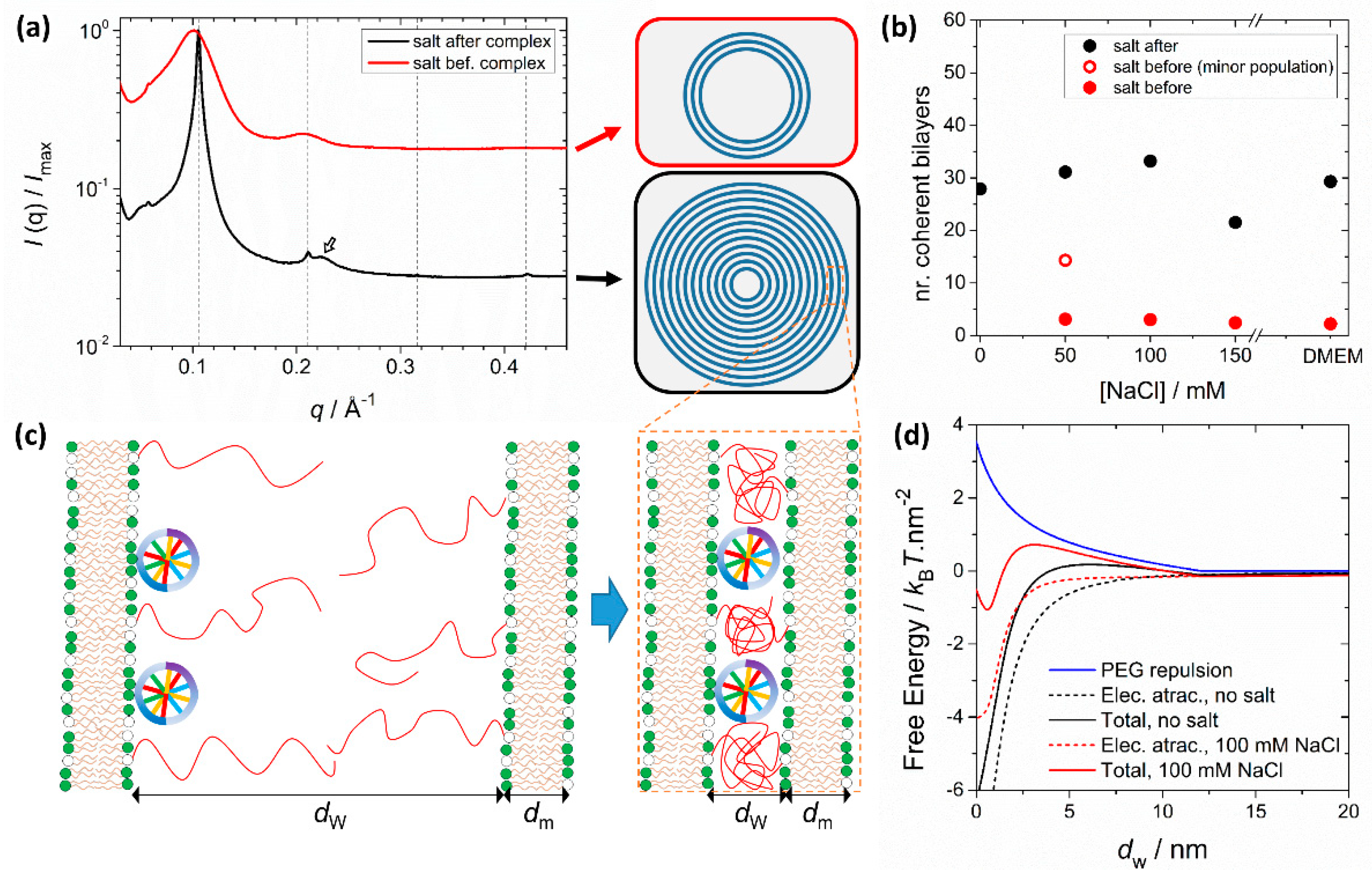
5. PEGylation—Improved Circulation Lifetime and Effects on the Particle Structure
5.1. PEGylation—Improved Colloidal Stability and Circulation Lifetime by Steric Stabilization
5.2. Size and Structure Modulation In Pre-Formed Lipid Assemblies
5.3. Solvent-Exchange and Monomolecular Nucleic Acid Lipid Particles
6. Particle Structure, Charge and Functionalization Influence on the Transfection Efficiency
6.1. Cell Uptake: Nonspecific Electrostatic Interactions and Targeting with Affinity Ligands
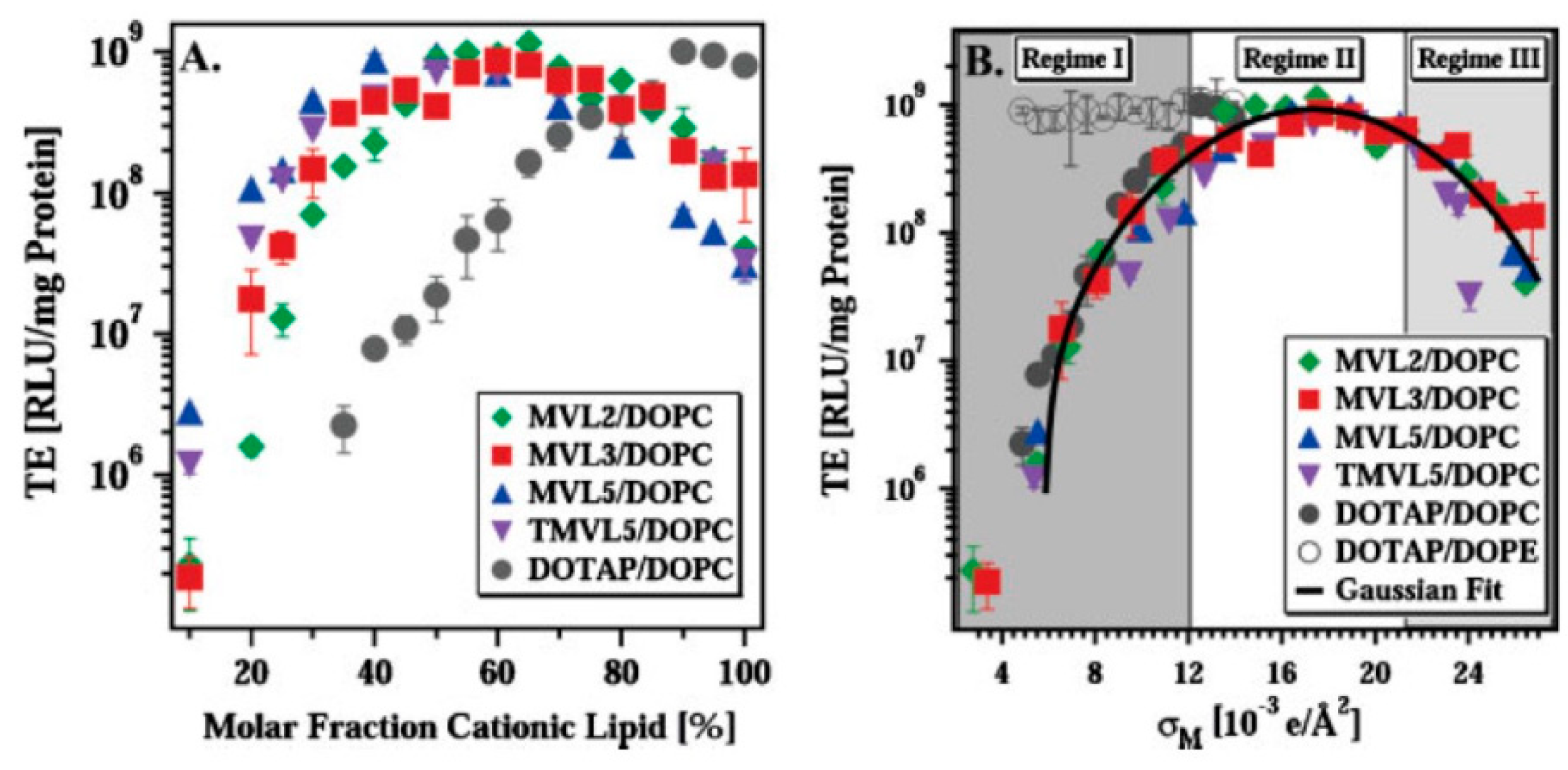
6.2. Endosomal Escape and Transfection Efficiency: Lipid Membrane Curvature and Charge Density
6.3. Exploring Intracellular Stimuli
7. Ionizable Cationic Lipids and Lipid Nanoparticles (LNPs)
8. Targeting the Tumor
8.1. Passive Targeting
8.2. Active Targeting
8.3. Exploiting Local Stimuli
9. Prospects
10. Conclusions
Funding
Conflicts of Interest
References
- Burrell, R.A.; McGranahan, N.; Bartek, J.; Swanton, C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature 2013, 501, 338–345. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Cancer Tomorrow. Available online: https://gco.iarc.fr/tomorrow/graphic-isotype (accessed on 1 September 2020).
