
Highly Multiplexed Single-Cell In Situ RNA and DNA Analysis by Consecutive Hybridization



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
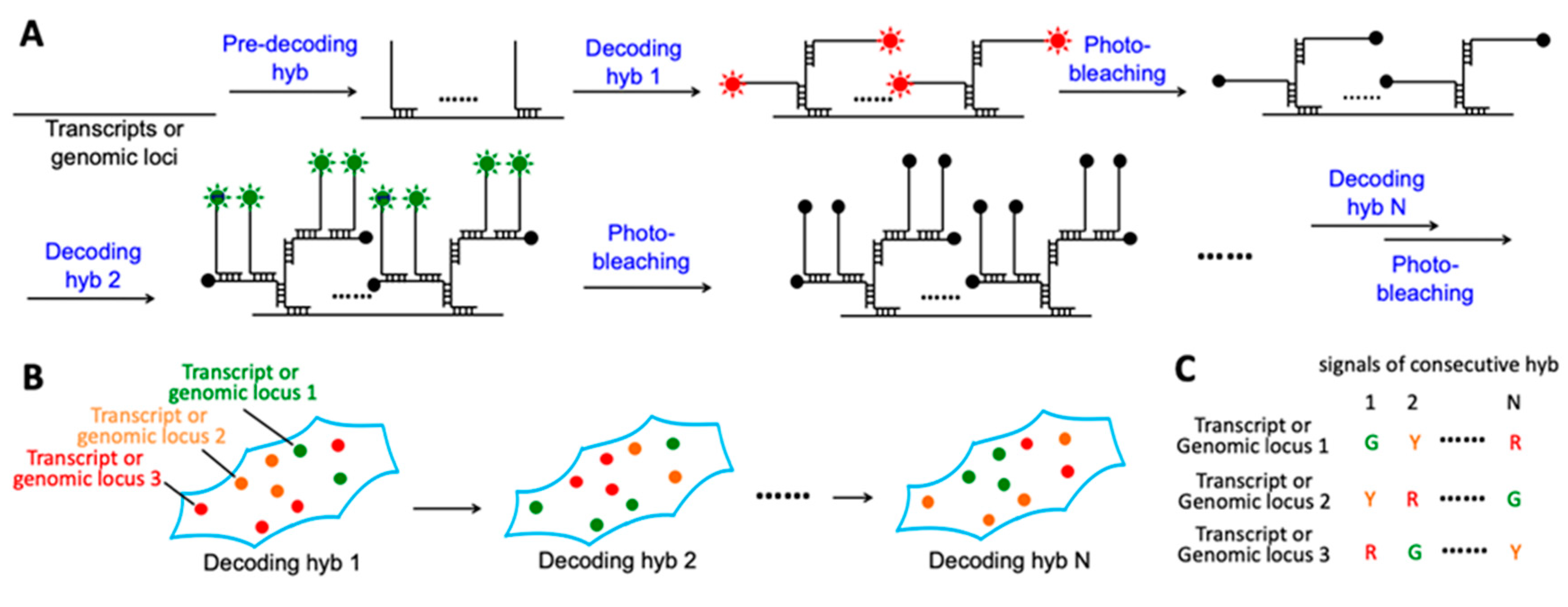
2.1. Platform Design
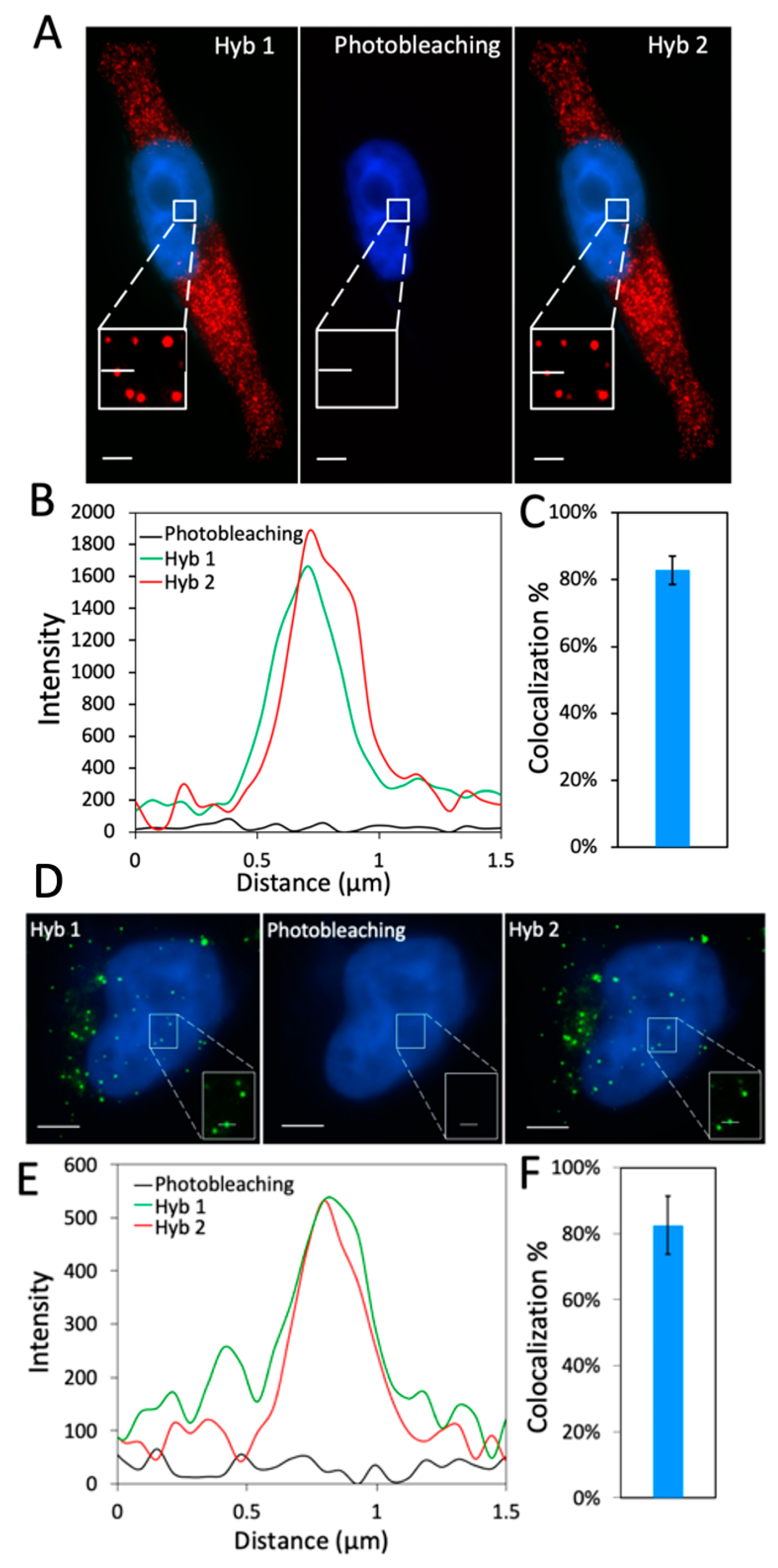
2.2. Efficiency and Effects of Photobleaching
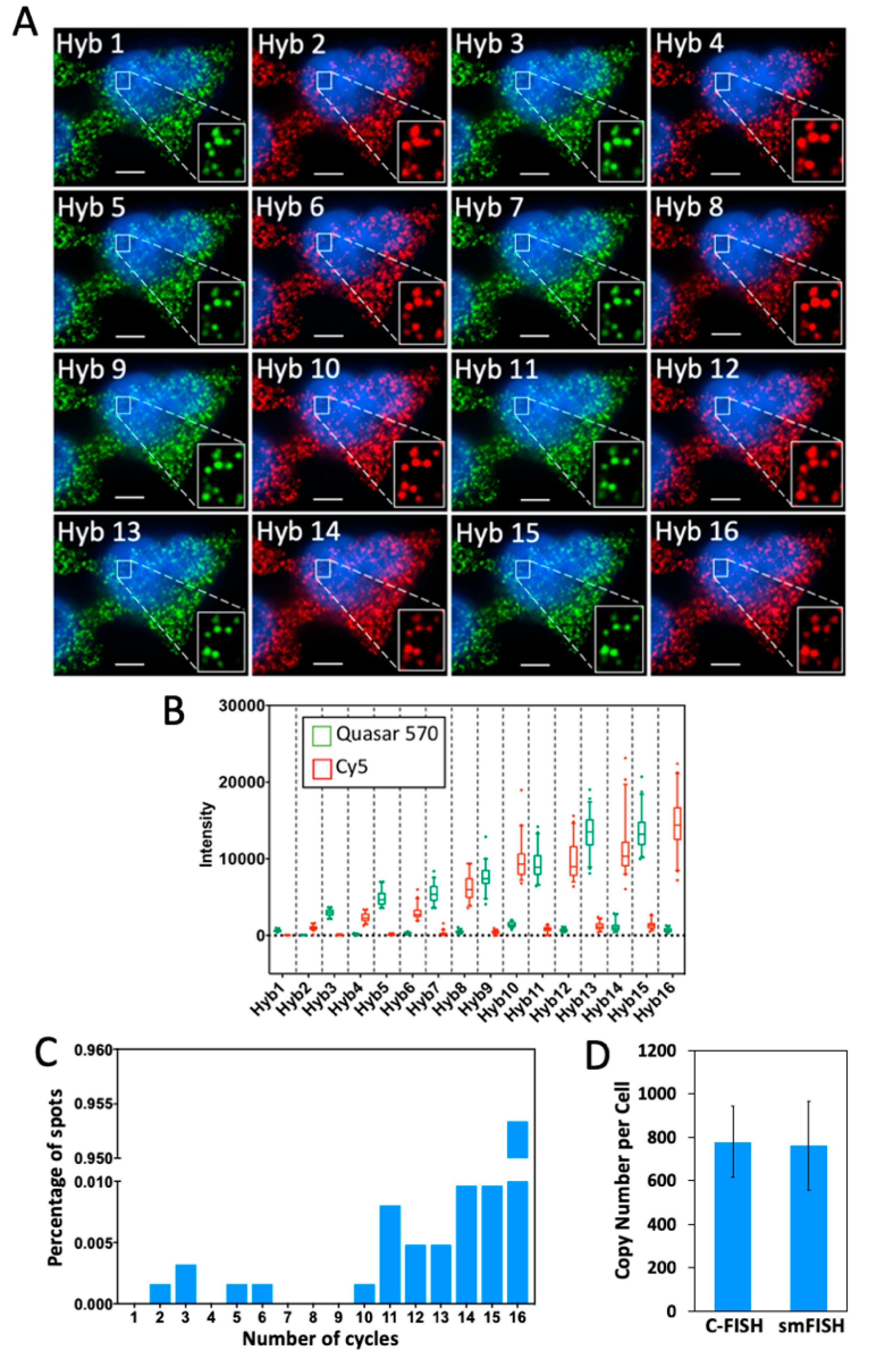
2.3. Multi-Cycle C-FISH for RNA Analysis
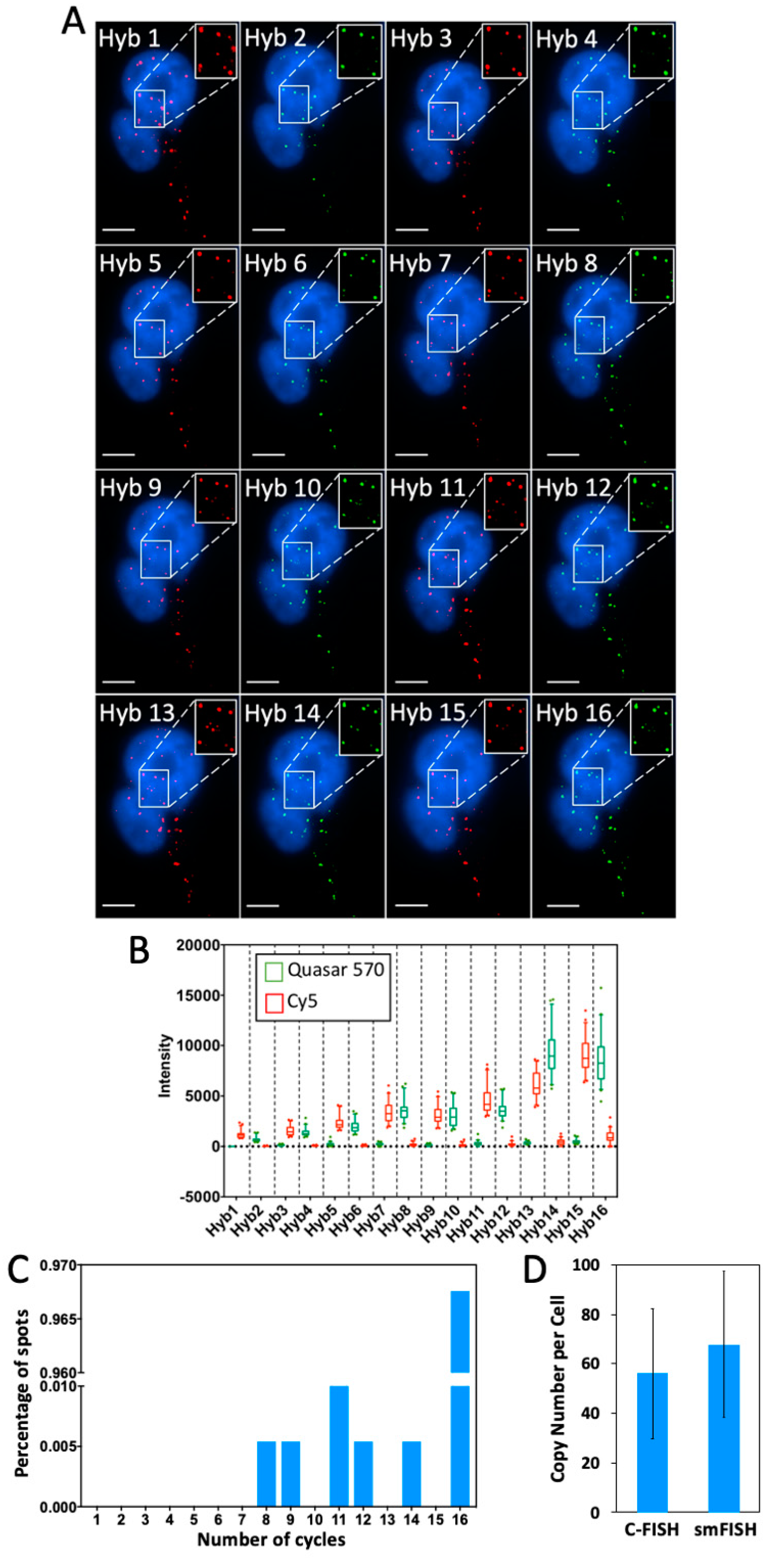
2.4. Multi-Cycle C-FISH for DNA Analysis
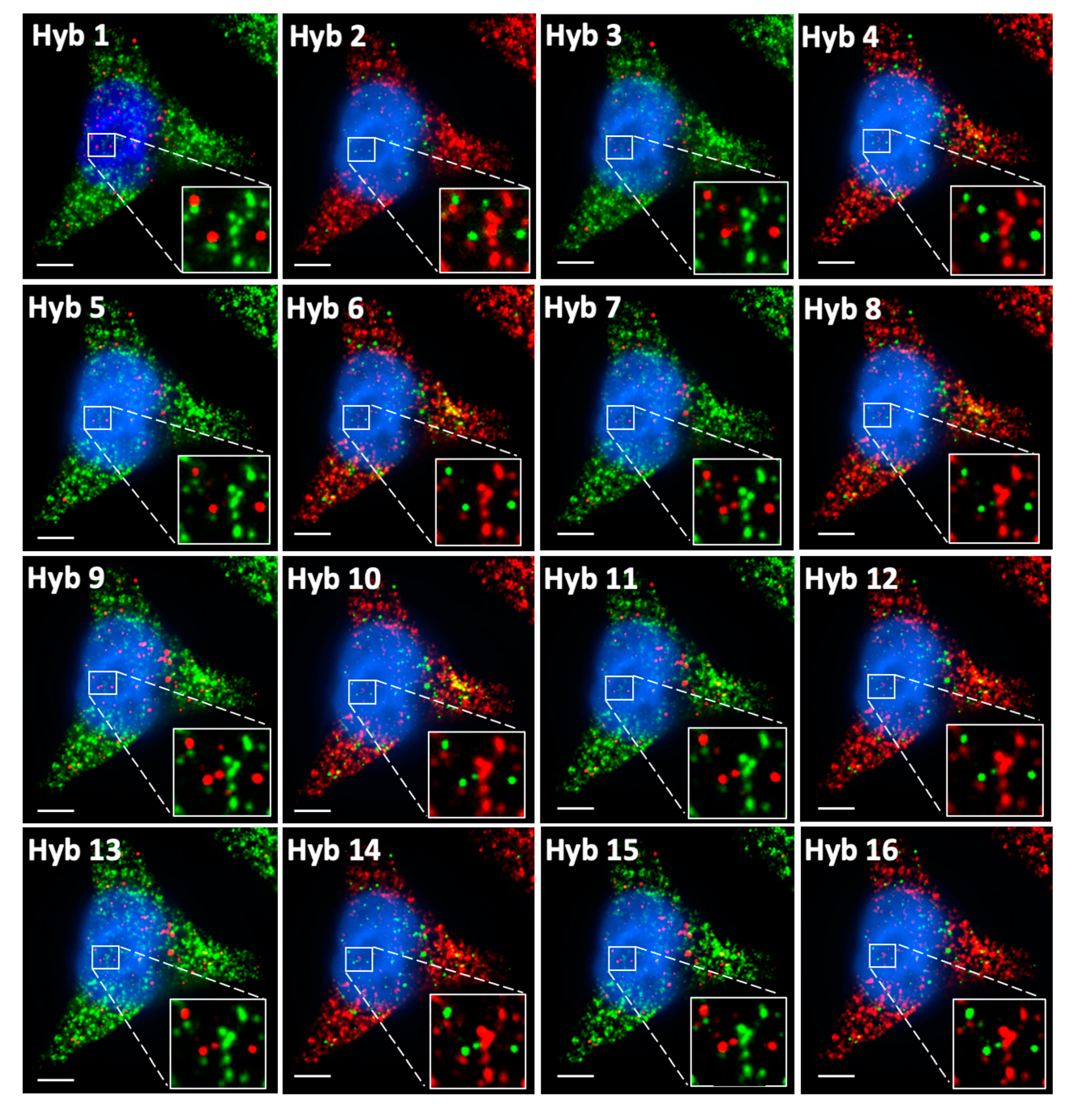
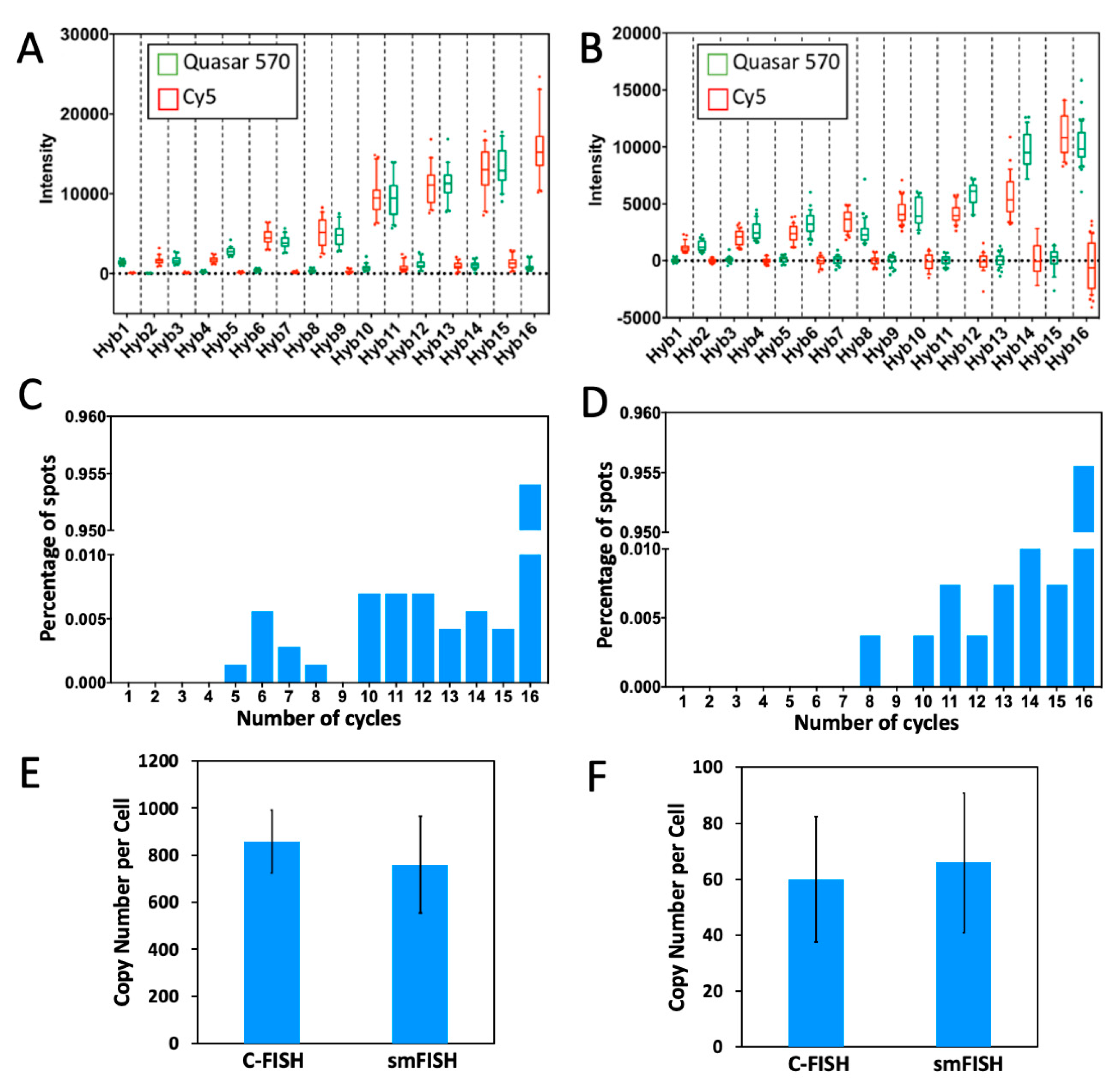
2.5. Multi-Cycle C-FISH for Multiplexed RNA Analysis
3. Discussion
4. Materials and Methods
4.1. General Information
4.2. Cell Culture
4.3. Cell Fixation
4.4. Probe Design
4.5. Probe Preparation
4.6. RNA Pre-Decoding Hybridization
4.7. DNA Pre-Decoding Hybridization
4.8. Consecutive RNA and DNA FISH
4.9. Consecutive FISH Imaging
4.10. Image Analysis
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Crosetto, N.; Bienko, M.; Van Oudenaarden, A. Spatially resolved transcriptomics and beyond. Nat. Rev. Genet. 2014, 16, 57–66. [Google Scholar] [CrossRef]