- Ewert, K.K.; Zidovska, A.; Ahmad, A.; Bouxsein, N.F.; Evans, H.M.; McAllister, C.S.; Samuel, C.E.; Safinya, C.R. Cationic Lipid–Nucleic Acid Complexes for Gene Delivery and Silencing: Pathways and Mechanisms for Plasmid DNA and siRNA. Top. Curr. Chem. 2010, 296, 191–226. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.; Kanasty, R.L.; Eltoukhy, A.A.; Vegas, A.J.; Dorkin, J.R.; Anderson, D.G. Non-viral vectors for gene-based therapy. Nat. Rev. Genet. 2014, 15, 541–555. [Google Scholar] [CrossRef]
- Das, S.K.; Menezes, M.E.; Bhatia, S.; Wang, X.-Y.; Emdad, L.; Sarkar, D.; Fisher, P.B. Gene Therapies for Cancer: Strategies, Challenges and Successes. J. Cell. Physiol. 2015, 230, 259–271. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, J.E.; Davis, M.E. Clinical experiences with systemically administered siRNA-based therapeutics in cancer. Nat. Rev. Drug Discov. 2015, 14, 843–856. [Google Scholar] [CrossRef]
- Ginn, S.L.; Amaya, A.K.; Alexander, I.E.; Edelstein, M.; Abedi, M.R. Gene therapy clinical trials worldwide to 2017: An update. J. Gene Med. 2018, 20, 1–16. [Google Scholar] [CrossRef]
- Kranz, L.M.; Diken, M.; Haas, H.; Kreiter, S.; Loquai, C.; Reuter, K.C.; Meng, M.; Fritz, D.; Vascotto, F.; Hefesha, H.; et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 2016, 534, 396–401. [Google Scholar] [CrossRef]
- Sahin, U.; Derhovanessian, E.; Miller, M.; Kloke, B.P.; Simon, P.; Löwer, M.; Bukur, V.; Tadmor, A.D.; Luxemburger, U.; Schrörs, B.; et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature 2017, 547, 222–226. [Google Scholar] [CrossRef]
- Sahin, U.; Türeci, Ö. Personalized vaccines for cancer immunotherapy. Science 2018, 359, 1355–1360. [Google Scholar] [CrossRef]
- Pardi, N.; Hogan, M.J.; Porter, F.W.; Weissman, D. mRNA vaccines-a new era in vaccinology. Nat. Rev. Drug Discov. 2018, 17, 261–279. [Google Scholar] [CrossRef]
- Pastor, F.; Berraondo, P.; Etxeberria, I.; Frederick, J.; Sahin, U.; Gilboa, E.; Melero, I. An RNA toolbox for cancer immunotherapy. Nat. Rev. Drug Discov. 2018, 17, 751–767. [Google Scholar] [CrossRef] [PubMed]
- Vormehr, M.; Türeci, Ö.; Sahin, U. Harnessing tumor mutations for truly individualized cancer vaccines. Annu. Rev. Med. 2019, 70, 395–407. [Google Scholar] [CrossRef] [PubMed]
- Riley, R.S.; June, C.H.; Langer, R.; Mitchell, M.J. Delivery technologies for cancer immunotherapy. Nat. Rev. Drug Discov. 2019, 18, 175–196. [Google Scholar] [CrossRef] [PubMed]
- Sahin, U.; Oehm, P.; Derhovanessian, E.; Jabulowsky, R.A.; Vormehr, M.; Gold, M.; Maurus, D.; Schwarck-Kokarakis, D.; Kuhn, A.N.; Omokoko, T.; et al. An RNA vaccine drives immunity in checkpoint-inhibitor-treated melanoma. Nature 2020, 585, 107–112. [Google Scholar] [CrossRef]
- Hewitt, S.L.; Bai, A.; Bailey, D.; Ichikawa, K.; Zielinski, J.; Karp, R.; Apte, A.; Arnold, K.; Zacharek, S.J.; Iliou, M.S.; et al. Durable anticancer immunity from intratumoral administration of IL-23, IL-36γ, and OX40L mRNAs. Sci. Transl. Med. 2019, 11, eaat9143. [Google Scholar] [CrossRef]
- Wang, T.; Upponi, J.R.; Torchilin, V.P. Design of multifunctional non-viral gene vectors to overcome physiological barriers: Dilemmas and strategies. Int. J. Pharm. 2012, 427, 3–20. [Google Scholar] [CrossRef]
- Matsumura, Y.; Maeda, H. A New Concept for Macromorecular Therapeutics in Cancer Chemothrapy: Mechanism of Tumoritropic Accumulation of Proteins and the Antitumor Agent Smancs. Cancer Res. 1986, 46, 6387–6392. [Google Scholar]
- Akinc, A.; Maier, M.A.; Manoharan, M.; Fitzgerald, K.; Jayaraman, M.; Barros, S.; Ansell, S.; Du, X.; Hope, M.J.; Madden, T.D.; et al. The Onpattro story and the clinical translation of nanomedicines containing nucleic acid-based drugs. Nat. Nanotechnol. 2019, 14, 1084–1087. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers break barriers in drug delivery: Endocytosis and endosomal escape of gene delivery vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef]
- Dunbar, C.E.; High, K.A.; Joung, J.K.; Kohn, D.B.; Ozawa, K.; Sadelain, M. Gene therapy comes of age. Science 2018, 359, eaan4672. [Google Scholar] [CrossRef]
- Felgner, P.L.; Gadek, T.R.; Holm, M.; Roman, R.; Chan, H.W.; Wenz, M.; Northrop, J.P.; Ringold, G.M.; Danielsen, M. Lipofection: A Highly efficient, lipid-mediated DNA-transfection procedure. Proc. Natl. Acad. Sci. USA 1987, 84, 7413–7417. [Google Scholar] [CrossRef] [PubMed]
- Felgner, P.L.; Rhodes, G. Gene therapeutics. Nature 1991, 349, 351–352. [Google Scholar] [CrossRef]
- Caracciolo, G.; Amenitsch, H. Cationic liposome/DNA complexes: From structure to interactions with cellular membranes. Eur. Biophys. J. 2012, 41, 815–829. [Google Scholar] [CrossRef] [PubMed]
- Majzoub, R.N.; Ewert, K.K.; Safinya, C.R. Cationic liposome—Nucleic acid nanoparticle assemblies with applications in gene delivery and gene silencing. Philos. Trans. R. Soc. A 2016, 374, 20150129. [Google Scholar] [CrossRef] [PubMed]
- Buck, J.; Grossen, P.; Cullis, P.R.; Huwyler, J.; Witzigmann, D. Lipid-Based DNA Therapeutics: Hallmarks of Non-Viral Gene Delivery. ACS Nano 2019, 13, 3754–3782. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.; Gonzalez-Duarte, A.; O’Riordan, W.D.; Yang, C.-C.; Ueda, M.; Kristen, A.V.; Tournev, I.; Schmidt, H.H.; Coelho, T.; Berk, J.L.; et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N. Engl. J. Med. 2018, 379, 11–21. [Google Scholar] [CrossRef]
- Dickerson, R.E.; Drew, H.R.; Conner, B.N.; Wing, R.M.; Fratini, A.V.; Kopka, M.L. The Anatomy of A-, B-, and Z-DNA. Science 1982, 216, 475–485. [Google Scholar] [CrossRef]
- Baumann, C.G.; Smith, S.B.; Bloomfield, V.A.; Bustamante, C. Ionic effects on the elasticity of single DNA molecules. Proc. Natl. Acad. Sci. USA 1997, 94, 6185–6190. [Google Scholar] [CrossRef]
- Gelbart, W.M.; Bruinsma, R.F.; Pincus, P.A.; Parsegian, V.A. DNA-Inspired Electrostatics. Phys. Today 2000, 53, 38–44. [Google Scholar] [CrossRef]
- Wong, G.C.L.; Pollack, L. Electrostatics of Strongly Charged Biological Polymers: Ion-Mediated Interactions and Self-Organization in Nucleic Acids and Proteins. Annu. Rev. Phys. Chem. 2010, 61, 171–189. [Google Scholar] [CrossRef]
- Manning, G.S. The molecular theory of polyelectrolyte solutions with applications to the electrostatic properties of polynucleotides. Q. Rev. Biophys. II 1978, 2, 179–246. [Google Scholar] [CrossRef] [PubMed]
- Bloomfield, V.A. DNA condensation. Curr. Opin. Struct. Biol. 1996, 6, 334–341. [Google Scholar] [CrossRef]
- Nakata, M.; Zanchetta, G.; Chapman, B.D.; Jones, C.D.; Cross, J.O.; Pindak, R.; Bellini, T.; Clark, N.A. End-to-End Stacking and Liquid Crystal Condensation of 6– to 20–Base Pair DNA Duplexes. Science 2007, 318, 1276–1279. [Google Scholar] [CrossRef]
- Bouxsein, N.F.; Leal, C.; McAllister, C.S.; Ewert, K.K.; Li, Y.; Samuel, C.E.; Safinya, C.R. Two-dimensional packing of short DNA with nonpairing overhangs in cationic liposome-DNA complexes: From Onsager nematics to columnar nematics with finite-length columns. J. Am. Chem. Soc. 2011, 133, 7585–7595. [Google Scholar] [CrossRef]
- Misra, S.K.; Naz, S.; Kondaiah, P.; Bhattacharya, S. A cationic cholesterol based nanocarrier for the delivery of p53-EGFP-C3 plasmid to cancer cells. Biomaterials 2014, 35, 1334–1346. [Google Scholar] [CrossRef] [PubMed]
- He, Z.-Y.; Deng, F.; Wei, X.-W.; Ma, C.-C.; Luo, M.; Zhang, P.; Sang, Y.-X.; Liang, X.; Liu, L.; Qin, H.-X.; et al. Ovarian cancer treatment with a tumor-targeting and gene expression-controllable lipoplex. Sci. Rep. 2016, 6, 23764. [Google Scholar] [CrossRef] [PubMed]
- Neves, S.; Faneca, H.; Bertin, S.; Konopka, K.; Düzgüneş, N.; Pierrefite-Carle, V.; Simões, S.; Pedroso De Lima, M.C. Transferrin lipoplex-mediated suicide gene therapy of oral squamous cell carcinoma in an immunocompetent murine model and mechanisms involved in the antitumoral response. Cancer Gene Ther. 2009, 16, 91–101. [Google Scholar] [CrossRef] [PubMed]
- Villaverde, M.S.; Combe, K.; Duchene, A.G.; Wei, M.X.; Glikin, G.C.; Finocchiaro, L.M.E. Suicide plus immune gene therapy prevents post-surgical local relapse and increases overall survival in an aggressive mouse melanoma setting. Int. Immunopharmacol. 2014, 22, 167–175. [Google Scholar] [CrossRef]
- Garu, A.; Moku, G.; Gulla, S.K.; Chaudhuri, A. Genetic immunization with in vivo dendritic cell-targeting liposomal DNA vaccine carrier induces long-lasting antitumor immune response. Mol. Ther. 2016, 24, 385–397. [Google Scholar] [CrossRef]
- Cornu, T.I.; Mussolino, C.; Cathomen, T. Refining strategies to translate genome editing to the clinic. Nat. Med. 2017, 23, 415–423. [Google Scholar] [CrossRef]
- Jubair, L.; Fallaha, S.; McMillan, N.A.J. Systemic Delivery of CRISPR/Cas9 Targeting HPV Oncogenes Is Effective at Eliminating Established Tumors. Mol. Ther. 2019, 27, 2091–2099. [Google Scholar] [CrossRef] [PubMed]
- Kebbekus, P.; Draper, D.E.; Hagermad, P. Persistence Length of RNA. Biochemistry 1995, 34, 4354–4357. [Google Scholar] [CrossRef] [PubMed]
- Abels, J.A.; Moreno-Herrero, F.; Van Der Heijden, T.; Dekker, C.; Dekker, N.H. Single-molecule measurements of the persistence length of double-stranded RNA. Biophys. J. 2005, 88, 2737–2744. [Google Scholar] [CrossRef] [PubMed]
- Pabit, S.A.; Qiu, X.; Lamb, J.S.; Li, L.; Meisburger, S.P.; Pollack, L. Both helix topology and counterion distribution contribute to the more effective charge screening in dsRNA compared with dsDNA. Nucleic Acids Res. 2009, 37, 3887–3896. [Google Scholar] [CrossRef] [PubMed]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent andspecific genetic interferenceby double-strandedRNA in Caenorhabditiselegans. Nature 1998, 391, 806–811. [Google Scholar] [CrossRef]
- Gomes-da-Silva, L.C.; Fonseca, N.A.; Moura, V.; de Lima, M.C.P.; Simões, S.; Moreira, J.N. Lipid-based nanoparticles for siRNA delivery in cancer therapy: Paradigms and challenges. Accounts Chem. Res. 2012, 45, 1163–1171. [Google Scholar] [CrossRef]
- Wu, S.Y.; Singhania, A.; Burgess, M.; Putral, L.N.; Kirkpatrick, C.; Davies, N.M.; McMillan, N.A.J. Systemic delivery of E6/7 siRNA using novel lipidic particles and its application with cisplatin in cervical cancer mouse models. Gene Ther. 2011, 18, 14–22. [Google Scholar] [CrossRef]
- Gomes-Da-Silva, L.C.; Ramalho, J.S.; Pedroso De Lima, M.C.; Simões, S.; Moreira, J.N. Impact of anti-PLK1 siRNA-containing F3-targeted liposomes on the viability of both cancer and endothelial cells. Eur. J. Pharm. Biopharm. 2013, 85, 356–364. [Google Scholar] [CrossRef]
- Tabernero, J.; Shapiro, G.I.; LoRusso, P.M.; Cervantes, A.; Schwartz, G.K.; Weiss, G.J.; Paz-Ares, L.; Cho, D.C.; Infante, J.R.; Alsina, M.; et al. First-in-Humans Trial of an RNA Interference Therapeutic Targeting VEGF and KSP in Cancer Patients with Liver Involvement. Cancer Discov. 2013, 3, 406–418. [Google Scholar] [CrossRef] [PubMed]
- Schultheis, B.; Strumberg, D.; Santel, A.; Vank, C.; Gebhardt, F.; Keil, O.; Lange, C.; Giese, K.; Kaufmann, J.; Khan, M.; et al. First-in-human phase I study of the liposomal RNA interference therapeutic Atu027 in patients with advanced solid tumors. J. Clin. Oncol. 2014, 32, 4141–4148. [Google Scholar] [CrossRef]
- Ozcan, G.; Ozpolat, B.; Coleman, R.L.; Sood, A.K.; Lopez-berestein, G. Preclinical and clinical development of siRNA-based therapeutics. Adv. Drug Deliv. Rev. 2015, 87, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Barata, P.; Sood, A.K.; Hong, D.S. RNA-targeted therapeutics in cancer clinical trials: Current status and future directions. Cancer Treat. Rev. 2016, 50, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Seol, Y.; Skinner, G.M.; Visscher, K.; Buhot, A.; Halperin, A. Stretching of Homopolymeric RNA Reveals Single-Stranded Helices and Base-Stacking. Phys. Rev. Lett. 2007, 98, 158103. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Meisburger, S.P.; Pabit, S.A.; Sutton, J.L.; Webb, W.W.; Pollack, L. Ionic strength-dependent persistence lengths of single-stranded RNA and DNA. Proc. Natl. Acad. Sci. USA 2012, 109, 799–804. [Google Scholar] [CrossRef]
- Jacobson, D.R.; McIntosh, D.B.; Stevens, M.J.; Rubinstein, M.; Saleh, O.A. Single-stranded nucleic acid elasticity arises from internal electrostatic tension. Proc. Natl. Acad. Sci. USA 2017, 114, 5095–5100. [Google Scholar] [CrossRef]
- Rosa, M.; Dias, R.; da Graça Miguel, M.; Lindman, B. DNA—Cationic surfactant interactions are different for double- and single-stranded DNA. Biomacromolecules 2005, 6, 2164–2171. [Google Scholar] [CrossRef]
- Liu, X.; Abbott, N.L. Characterization of the nanostructure of complexes formed by single- or double-stranded oligonucleotides with a cationic surfactant. J. Phys. Chem. B 2010, 114, 15554–15564. [Google Scholar] [CrossRef]
- Neumann, T.; Gajria, S.; Bouxsein, N.F.; Jaeger, L.; Tirrell, M. Structural responses of DNA-DDAB films to varying hydration and temperature. J. Am. Chem. Soc. 2010, 132, 7025–7037. [Google Scholar] [CrossRef]
- Cuomo, F.; Mosca, M.; Murgia, S.; Avino, P.; Ceglie, A.; Lopez, F. Evidence for the role of hydrophobic forces on the interactions of nucleotide-monophosphates with cationic liposomes. J. Colloid Interface Sci. 2013, 410, 146–151. [Google Scholar] [CrossRef]
- Sahin, U.; Karikó, K.; Türeci, Ö. mRNA-based therapeutics-developing a new class of drugs. Nat. Rev. Drug Discov. 2014, 13, 759–780. [Google Scholar] [CrossRef]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Finn, J.D.; Smith, A.R.; Patel, M.C.; Shaw, L.; Youniss, M.R.; van Heteren, J.; Dirstine, T.; Ciullo, C.; Lescarbeau, R.; Seitzer, J.; et al. A Single Administration of CRISPR/Cas9 Lipid Nanoparticles Achieves Robust and Persistent In Vivo Genome Editing. Cell Rep. 2018, 22, 2227–2235. [Google Scholar] [CrossRef] [PubMed]
- Reinhard, K.; Rengstl, B.; Oehm, P.; Michel, K.; Billmeier, A.; Hayduk, N.; Klein, O.; Kuna, K.; Ouchan, Y.; Wöll, S.; et al. An RNA vaccine drives expansion and efficacy of claudin-CAR-T cells against solid tumors. Science 2020, 367, 446–453. [Google Scholar] [CrossRef] [PubMed]
- Bartel, D.P. MicroRNAs: Target Recognition and Regulatory Functions. Cell 2009, 136, 215–233. [Google Scholar] [CrossRef]
- Chen, Y.; Gao, D.-Y.; Huang, L. In vivo delivery of miRNAs for cancer therapy: Challenges and strategies. Adv. Drug Deliv. Rev. 2015, 81, 128–141. [Google Scholar] [CrossRef]
- Lee, S.W.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef]
- Costa, P.M.; Cardoso, A.L.; Custódia, C.; Cunha, P.; Pereira de Almeida, L.; Pedroso de Lima, M.C. MiRNA-21 silencing mediated by tumor-targeted nanoparticles combined with sunitinib: A new multimodal gene therapy approach for glioblastoma. J. Control. Release 2015, 207, 31–39. [Google Scholar] [CrossRef]
- Esposito, C.L.; Nuzzo, S.; Kumar, S.A.; Rienzo, A.; Lawrence, C.L.; Pallini, R.; Shaw, L.; Alder, J.E.; Ricci-Vitiani, L.; Catuogno, S.; et al. A combined microRNA-based targeted therapeutic approach to eradicate glioblastoma stem-like cells. J. Control. Release 2016, 238, 43–57. [Google Scholar] [CrossRef]
- Maroof, H.; Islam, F.; Dong, L.; Ajjikuttira, P.; Gopalan, V.; McMillan, N.A.J.; Lam, A.K. Liposomal Delivery of miR-34b-5p Induced Cancer Cell Death in Thyroid Carcinoma. Cells 2018, 7, 265. [Google Scholar] [CrossRef]