- Guo, J.; Yu, L.; Turro, N.J.; Ju, J. An integrated system for DNA sequencing by synthesis using novel nucleotide analogues. Acc. Chem. Res. 2010, 43, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Hoheisel, J.D. Microarray technology: Beyond transcript profiling and genotype analysis. Nat. Rev. Genet. 2006, 7, 200–210. [Google Scholar] [CrossRef]
- Speicher, M.R.; Ballard, S.G.; Ward, D.C. Karyotyping human chromosomes by combinatorial multi-fluor FISH. Nat. Genet. 1996, 12, 368–375. [Google Scholar] [CrossRef] [PubMed]
- Schröck, E.; Du Manoir, S.; Veldman, T.; Schoell, B.; Wienberg, J.; Ferguson-Smith, M.A.; Ning, Y.; Ledbetter, D.H.; Bar-Am, I.; Soenksen, D.; et al. Multicolor spectral karyotyping of human chromosomes. Science 1996, 273, 494–497. [Google Scholar] [CrossRef] [PubMed]
- Tanke, H.J.; Wiegant, J.; Van Gijlswijk, R.P.M.; Bezrookove, V.; Pattenier, H.; Heetebrij, R.J.; Talman, E.G.; Raap, A.K.; Vrolijk, J. New strategy for multi-colour fluorescence in situ hybridisation: COBRA: COmbined Binary RAtio labelling. Eur. J. Hum. Genet. 1999, 7, 2–11. [Google Scholar] [CrossRef]
- Henegariu, O.; Heerema, N.A.; Bray-Ward, P.; Ward, D.C. Colour-changing karyotyping: An alternative to M-FISH/SKY. Nat. Genet. 1999, 23, 263–264. [Google Scholar] [CrossRef] [PubMed]