- Hanna, J.; Hossain, G.S.; Kocerha, J. The potential for microRNA therapeutics and clinical research. Front. Genet. 2019, 10, 478. [Google Scholar] [CrossRef]
- Van Zandwijk, N.; Pavlakis, N.; Kao, S.C.; Linton, A.; Boyer, M.J.; Clarke, S.; Huynh, Y.; Chrzanowska, A.; Fulham, M.J.; Bailey, D.L.; et al. Safety and activity of microRNA-loaded minicells in patients with recurrent malignant pleural mesothelioma: A first-in-man, phase 1, open-label, dose-escalation study. Lancet Oncol. 2017, 18, 1386–1396. [Google Scholar] [CrossRef]
- Marques, E.F.; Silva, B.F.B. Surfactant Self-Assembly. Encycl. Colloid Interface Sci. 2013, 1202–1241. [Google Scholar] [CrossRef]
- Helfrich, W. Elastic Properties of Lipid Bilayers: Theory and Possible Experiments. Z. Nat. C 1973, 28c, 693–703. [Google Scholar] [CrossRef]
- Faizi, H.A.; Frey, S.L.; Steinkühler, J.; Dimova, R.; Vlahovska, P.M. Bending rigidity of charged lipid bilayer membranes. Soft Matter 2019, 15, 6006–6013. [Google Scholar] [CrossRef]
- Khan, A.; Marques, E.F. Synergism and polymorphism in mixed surfactant systems. Curr. Opin. Colloid Interface Sci. 2000, 4, 4002–4410. [Google Scholar] [CrossRef]
- Silva, B.F.B.; Marques, E.F.; Olsson, U.; Linse, P. Size, Shape, and Charge of Salt-Free Catanionic Microemulsion Droplets: A Small-Angle Neutron Scattering and Modeling Study. J. Phys. Chem. B 2009, 113, 10230–10239. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.K.; Ahmad, A.; Evans, H.M.; Safinya, C.R. Cationic lipid-DNA complexes for non-viral gene therapy: Relating supramolecular structures to cellular pathways. Expert Opin. Biol. Ther. 2005, 5, 33–53. [Google Scholar] [CrossRef]
- Rädler, J.O.; Koltover, I.; Salditt, T.; Safinya, C.R. Structure of DNA-cationic liposome complexes: DNA intercalation in multilamellar membranes in distinct interhelical packing regimes. Science 1997, 275, 810–814. [Google Scholar] [CrossRef]
- Harries, D.; May, S.; Gelbart, W.M.; Ben-Shaul, A. Structure, Stability, and Thermodynamics of Lamellar DNA-Lipid Complexes. Biophys. J. 1998, 75, 159–173. [Google Scholar] [CrossRef]
- Harries, D.; May, S.; Ben-shaul, A. Counterion release in membrane–biopolymer interactions. Soft Matter 2013, 9, 9268–9284. [Google Scholar] [CrossRef]
- Barreleiro, P.C.A.; Olofsson, G.; Alexandridis, P. Interaction of DNA with cationic vesicles: A calorimetric study. J. Phys. Chem. B 2000, 104, 7795–7802. [Google Scholar] [CrossRef]
- Wagner, K.; Harries, D.; May, S.; Kahl, V.; Rädler, J.O.; Ben-Shaul, A. Direct evidence for counterion release upon cationic lipid-DNA condensation. Langmuir 2000, 16, 303–306. [Google Scholar] [CrossRef]
- Liang, H.; Harries, D.; Wong, G.C.L. Polymorphism of DNA-anionic liposome complexes reveals hierarchy of ion-mediated interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 11173–11178. [Google Scholar] [CrossRef] [PubMed]
- Mcmanus, J.J.; Rädler, J.O.; Dawson, K.A. Does Calcium Turn a Zwitterionic Lipid Cationic? J. Phys. Chem. B 2003, 107, 9869–9875. [Google Scholar] [CrossRef]
- McManus, J.J.; Rädler, J.O.; Dawson, K.A. Observation of a rectangular columnar phase in a DNA-calcium-zwitterionic lipid complex. J. Am. Chem. Soc. 2004, 126, 15966–15967. [Google Scholar] [CrossRef] [PubMed]
- DeHaseth, P.L.; Lohman, T.M.; Record, M.T. Nonspecific Interaction of lac Repressor with DNA: An Association Reaction Driven by Counterion Release. Biochemistry 1977, 16, 4783–4790. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Safinya, C.R. Phase diagram, stability, and overcharging of lamellar cationic lipid-DNA self-assembled complexes. Biophys. J. 1999, 77, 915–924. [Google Scholar] [CrossRef]
- Jonsson, M.; Linse, P. Polyelectrolyte-macroion complexation. I. Effect of linear charge density, chain length, and macroion charge. J. Chem. Phys. 2001, 115, 3406–3418. [Google Scholar] [CrossRef]
- Jonsson, M.; Linse, P. Polyelectrolyte-macroion complexation. II. Effect of chain flexibility. J. Chem. Phys. 2001, 115, 10975–10985. [Google Scholar] [CrossRef]
- Stornes, M.; Linse, P.; Dias, R.S. Monte Carlo Simulations of Complexation between Weak Polyelectrolytes and a Charged Nanoparticle. Influence of Polyelectrolyte Chain Length and Concentration. Macromolecules 2017, 50, 5978–5988. [Google Scholar] [CrossRef]
- Park, S.Y.; Bruinsma, R.F.; Gelbart, W.M. Spontaneous overcharging of macro-ion complexes. Europhys. Lett. 1999, 46, 454–460. [Google Scholar] [CrossRef]
- Majzoub, R.N.; Chan, C.-L.; Ewert, K.K.; Silva, B.F.B.; Liang, K.S.; Jacovetty, E.L.; Carragher, B.; Potter, C.S.; Safinya, C.R. Uptake and transfection efficiency of PEGylated cationic liposome-DNA complexes with and without RGD-tagging. Biomaterials 2014, 35, 4996–5005. [Google Scholar] [CrossRef] [PubMed]
- Safinya, C.R.; Ewert, K.K.; Majzoub, R.N.; Leal, C. Cationic liposome–nucleic acid complexes for gene delivery and gene silencing. New J. Chem. 2014, 38, 5164–5172. [Google Scholar] [CrossRef] [PubMed]
- Raviv, U.; Needleman, D.J.; Li, Y.; Miller, H.P.; Wilson, L.; Safinya, C.R. Cationic-liposome microtubule complexes: Pathways to the formation of two-state lipid-protein nanotubes with open or closed ends. Proc. Natl. Acad. Sci. USA 2005, 102, 11167–11172. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.; Kim, H.; Leal, C. Self-organization of nucleic acids in lipid constructs. Curr. Opin. Colloid Interface Sci. 2016, 26, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Ewert, K.K.; Evans, H.M.; Zidovska, A.; Bouxsein, N.F.; Ahmad, A.; Safinya, C.R. A columnar phase of dendritic lipid-based cationic liposome-DNA complexes for gene delivery: Hexagonally ordered cylindrical micelles embedded in a DNA honeycomb lattice. J. Am. Chem. Soc. 2006, 128, 3998–4006. [Google Scholar] [CrossRef]
- Koltover, I.; Salditt, T.; Rädler, J.O.; Safinya, C.R. An Inverted Hexagonal Phase of Cationic Liposome-DNA Complexes Related to DNA Release and Delivery. Science 1998, 281, 78–81. [Google Scholar] [CrossRef]
- Lasic, D.D.; Strey, H.; Stuart, M.C.A.; Podgornik, R.; Frederik, P.M. The Structure of DNA-Liposome Complexes. J. Am. Chem. Soc. 1997, 119, 832–833. [Google Scholar] [CrossRef]
- Battersby, B.J.; Grimm, R.; Huebner, S.; Cevc, G. Evidence for three-dimensional interlayer correlations in cationic lipid-DNA complexes as observed by cryo-electron microscopy. Biochim. Biophys. Acta 1998, 1372, 379–383. [Google Scholar] [CrossRef]
- Schmutz, M.; Durand, D.; Debin, A.; Palvadeau, Y.; Etienne, A.; Thierry, A.R. DNA packing in stable lipid complexes designed for gene transfer imitates DNA compaction in bacteriophage. Proc. Natl. Acad. Sci. USA 1999, 96, 12293–12298. [Google Scholar] [CrossRef]
- Salditt, T.; Koltover, I.; Rädler, J.O.; Safinya, C.R. Two-dimensional smectic ordering of linear DNA chains in self-assembled dna-cationic liposome mixtures. Phys. Rev. Lett. 1997, 79, 2582–2585. [Google Scholar] [CrossRef]
- Gómez-Varela, A.I.; Gaspar, R.; Miranda, A.; Assis, J.L.; Valverde, R.R.H.F.; Einicker-Lamas, M.; Silva, B.F.B.; De Beule, P.A.A. Fluorescence Cross-Correlation Spectroscopy as a valuable tool to characterize cationic liposome-DNA nanoparticle assembly. J. Biophotonics 2020, e202000200. [Google Scholar] [CrossRef] [PubMed]
- Koltover, I.; Wagner, K.; Safinya, C.R. DNA Condensation in Two-Dimensions. Proc. Natl. Acad. Sci. USA 2000, 97, 14046–14052. [Google Scholar] [CrossRef]
- Artzner, F.; Zantl, R.; Rapp, G.; Rädler, J.O. Observation of a Rectangular Columnar Phase in Condensed Lamellar Cationic Lipid-DNA Complexes. Phys. Rev. Lett. 1998, 81, 5015–5018. [Google Scholar] [CrossRef]
- Koynova, R.; MacDonald, R.C. Columnar DNA superlattices in lamellar o-ethylphosphatidylcholine lipoplexes: Mechanism of the gel-liquid crystalline lipid phase transition. Nano Lett. 2004, 4, 1475–1479. [Google Scholar] [CrossRef]
- Bouxsein, N.F.; Leal, C.; Mcallister, C.S.; Li, Y.; Ewert, K.K.; Samuel, C.E.; Safinya, C.R. 3D Columnar Phase of Stacked Short DNA Organized by Coherent Membrane Undulations. Langmuir 2019, 35, 11891–11901. [Google Scholar] [CrossRef] [PubMed]
- Schiessel, H.; Aranda-Espinoza, H. Electrostatically induced undulations of lamellar DNA-lipid complexes. Eur. Phys. J. E 2001, 5, 499–506. [Google Scholar] [CrossRef][Green Version]
- Dan, N.; Danino, D. Structure and kinetics of lipid-nucleic acid complexes. Adv. Colloid Interface Sci. 2014, 205, 230–239. [Google Scholar] [CrossRef]
- Majzoub, R.N.; Ewert, K.K.; Jacovetty, E.L.; Carragher, B.; Potter, C.S.; Li, Y.; Safinya, C.R. Patterned Threadlike Micelles and DNA-Tethered Nanoparticles: A Structural Study of PEGylated Cationic Liposome-DNA Assemblies. Langmuir 2015, 31, 7073–7083. [Google Scholar] [CrossRef]
- Huebner, S.; Battersby, B.J.; Grimm, R.; Cevc, G. Lipid-DNA Complex Formation: Reorganization and Rupture of Lipid Vesicles in the Presence of DNA As Observed by Cryoelectron Microscopy. Biophys. J. 1999, 76, 3158–3166. [Google Scholar] [CrossRef]
- Aytar, B.S.; Muller, J.P.E.; Golan, S.; Kondo, Y.; Talmon, Y.; Abbott, N.L.; Lynn, D.M. Chemical oxidation of a redox-active, ferrocene-containing cationic lipid: Influence on interactions with DNA and characterization in the context of cell transfection. J. Colloid Interface Sci. 2012, 387, 56–64. [Google Scholar] [CrossRef]
- Desigaux, L.; Sainlos, M.; Lambert, O.; Chevre, R.; Letrou-Bonneval, E.; Vigneron, J.P.; Lehn, P.; Lehn, J.-M.; Pitard, B. Self-assembled lamellar complexes of siRNA with lipidic aminoglycoside derivatives promote efficient siRNA delivery and interference. Proc. Natl. Acad. Sci. USA 2007, 104, 16534–16539. [Google Scholar] [CrossRef]
- Pozharski, E.V.; MacDonald, R.C. Single lipoplex study of cationic lipoid-DNA, self-assembled complexes. Mol. Pharm. 2007, 4, 962–974. [Google Scholar] [CrossRef] [PubMed]
- Barreleiro, P.C.A.; May, R.P.; Lindman, B. Mechanism of formation of DNA—Cationic vesicle complexes. Faraday Discuss. 2002, 122, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Koynova, R.; Tenchov, B. Cationic lipids: Molecular structure/transfection activity relationships and interactions with biomembranes. Top. Curr. Chem. 2010, 296, 51–93. [Google Scholar] [CrossRef]
- Silva, B.F.B.; Majzoub, R.M.; Chan, C.-L.; Li, Y.; Olsson, U.; Safinya, C.R. PEGylated cationic liposome–DNA complexation in brine is pathway-dependent. Biochim. Biophys. Acta Biomembr. 2014, 1838, 398–412. [Google Scholar] [CrossRef] [PubMed]
- Bouxsein, N.F.; McAllister, C.S.; Ewert, K.K.; Samuel, C.E.; Safinya, C.R. Structure and gene silencing activities of monovalent and pentavalent cationic lipid vectors complexed with siRNA. Biochemistry 2007, 46, 4785–4792. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Tam, Y.Y.C.; Cullis, P.R. On the role of helper lipids in lipid nanoparticle formulations of siRNA. Nanoscale 2019, 11, 21733–21739. [Google Scholar] [CrossRef]
- Junquera, E.; Aicart, E. Recent progress in gene therapy to deliver nucleic acids with multivalent cationic vectors. Adv. Colloid Interface Sci. 2016, 233, 161–175. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Darjuan, M.M.; Mercer, J.E.; Chen, S.; Van Der Meel, R.; Thewalt, J.L.; Tam, Y.Y.C.; Cullis, P.R. On the Formation and Morphology of Lipid Nanoparticles Containing Ionizable Cationic Lipids and siRNA. ACS Nano 2018, 12, 4787–4795. [Google Scholar] [CrossRef]
- Thierry, A.R.; Norris, V.; Molina, F.; Schmutz, M. Lipoplex nanostructures reveal a general self-organization of nucleic acids. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 385–394. [Google Scholar] [CrossRef] [PubMed]
- Ziller, A.; Nogueira, S.S.; Hühn, E.; Funari, S.S.; Brezesinski, G.; Hartmann, H.; Sahin, U.; Haas, H.; Langguth, P. Incorporation of mRNA in Lamellar Lipid Matrices for Parenteral Administration. Mol. Pharm. 2018, 15, 642–651. [Google Scholar] [CrossRef] [PubMed]
- Arteta, M.Y.; Kjellman, T.; Bartesaghi, S.; Wallin, S.; Wu, X.; Kvist, A.J.; Dabkowska, A.; Székely, N.; Radulescu, A.; Bergenholtz, J.; et al. Successful reprogramming of cellular protein production through mRNA delivered by functionalized lipid nanoparticles. Proc. Natl. Acad. Sci. USA 2018, 115, E3351–E3360. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Ashwanikumar, N.; Robinson, E.; Xia, Y.; Mihai, C.; Griffith, J.P.; Hou, S.; Esposito, A.A.; Ketova, T.; Welsher, K.; et al. Naturally-occurring cholesterol analogues in lipid nanoparticles induce polymorphic shape and enhance intracellular delivery of mRNA. Nat. Commun. 2020, 11, 983. [Google Scholar] [CrossRef]
- Weisman, S.; Hirsch-Lerner, D.; Barenholz, Y.; Talmon, Y. Nanostructure of cationic lipid-oligonucleotide complexes. Biophys. J. 2004, 87, 609–614. [Google Scholar] [CrossRef]
- Rozenfeld, J.H.K.; Duarte, E.L.; Barbosa, L.R.S.; Lamy, M.T. The effect of an oligonucleotide on the structure of cationic DODAB vesicles. Phys. Chem. Chem. Phys. 2015, 17, 7498–7506. [Google Scholar] [CrossRef]
- Ram-On, M.; Cohen, Y.; Talmon, Y. Effect of polyelectrolyte stiffness and solution pH on the nanostructure of complexes formed by cationic amphiphiles and negatively charged polyelectrolytes. J. Phys. Chem. B 2016, 120, 5907–5915. [Google Scholar] [CrossRef]
- Zhou, S.; Liang, D.; Burger, C.; Yeh, F.; Chu, B. Nanostructures of complexes formed by calf thymus DNA interacting with cationic surfactants. Biomacromolecules 2004, 5, 1256–1261. [Google Scholar] [CrossRef]
- Lin, A.; Slack, N.; Ahmad, A.; George, C.; Samuel, C.; Safinya, C.R. Three-dimensional Imaging of Lipid Gene-Carriers: Membrane Charge Density Controls Universal Transfection Behavior in Lamellar Cationic Liposome-DNA Complexes. Biophys. J. 2003, 84, 3307–3316. [Google Scholar] [CrossRef]
- Semple, S.C.; Akinc, A.; Chen, J.; Sandhu, A.P.; Mui, B.L.; Cho, C.K.; Sah, D.W.Y.; Stebbing, D.; Crosley, E.J.; Yaworski, E.; et al. Rational design of cationic lipids for siRNA delivery. Nat. Biotechnol. 2010, 28, 172–176. [Google Scholar] [CrossRef]
- Leal, C.; Ewert, K.K.; Bouxsein, N.F.; Shirazi, R.S.; Li, Y.; Safinya, C.R. Stacking of short DNA induces the gyroid cubic-to-inverted hexagonal phase transition in lipid-DNA complexes. Soft Matter 2013, 9, 795–804. [Google Scholar] [CrossRef]
- Leal, C.; Bouxsein, N.F.; Ewert, K.K.; Safinya, C.R. Highly Efficient Gene Silencing Activity of siRNA Embedded in a Nanostructured Gyroid Cubic Lipid Matrix. J. Am. Chem. Soc. 2010, 132, 16841–16847. [Google Scholar] [CrossRef] [PubMed]
- Danino, D.; Kesselman, E.; Saper, G.; Petrache, H.I.; Harries, D. Osmotically induced reversible transitions in lipid-DNA mesophases. Biophys. J. 2009, 96, L43–L45. [Google Scholar] [CrossRef][Green Version]
- Seddon, J.M.; Templer, R.H. Cubic phases of self-assembled amphiphilic aggregates. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 1993, 344, 377–401. [Google Scholar] [CrossRef]
- Larsson, K. Aqueous dispersions of cubic lipid-water phases. Curr. Opin. Colloid Interface Sci. 2000, 5, 64–69. [Google Scholar] [CrossRef]
- Chang, D.P.; Barauskas, J.; Dabkowska, A.P.; Wadsäter, M.; Tiberg, F.; Nylander, T. Non-lamellar lipid liquid crystalline structures at interfaces. Adv. Colloid Interface Sci. 2015, 222, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Song, Z.; Leal, C. Super-swelled lyotropic single crystals. Proc. Natl. Acad. Sci. USA 2017, 114, 10834–10839. [Google Scholar] [CrossRef] [PubMed]