- Raj, A.; Van Den Bogaard, P.; Rifkin, S.A.; Van Oudenaarden, A.; Tyagi, S. Imaging individual mRNA molecules using multiple singly labeled probes. Nat. Methods 2008, 5, 877–879. [Google Scholar] [CrossRef]
- Franzini, R.M.; Kool, E.T. Efficient nucleic acid detection by templated reductive quencher release. J. Am. Chem. Soc. 2009, 131, 16021–16023. [Google Scholar] [CrossRef]
- Levesque, M.J.; Raj, A. Single-chromosome transcriptional profiling reveals chromosomal gene expression regulation. Nat. Methods 2013, 10, 246–248. [Google Scholar] [CrossRef] [PubMed]
- Levsky, J.M.; Shenoy, S.M.; Pezo, R.C.; Singer, R.H. Single-cell gene expression profiling. Science 2002, 297, 836–840. [Google Scholar] [CrossRef] [PubMed]
- Lubeck, E.; Cai, L. Single-cell systems biology by super-resolution imaging and combinatorial labeling. Nat. Methods 2012, 9, 743–748. [Google Scholar] [CrossRef]
- Xiao, L.; Guo, J. Multiplexed single-cell in situ RNA analysis by reiterative hybridization. Anal. Methods 2015, 7, 7290–7295. [Google Scholar] [CrossRef]
- Xiao, L.; Guo, J. Single-Cell in Situ RNA Analysis with Switchable Fluorescent Oligonucleotides. Front. Cell Dev. Biol. 2018, 6, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Mondal, M.; Liao, R.; Nazaroff, C.D.; Samuel, A.D.; Guo, J. Highly