- Angelov, B.; Angelova, A.; Filippov, S.K.; Narayanan, T.; Drechsler, M.; Štěpánek, P.; Couvreur, P.; Lesieur, S. DNA/fusogenic lipid nanocarrier assembly: Millisecond structural dynamics. J. Phys. Chem. Lett. 2013, 4, 1959–1964. [Google Scholar] [CrossRef]
- Leal, C.; Ewert, K.K.; Shirazi, R.S.; Bouxsein, N.F.; Safinya, C.R. Nanogyroids Incorporating Multivalent Lipids: Enhanced Membrane Charge Density and Pore Forming Ability for Gene Silencing. Langmuir 2011, 27, 7691–7697. [Google Scholar] [CrossRef]
- Hyde, S.T. Microstructure of Bicontinuous Surfactant Aggregates. J. Phys. Chem. 1989, 93, 1458–1464. [Google Scholar] [CrossRef]
- Seddon, J.M.; Templer, R.H. Polymorphism of Lipid-Water Systems. In Handbook of Biological Physics; Lipowsky, R., Sackmann, E., Eds.; Elsevier Science B. V.: Amsterdam, The Netherlands, 1995; pp. 97–160. ISBN 978-0-444-81975-8. [Google Scholar]
- Bilalov, A.; Olsson, U.; Lindman, B. A cubic DNA-lipid complex. Soft Matter 2009, 5, 3827–3830. [Google Scholar] [CrossRef]
- Bilalov, A.; Olsson, U.; Lindman, B. Complexation between DNA and surfactants and lipids: Phase behavior and molecular organization. Soft Matter 2012, 8, 11022–11033. [Google Scholar] [CrossRef]
- Bilalov, A.; Olsson, U.; Lindman, B. DNA–lipid self-assembly: Phase behavior and phase structures of a DNA–surfactant complex mixed with lecithin and water. Soft Matter 2011, 7, 730–742. [Google Scholar] [CrossRef]
- Zidovska, A.; Evans, H.M.; Ewert, K.K.; Quispe, J.; Carragher, B.; Potter, C.S.; Safinya, C.R. Liquid Crystalline Phases of Dendritic Lipid–DNA Self-Assemblies: Lamellar, Hexagonal and DNA Bundles. J. Phys. Chem. B 2009, 113, 3694–3703. [Google Scholar] [CrossRef] [PubMed]
- Ghirlando, R.; Wachtel, E.J.; Arad, T.; Minsky, A. DNA Packaging Induced by Micellar Aggregates: A Novel in Vitro DNA Condensation System. Biochemistry 1992, 31, 7110–7119. [Google Scholar] [CrossRef] [PubMed]
- Mel’nikov, S.M.; Sergeyev, V.G.; Yoshikawa, K.; Takahashi, H.; Hatta, I. Cooperativity or phase transition? Unfolding transition of DNA cationic surfactant complex. J. Chem. Phys. 1997, 107, 6917–6924. [Google Scholar] [CrossRef]
- Krishnaswamy, R.; Raghunathan, V.A.; Sood, A.K. Reentrant phase transitions of DNA-surfactant complexes. Phys. Rev. E 2004, 69, 031905. [Google Scholar] [CrossRef]
- Krishnaswamy, R.; Pabst, G.; Rappolt, M.; Raghunathan, V.A.; Sood, A.K. Structure of DNA-CTAB-hexanol complexes. Phys. Rev. E 2006, 73, 031904. [Google Scholar] [CrossRef]
- Leal, C.; Moniri, E.; Pegado, K.; Wennerström, H. Electrostatic Attraction between DNA and a Cationic Surfactant Aggregate. The Screening Effect of Salt. J. Phys. Chem. B 2007, 111, 5999–6005. [Google Scholar] [CrossRef]
- Radhakrishnan, A.V.; Ghosh, S.K.; Pabst, G.; Raghunathan, V.A.; Sood, A.K. Tuning DNA-amphiphile condensate architecture with strongly binding counterions. Proc. Natl. Acad. Sci. USA 2012, 109, 6394–6398. [Google Scholar] [CrossRef]
- Uhríková, D.; Zajac, I.; Dubničková, M.; Pisárčik, M.; Funari, S.S.; Rapp, G.; Balgavý, P. Interaction of gemini surfactants butane-1,4-diyl-bis (alkyldimethylammonium bromide) with DNA. Colloids Surfaces B Biointerfaces 2005, 42, 59–68. [Google Scholar] [CrossRef] [PubMed]
- Foldvari, M.; Badea, I.; Wettig, S.; Verrall, R.; Bagonluri, M. Structural characterization of novel gemini non-viral DNA delivery systems for cutaneous gene therapy. J. Exp. Nanosci. 2006, 1, 165–176. [Google Scholar] [CrossRef]
- Hubčík, L.; Galliková, D.; Pullmannová1, P.; Lacinová, L.; Sulová, Z.; Hanulová, M.; Funari, S.S.; Devínsky, F.; Uhríková, D. DNA–DOPE–gemini surfactants complexes at low surface charge density: From structure to transfection efficiency. Gen. Physiol. Biophys. 2018, 37, 57–69. [Google Scholar] [CrossRef]
- Subramanian, G.; Hjelm, R.P.; Deming, T.J.; Smith, G.S.; Li, Y.; Safinya, C.R. Structure of complexes of cationic lipids and poly(glutamic acid) polypeptides: A pinched lamellar phase. J. Am. Chem. Soc. 2000, 122, 26–34. [Google Scholar] [CrossRef]
- Piculell, L.; Norrman, J.; Svensson, A.V.; Lynch, I.; Bernardes, J.S.; Loh, W. Ionic surfactants with polymeric counterions. Adv. Colloid Interface Sci. 2009, 147–148, 228–236. [Google Scholar] [CrossRef]
- Chiappisi, L.; Hoffmann, I.; Gradzielski, M. Complexes of oppositely charged polyelectrolytes and surfactants—Recent developments in the field of biologically derived polyelectrolytes. Soft Matter 2013, 9, 3896–3909. [Google Scholar] [CrossRef]
- Falsini, S.; Ristori, S.; Ciani, L.; Di Cola, E.; Supuran, C.T.; Arcangeli, A.; In, M. Time resolved SAXS to study the complexation of siRNA with cationic micelles of divalent surfactants. Soft Matter 2014, 10, 2226–2233. [Google Scholar] [CrossRef]
- Andrzejewska, W.; Pietralik, Z.; Skupin, M.; Kozak, M. Structural studies of the formation of lipoplexes between siRNA and selected bis-imidazolium gemini surfactants. Colloids Surfaces B Biointerfaces 2016, 146, 598–606. [Google Scholar] [CrossRef]
- Zelphati, O.; Uyechi, L.S.; Barron, L.G.; Szoka, F.C., Jr. Effect of serum components on the physico-chemical properties of cationic lipid/oligonucleotide complexes and on their interactions with cells. Biochim. Biophys. Acta 1998, 1390, 119–133. [Google Scholar] [CrossRef]
- Allen, T.M.; Chonn, A. Large unilamellar liposomes with low uptake into the reticuloendothelial system. FEBS Lett. 1987, 223, 42–46. [Google Scholar] [CrossRef]
- Gabizon, A.; Papahadjopoulos, D. Liposome formulations with prolonged circulation time in blood and enhanced uptake in tumors. Proc. Natl. Acad. Sci. USA 1988, 85, 6949–6953. [Google Scholar] [CrossRef] [PubMed]
- Klibanov, A.L.; Maruyama, K.; Torchilin, V.P.; Huang, L. Amphipathic polyethyleneglycols effectively prolong the circulation time of liposomes. FEBS Lett. 1990, 268, 235–237. [Google Scholar] [CrossRef]
- Blume, G.; Cevc, G. Liposomes for sustained drug release in vivo. Biochim. Biophys. Acta 1990, 1029, 91–97. [Google Scholar] [CrossRef]
- Papahadjopoulos, D.; Allen, T.M.; Gabizon, A.; Mayhew, E.; Matthay, K.; Huang, S.K.; Lee, K.-D.; Woodle, M.C.; Lasic, D.D.; Redemann, C.; et al. Sterically stabilized liposomes: Improvements in pharmacokinetics and antitumor therapeutic efficacy. Proc. Natl. Acad. Sci. USA 1991, 88, 11460–11464. [Google Scholar] [CrossRef]
- Kuhl, T.L.; Leckband, D.E.; Lasic, D.D.; Israelachvili, J.N. Modulation of Interaction Forces Between Bilayers Exposing Short-Chained Ethylene Oxide Headgroups. Biophys. J. 1994, 66, 1479–1488. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Hristova, K.; Needham, D.; McIntosh, T.J. Range and Magnitude of the Steric Pressure Between Bilayers Containing Phospholipids with Covalently Attached Poly(ethylene glycol). Biophys. J. 1995, 68, 1921–1936. [Google Scholar] [CrossRef]
- Martin-Herranz, A.; Ahmad, A.; Evans, H.M.; Ewert, K.; Schulze, U.; Safinya, C.R. Surface Functionalized Cationic Lipid-DNA Complexes for Gene Delivery: PEGylated Lamellar Complexes Exhibit Distinct DNA-DNA Interaction Regimes. Biophys. J. 2004, 86, 1160–1168. [Google Scholar] [CrossRef][Green Version]
- Aissaoui, A.; Chami, M.; Hussein, M.; Miller, A.D. Efficient topical delivery of plasmid DNA to lung in vivo mediated by putative triggered, PEGylated pDNA nanoparticles. J. Control. Release 2011, 154, 275–284. [Google Scholar] [CrossRef]
- Wonder, E.; Simón-Gracia, L.; Scodeller, P.; Majzoub, R.N.; Kotamraju, V.R.; Ewert, K.K.; Teesalu, T.; Safinya, C.R. Competition of charge-mediated and specific binding by peptide-tagged cationic liposome–DNA nanoparticles in vitro and in vivo. Biomaterials 2018, 166, 52–63. [Google Scholar] [CrossRef]
- Morille, M.; Passirani, C.; Dufort, S.; Bastiat, G.; Pitard, B.; Coll, J.L.; Benoit, J.-P. Tumor transfection after systemic injection of DNA lipid nanocapsules. Biomaterials 2011, 32, 2327–2333. [Google Scholar] [CrossRef]
- Ho, E.A.; Osooly, M.; Strutt, D.; Masin, D.; Yang, Y.; Yan, H.; Bally, M. Characterization of Long-Circulating Cationic Nanoparticle Formulations Consisting of a Two-Stage PEGylation Step for the Delivery of siRNA in a Breast Cancer Tumor Model. J. Pharm. Biomed. Anal. 2013, 102, 227–236. [Google Scholar] [CrossRef]
- Oliveira, A.C.N.; Raemdonck, K.; Martens, T.; Rombouts, K.; Simón-Vázquez, R.; Botelho, C.; Lopes, I.; Lúcio, M.; González-Fernández, Á.; Real Oliveira, M.E.C.D.; et al. Stealth monoolein-based nanocarriers for delivery of siRNA to cancer cells. Acta Biomater. 2015, 25, 216–229. [Google Scholar] [CrossRef] [PubMed]
- Krzysztoń, R.; Salem, B.; Lee, D.J.; Schwake, G.; Wagner, E.; Rädler, J.O. Microfluidic self-assembly of folate-targeted monomolecular siRNA-lipid nanoparticles. Nanoscale 2017, 9, 7442–7453. [Google Scholar] [CrossRef] [PubMed]
- Hatakeyama, H.; Akita, H.; Harashima, H. The polyethyleneglycol dilemma: Advantage and disadvantage of PEGylation of liposomes for systemic genes and nucleic acids delivery to tumors. Biol. Pharm. Bull. 2013, 36, 892–899. [Google Scholar] [CrossRef] [PubMed]
- Romberg, B.; Hennink, W.E.; Storm, G. Sheddable coatings for long-circulating nanoparticles. Pharm. Res. 2008, 25, 55–71. [Google Scholar] [CrossRef]
- Chan, C.-L.; Majzoub, R.N.; Shirazi, R.S.; Ewert, K.K.; Chen, Y.-J.; Liang, K.S.; Safinya, C.R. Endosomal escape and transfection efficiency of PEGylated cationic liposome-DNA complexes prepared with an acid-labile PEG-lipid. Biomaterials 2012, 33, 4928–4935. [Google Scholar] [CrossRef]
- MacLachlan, I.; Cullis, P. Diffusible-PEG-Lipid Stabilized Plasmid Lipid Particles. Adv. Genet. 2005, 53PA, 157–188. [Google Scholar] [CrossRef]
- Belliveau, N.M.; Huft, J.; Lin, P.J.; Chen, S.; Leung, A.K.; Leaver, T.J.; Wild, A.W.; Lee, J.B.; Taylor, R.J.; Tam, Y.K.; et al. Microfluidic Synthesis of Highly Potent Limit-size Lipid Nanoparticles for In Vivo Delivery of siRNA. Mol. Ther. Nucleic Acids 2012, 1, e37. [Google Scholar] [CrossRef]
- Gindy, M.E.; DiFelice, K.; Kumar, V.; Prud’Homme, R.K.; Celano, R.; Haas, R.M.; Smith, J.S.; Boardman, D. Mechanism of macromolecular structure evolution in self-assembled lipid nanoparticles for siRNA delivery. Langmuir 2014, 30, 4613–4622. [Google Scholar] [CrossRef]
- Kim, H.; Leal, C. Cuboplexes: Topologically Active siRNA Delivery. ACS Nano 2015, 9, 10214–10226. [Google Scholar] [CrossRef]
- Kim, H.; Sung, J.; Chang, Y.; Alfeche, A.; Leal, C. Microfluidics Synthesis of Gene Silencing Cubosomes. ACS Nano 2018, 12, 9196–9205. [Google Scholar] [CrossRef]
- Leung, A.K.K.; Hafez, I.M.; Baoukina, S.; Belliveau, N.M.; Zhigaltsev, I.V.; Afshinmanesh, E.; Tieleman, D.P.; Hansen, C.L.; Hope, M.J.; Cullis, P.R. Lipid nanoparticles containing siRNA synthesized by microfluidic mixing exhibit an electron-dense nanostructured core. J. Phys. Chem. C 2012, 116, 18440–18450. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Love, K.T.; Chen, Y.; Eltoukhy, A.A.; Kastrup, C.; Sahay, G.; Jeon, A.; Dong, Y.; Whitehead, K.A.; Anderson, D.G. Rapid Discovery of Potent siRNA-Containing Lipid Nanoparticles Enabled by Controlled Microfluidic Formulation. J. Am. Chem. Soc. 2012, 134, 6948–6951. [Google Scholar] [CrossRef]
- Rudorf, S.; Rädler, J.O. Self-assembly of stable monomolecular nucleic acid lipid particles with a size of 30 nm. J. Am. Chem. Soc. 2012, 134, 11652–11658. [Google Scholar] [CrossRef]
- Rehman, Z.; Zuhorn, I.S.; Hoekstra, D. How cationic lipids transfer nucleic acids into cells and across cellular membranes: Recent advances. J. Control. Release 2013, 166, 46–56. [Google Scholar] [CrossRef]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer nanotechnology: The impact of passive and active targeting in the era of modern cancer biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef]
- Sahay, G.; Querbes, W.; Alabi, C.; Eltoukhy, A.; Sarkar, S.; Zurenko, C.; Karagiannis, E.; Love, K.; Chen, D.; Zoncu, R.; et al. Efficiency of siRNA delivery by lipid nanoparticles is limited by endocytic recycling. Nat. Biotechnol. 2013, 31, 653–658. [Google Scholar] [CrossRef] [PubMed]
- Majzoub, R.N.; Wonder, E.; Ewert, K.K.; Kotamraju, V.R.; Teesalu, T.; Safinya, C.R. Rab11 and Lysotracker Markers Reveal Correlation between Endosomal Pathways and Transfection Efficiency of Surface-Functionalized Cationic Liposome-DNA Nanoparticles. J. Phys. Chem. B 2016, 120, 6439–6453. [Google Scholar] [CrossRef] [PubMed]
- Hafez, I.M.; Cullis, P.R. Roles of lipid polymorphism in intracellular delivery. Adv. Drug Deliv. Rev. 2001, 47, 139–148. [Google Scholar] [CrossRef]
- Hafez, I.M.; Maurer, N.; Cullis, P.R. On the mechanism whereby cationic lipids promote intracellular delivery of polynucleic acids. Gene Ther. 2001, 8, 1188–1196. [Google Scholar] [CrossRef]
- Ahmad, A.; Evans, H.M.; Ewert, K.; George, C.X.; Samuel, C.E.; Safinya, C.R. New multivalent cationic lipids reveal bell curve for transfection efficiency versus membrane charge density: Lipid–DNA complexes for gene delivery. J. Gene Med. 2005, 7, 739–748. [Google Scholar] [CrossRef] [PubMed]
- Rejman, J.; Oberle, V.; Zuhorn, I.S.; Hoekstra, D. Size-dependent internalization of particles via the pathways of clathrin- and caveolae-mediated endocytosis. Biochem. J. 2004, 377, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Gilleron, J.; Querbes, W.; Zeigerer, A.; Borodovsky, A.; Marsico, G.; Schubert, U.; Manygoats, K.; Seifert, S.; Andree, C.; Stöter, M.; et al. Image-based analysis of lipid nanoparticle–mediated siRNA delivery, intracellular trafficking and endosomal escape. Nat. Biotechnol. 2013, 31, 638–646. [Google Scholar] [CrossRef]
- Majzoub, R.N.; Chan, C.-L.; Ewert, K.K.; Silva, B.F.B.; Liang, K.S.; Safinya, C.R. Fluorescence microscopy colocalization of lipid-nucleic acid nanoparticles with wildtype and mutant Rab5-GFP: A platform for investigating early endosomal events. Biochim. Biophys. Acta Biomembr. 2015, 1848, 1308–1318. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, R.S.; Ewert, K.K.; Leal, C.; Majzoub, R.N.; Bouxsein, N.F.; Safinya, C.R. Synthesis and characterization of degradable multivalent cationic lipids with disulfide-bond spacers for gene delivery. Biochim. Biophys. Acta 2011, 1808, 2156–2166. [Google Scholar] [CrossRef] [PubMed]
- Shirazi, R.S.; Ewert, K.K.; Silva, B.F.B.; Leal, C.; Li, Y.; Safinya, C.R. Structural evolution of environmentally responsive cationic liposome-DNA complexes with a reducible lipid linker. Langmuir 2012, 28, 10495–10503. [Google Scholar] [CrossRef] [PubMed]
- Lv, H.; Zhang, S.; Wang, B.; Cui, S.; Yan, J. Toxicity of cationic lipids and cationic polymers in gene delivery. J. Control. Release 2006, 114, 100–109. [Google Scholar] [CrossRef]
- Khalil, I.A.; Younis, M.A.; Kimura, S.; Harashima, H. Lipid Nanoparticles for Cell-Specific in Vivo Targeted Delivery of Nucleic Acids. Biol. Pharm. Bull. 2020, 43, 584–595. [Google Scholar] [CrossRef]
- Semple, S.C.; Klimuk, S.K.; Harasym, T.O.; Dos Santos, N.; Ansell, S.M.; Wong, K.F.; Maurer, N.; Stark, H.; Cullis, P.R.; Hope, M.J.; et al. Efficient encapsulation of antisense oligonucleotides in lipid vesicles using ionizable aminolipids: Formation of novel small multilamellar vesicle structures. Biochim. Biophys. Acta Biomembr. 2001, 1510, 152–166. [Google Scholar] [CrossRef]
- Heyes, J.; Palmer, L.; Bremner, K.; MacLachlan, I. Cationic lipid saturation influences intracellular delivery of encapsulated nucleic acids. J. Control. Release 2005, 107, 276–287. [Google Scholar] [CrossRef]