multiplexed single-cell in situ RNA and DNA analysis with bioorthogonal cleavable fluorescent oligonucleotides. Chem. Sci. 2018, 9, 2909–2917. [Google Scholar] [CrossRef]
- Shaffer, S.M.; Dunagin, M.C.; Torborg, S.R.; Torre, E.A.; Emert, B.; Krepler, C.; Beqiri, M.; Sproesser, K.; Brafford, P.A.; Xiao, M.; et al. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 2017, 546, 431–435. [Google Scholar] [CrossRef]
- Ke, R.; Mignardi, M.; Pacureanu, A.; Svedlund, J.; Botling, J.; Wählby, C.; Nilsson, M. In situ sequencing for RNA analysis in preserved tissue and cells. Nat. Methods 2013, 10, 857–860. [Google Scholar] [CrossRef]
- Lee, J.H.; Daugharthy, E.R.; Scheiman, J.; Kalhor, R.; Yang, J.L.; Ferrante, T.C.; Terry, R.; Jeanty, S.S.F.; Li, C.; Amamoto, R.; et al. Highly multiplexed subcellular RNA sequencing in situ. Science 2014, 343, 1360–1363. [Google Scholar] [CrossRef]
- Lubeck, E.; Coskun, A.F.; Zhiyentayev, T.; Ahmad, M.; Cai, L. Single-cell in situ RNA profiling by sequential hybridization. Nat. Methods 2014, 11, 360–361. [Google Scholar] [CrossRef]
- Shah, S.; Takei, Y.; Zhou, W.; Lubeck, E.; Yun, J.; Eng, C.H.L.; Koulena, N.; Cronin, C.; Karp, C.; Liaw, E.J.; et al. Dynamics and Spatial Genomics of the Nascent Transcriptome by Intron seqFISH. Cell 2018, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Eng, C.H.L.; Lawson, M.; Zhu, Q.; Dries, R.; Koulena, N.; Takei, Y.; Yun, J.; Cronin, C.; Karp, C.; Yuan, G.C.; et al. Transcriptome-scale super-resolved imaging in tissues by RNA seqFISH+. Nature 2019, 568, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.H.; Boettiger, A.N.; Moffitt, J.R.; Wang, S.; Zhuang, X. Spatially resolved, highly multiplexed RNA profiling in single cells. Science 2015, 1363, 1360–1363. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Hao, J.; Wang, G.; Chen, K.H.; Babcock, H.P.; Zhuang, X. High-throughput single-cell gene-expression profiling with multiplexed error-robust fluorescence in situ hybridization. Proc. Natl. Acad. Sci. USA 2016, 113, 11046–11051. [Google Scholar] [CrossRef] [PubMed]
- Moffitt, J.R.; Hao, J.; Bambah-Mukku, D.; Lu, T.; Dulac, C.; Zhuang, X. High-performance multiplexed fluorescence in situ hybridization in culture and tissue with matrix imprinting and clearing. Proc. Natl. Acad. Sci. USA 2016, 113, 14456–14461. [Google Scholar] [CrossRef]