- Oberli, M.A.; Reichmuth, A.M.; Dorkin, J.R.; Mitchell, M.J.; Fenton, O.S.; Jaklenec, A.; Anderson, D.G.; Langer, R.; Blankschtein, D. Lipid Nanoparticle Assisted mRNA Delivery for Potent Cancer Immunotherapy. Nano Lett. 2017, 17, 1326–1335. [Google Scholar] [CrossRef]
- Kauffman, K.J.; Webber, M.J.; Anderson, D.G. Materials for non-viral intracellular delivery of messenger RNA therapeutics. J. Control. Release 2016, 240, 227–234. [Google Scholar] [CrossRef]
- Leung, A.K.K.; Tam, Y.Y.C.; Chen, S.; Hafez, I.M.; Cullis, P.R. Microfluidic Mixing: A General Method for Encapsulating Macromolecules in Lipid Nanoparticle Systems. J. Phys. Chem. B 2015, 119, 8698–8706. [Google Scholar] [CrossRef] [PubMed]
- Kulkarni, J.A.; Witzigmann, D.; Leung, J.; Van Der Meel, R.; Zaifman, J.; Darjuan, M.M.; Grisch-Chan, H.M.; Thöny, B.; Tam, Y.Y.C.; Cullis, P.R. Fusion-dependent formation of lipid nanoparticles containing macromolecular payloads. Nanoscale 2019, 11, 9023–9031. [Google Scholar] [CrossRef] [PubMed]
- Jayaraman, M.; Ansell, S.M.; Mui, B.L.; Tam, Y.K.; Chen, J.; Du, X.; Butler, D.; Eltepu, L.; Matsuda, S.; Narayanannair, J.K.; et al. Maximizing the potency of siRNA lipid nanoparticles for hepatic gene silencing in vivo. Angew. Chemie Int. Ed. 2012, 51, 8529–8533. [Google Scholar] [CrossRef]
- Sato, Y.; Hashiba, K.; Sasaki, K.; Maeki, M.; Tokeshi, M.; Harashima, H. Understanding structure-activity relationships of pH-sensitive cationic lipids facilitates the rational identification of promising lipid nanoparticles for delivering siRNAs in vivo. J. Control. Release 2019, 295, 140–152. [Google Scholar] [CrossRef] [PubMed]
- Ramishetti, S.; Hazan-Halevy, I.; Palakuri, R.; Chatterjee, S.; Naidu Gonna, S.; Dammes, N.; Freilich, I.; Kolik Shmuel, L.; Danino, D.; Peer, D. A Combinatorial Library of Lipid Nanoparticles for RNA Delivery to Leukocytes. Adv. Mater. 2020, 32, 1906128. [Google Scholar] [CrossRef] [PubMed]
- Billingsley, M.M.; Singh, N.; Ravikumar, P.; Zhang, R.; June, C.H.; Mitchell, M.J. Ionizable Lipid Nanoparticle-Mediated mRNA Delivery for Human CAR T Cell Engineering. Nano Lett. 2020, 20, 1578–1589. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Querbes, W.; De, S.; Qin, J.; Frank-Kamenetsky, M.; Jayaprakash, K.N.; Jayaraman, M.; Rajeev, K.G.; Cantley, W.L.; Dorkin, J.R.; et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010, 18, 1357–1364. [Google Scholar] [CrossRef]
- Kulkarni, J.A.; Cullis, P.R.; Van Der Meel, R. Lipid Nanoparticles Enabling Gene Therapies: From Concepts to Clinical Utility. Nucleic Acid Ther. 2018, 28, 146–157. [Google Scholar] [CrossRef]
- Rehman, Z.U.; Hoekstra, D.; Zuhorn, I.S. Mechanism of Polyplex- and Lipoplex-Mediated Delivery of Nucleic Acids: Real-Time Visualization of Transient Membrane Destabilization without Endosomal Lysis. ACS Nano 2013, 7, 3767–3777. [Google Scholar] [CrossRef] [PubMed]
- Zidovska, A.; Evans, H.M.; Ahmad, A.; Ewert, K.K.; Safinya, C.R. The Role of Cholesterol and Structurally Related Molecules in Enhancing Transfection of Cationic Liposome—DNA Complexes. J. Phys. Chem. B 2009, 113, 5208–5216. [Google Scholar] [CrossRef][Green Version]
- Peer, D.; Karp, J.M.; Hong, S.; Farokhzad, O.C.; Margalit, R.; Langer, R. Nanocarriers as an emerging platform for cancer therapy. Nat. Nanotechnol. 2007, 2, 751–760. [Google Scholar] [CrossRef] [PubMed]
- Danhier, F.; Feron, O.; Préat, V. To exploit the tumor microenvironment: Passive and active tumor targeting of nanocarriers for anti-cancer drug delivery. J. Control. Release 2010, 148, 135–146. [Google Scholar] [CrossRef] [PubMed]
- Deshpande, P.P.; Biswas, S.; Torchilin, V.P. Current trends in the use of liposomes for tumor targeting. Nanomedicine 2013, 8, 1509–1528. [Google Scholar] [CrossRef]
- Golombek, S.K.; May, J.N.; Theek, B.; Appold, L.; Drude, N.; Kiessling, F.; Lammers, T. Tumor targeting via EPR: Strategies to enhance patient responses. Adv. Drug Deliv. Rev. 2018, 130, 17–38. [Google Scholar] [CrossRef]
- Lammers, T.; Kiessling, F.; Hennink, W.E.; Storm, G. Drug targeting to tumors: Principles, pitfalls and (pre-) clinical progress. J. Control. Release 2012, 161, 175–187. [Google Scholar] [CrossRef]
- Van der Meel, R.; Sulheim, E.; Shi, Y.; Kiessling, F.; Mulder, W.J.M.; Lammers, T. Smart cancer nanomedicine. Nat. Nanotechnol. 2019, 14, 1007–1017. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef]
- Egeblad, M.; Werb, Z. New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2002, 2, 161–174. [Google Scholar] [CrossRef]
- Bruun, J.; Larsen, T.B.; Jølck, R.I.; Eliasen, R.; Holm, R.; Gjetting, T.; Andresen, T.L. Investigation of enzyme-sensitive lipid nanoparticles for delivery of siRNA to blood–brain barrier and glioma cells. Int. J. Nanomed. 2015, 10, 5995–6008. [Google Scholar] [CrossRef]
- Hatakeyama, H.; Akita, H.; Ito, E.; Hayashi, Y.; Oishi, M.; Nagasaki, Y.; Danev, R.; Nagayama, K.; Kaji, N.; Kikuchi, H.; et al. Systemic delivery of siRNA to tumors using a lipid nanoparticle containing a tumor-specific cleavable PEG-lipid. Biomaterials 2011, 32, 4306–4316. [Google Scholar] [CrossRef]
- Wan, Y.; Han, J.; Fan, G.; Zhang, Z.; Gong, T.; Sun, X. Enzyme-responsive liposomes modified adenoviral vectors for enhanced tumor cell transduction and reduced immunogenicity. Biomaterials 2013, 34, 3020–3030. [Google Scholar] [CrossRef]
- Han, B.; Qu, C.; Park, K.; Konieczny, S.F.; Korc, M. Recapitulation of complex transport and action of drugs at the tumor microenvironment using tumor-microenvironment-on-chip. Cancer Lett. 2016, 380, 319–329. [Google Scholar] [CrossRef]
- Wan, L.; Neumann, C.A.; LeDuc, P.R. Tumor-on-a-chip for integrating a 3D tumor microenvironment: Chemical and mechanical factors. Lab Chip 2020, 20, 873–888. [Google Scholar] [CrossRef]
- Koh, C.G.; Zhang, X.; Liu, S.; Golan, S.; Yu, B.; Yang, X.; Guan, J.; Jin, Y.; Talmon, Y.; Muthusamy, N.; et al. Delivery of antisense oligodeoxyribonucleotide lipopolyplex nanoparticles assembled by microfluidic hydrodynamic focusing. J. Control. Release 2010, 141, 62–69. [Google Scholar] [CrossRef]
- Balbino, T.A.; Azzoni, A.R.; de la Torre, L.G. Microfluidic devices for continuous production of pDNA/cationic liposome complexes for gene delivery and vaccine therapy. Colloids Surfaces B Biointerfaces 2013, 111, 203–210. [Google Scholar] [CrossRef]
- Balbino, T.A.; Serafin, J.M.; Malfatti-Gasperini, A.A.; De Oliveira, C.L.P.; Cavalcanti, L.P.; De Jesus, M.B.; De La Torre, L.G. Microfluidic Assembly of pDNA/Cationic Liposome Lipoplexes with High pDNA Loading for Gene Delivery. Langmuir 2016, 32, 1799–1807. [Google Scholar] [CrossRef]
- Kowalski, P.S.; Rudra, A.; Miao, L.; Anderson, D.G. Delivering the Messenger: Advances in Technologies for Therapeutic mRNA Delivery. Mol. Ther. 2019, 27, 710–728. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gaspar, R.; Coelho, F.; Silva, B.F.B. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules 2020, 25, 5006. https://doi.org/10.3390/molecules25215006
Gaspar R, Coelho F, Silva BFB. Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules. 2020; 25(21):5006. https://doi.org/10.3390/molecules25215006
Chicago/Turabian StyleGaspar, Ricardo, Filipe Coelho, and Bruno F. B. Silva. 2020. "Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment" Molecules 25, no. 21: 5006. https://doi.org/10.3390/molecules25215006
APA StyleGaspar, R., Coelho, F., & Silva, B. F. B. (2020). Lipid-Nucleic Acid Complexes: Physicochemical Aspects and Prospects for Cancer Treatment. Molecules, 25(21), 5006. https://doi.org/10.3390/molecules25215006