- Winkler, R.; Perner, B.; Rapp, A.; Durm, M.; Cremer, C.; Greulich, K.O.; Hausmann, M. Labelling quality and chromosome morphology after low temperature FISH analysed by scanning far-field and near-field optical microscopy. J. Microsc. 2003, 209, 23–33. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- Guo, J.; Wang, S.; Dai, N.; Teo, Y.N.; Kool, E.T. Multispectral labeling of antibodies with polyfluorophores on a DNA backbone and application in cellular imaging. Proc. Natl. Acad. Sci. USA 2011, 108, 3493–3498. [Google Scholar] [CrossRef]
- Dai, N.; Guo, J.; Teo, Y.N.; Kool, E.T. Protease probes built from DNA: Multispectral fluorescent DNA-peptide conjugates as caspase chemosensors. Angew. Chem. Int. Ed. 2011, 50, 5105–5109. [Google Scholar] [CrossRef]
- Wang, S.; Guo, J.; Ono, T.; Kool, E.T. DNA Polyfluorophores for Real-Time Multicolor Tracking of Dynamic Biological Systems. Angew. Chem. Int. Ed. 2012, 51, 7176–7180. [Google Scholar] [CrossRef]
- Garini, Y.; Young, I.T.; McNamara, G. Spectral imaging: Principles and applications. Cytometry A 2006, 69, 735–747. [Google Scholar] [CrossRef] [PubMed]
- Huang, B.; Jones, S.A.; Brandenburg, B.; Zhuang, X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nat. Methods 2008, 5, 1047–1052. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Wassie, A.T.; Cote, A.; Sinha, A.; Alon, S.; Asano, S.; Daugharthy, E.R.; Chang, J.-B.; Marblestone, A.; Church, G.M.; et al. Nanoscale Imaging of RNA with Expansion Microscopy. Nat. Methods 2016, 13, 679–684. [Google Scholar] [CrossRef]
- Xu, Q.; Schlabach, M.R.; Hannon, G.J.; Elledge, S.J. Design of 240,000 orthogonal 25mer DNA barcode probes. Proc. Natl. Acad. Sci. USA 2009, 106, 2289–2294. [Google Scholar] [CrossRef]
- Murgha, Y.E.; Rouillard, J.M.; Gulari, E. Methods for the preparation of large quantities of complex single-stranded oligonucleotide libraries. PLoS ONE 2014, 9, e94752. [Google Scholar] [CrossRef]
- Mondal, M.; Liao, R.; Guo, J. Highly multiplexed single-cell protein analysis. Chem. Eur. J. 2018, 24, 7083–7091. [Google Scholar] [CrossRef] [PubMed]
- Pham, T.; Tyagi, A.; Wang, Y.S.; Guo, J. Single-cell proteomic analysis. Wiley Interdiscip. Rev. Syst. Biol. Med. 2020, e1503. [Google Scholar] [CrossRef] [PubMed]
- Beliveau, B.J.; Joyce, E.F.; Apostolopoulos, N.; Yilmaz, F.; Fonseka, C.Y.; McCole, R.B.; Chang, Y.; Li, J.B.; Senaratne, T.N.; Williams, B.R.; et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proc. Natl. Acad. Sci. USA 2012, 109, 21301–21306. [Google Scholar] [CrossRef]
- Olivo-Marin, J.-C. Extraction of spots in biological images using multiscale products. Pattern Recognit. 2002, 35, 1989–1996. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compounds, such as the pre-decoding and decoding probes, are available from the authors. |








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiao, L.; Liao, R.; Guo, J. Highly Multiplexed Single-Cell In Situ RNA and DNA Analysis by Consecutive Hybridization. Molecules 2020, 25, 4900. https://doi.org/10.3390/molecules25214900
Xiao L, Liao R, Guo J. Highly Multiplexed Single-Cell In Situ RNA and DNA Analysis by Consecutive Hybridization. Molecules. 2020; 25(21):4900. https://doi.org/10.3390/molecules25214900
Chicago/Turabian StyleXiao, Lu, Renjie Liao, and Jia Guo. 2020. "Highly Multiplexed Single-Cell In Situ RNA and DNA Analysis by Consecutive Hybridization" Molecules 25, no. 21: 4900. https://doi.org/10.3390/molecules25214900
APA StyleXiao, L., Liao, R., & Guo, J. (2020). Highly Multiplexed Single-Cell In Situ RNA and DNA Analysis by Consecutive Hybridization. Molecules, 25(21), 4900. https://doi.org/10.3390/molecules25214900