
Rotational Spectroscopy Meets Quantum Chemistry for Analyzing Substituent Effects on Non-Covalent Interactions: The Case of the Trifluoroacetophenone-Water Complex

 , , and
, , and 
Abstract

1. Introduction
- To investigate whether trifluoromethylation plays a role in tuning the NCIs established by TFAP and water with respect to the 1:1 AP-water complex [35].
- To describe the interactions and the contributions involved in the stabilization of the complex using different quantum-chemical energy partitioning techniques.
2. Results and Discussion
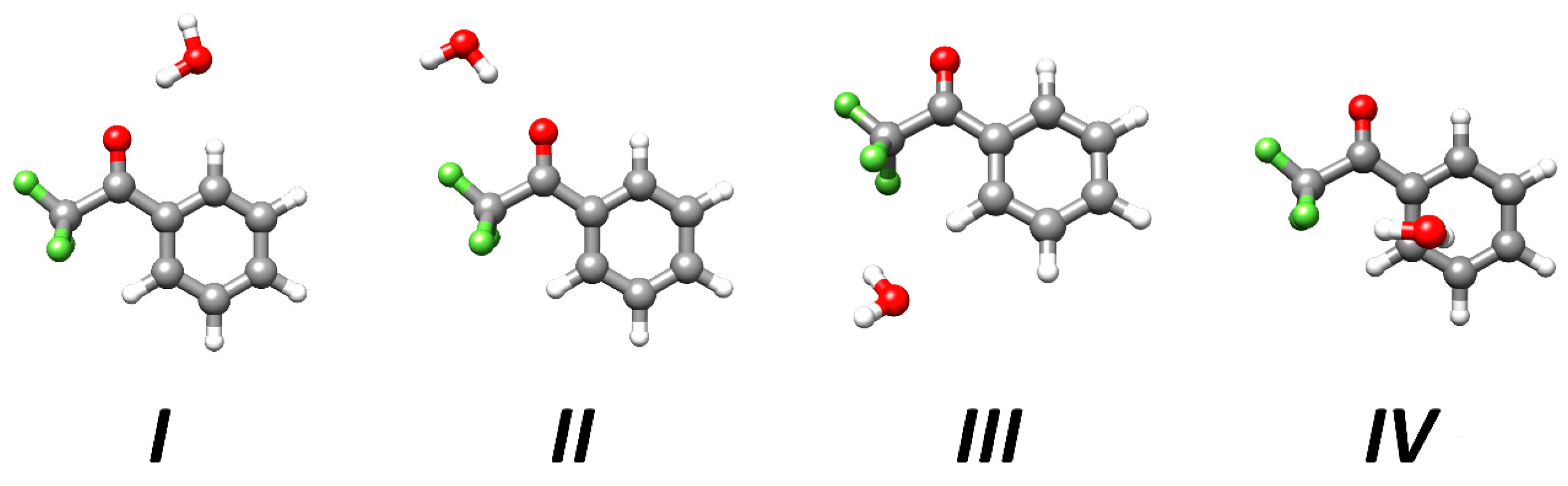
2.1. Structures and Energetics of the Low-Lying Isomers
2.2. Rotational Spectroscopy Investigation
2.3. Semi-Experimental Equilibrium Structure
2.4. Bond Analysis
- (1)
- LP(1) OC=O-BD*(1) O-HHB,water: 10.0 kJ·mol−1, and LP(2) OC=O-BD*(1) O-HHB,water: 10.8 kJ·mol−1 vs. LP(2) Owater-BD*(1) C-HTFAP: 4.9 kJ·mol−1;
- (2)
- BD(1) O-HHB,water-BD*(1) C-HTFAP: 1.1 kJ·mol−1.
2.5. 2,2,2-Trifluoroacetophenone vs. Acetophenone: Effect of Fluorination on the Complex Structure
- The aromatic C-H groups present a more positive electrostatic potential.
- The electron density above the aromatic ring and on the outer carbonyl oxygen is reduced.
- The positive electrostatic potential around the -CH3 group in AP becomes negative around the -CF3 group in TFAP.
3. Materials and Methods
3.1. Experimental Details
3.2. Theoretical Methodology
3.2.1. Isomers Characterization
3.2.2. Semi-Experimental Equilibrium Structure
- (1)
- (2)
- Determination of the semi-experimental equilibrium structure of the TFAP-W complex by using the “template approach” to fix the internal coordinates of the two partners in the complex, and fitting only the inter-molecular parameters.
3.2.3. Bond Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cocinero, E.J.; Lesarri, A.; Écija, P.; Cimas, Á.; Davis, B.G.; Basterretxea, F.J.; Fernández, J.A.; Castaño, F. Free Fructose Is Conformationally Locked. J. Am. Chem. Soc. 2013, 135, 2845–2852. [Google Scholar] [CrossRef] [PubMed]
- Burke, N.L.; DeBlase, A.F.; Redwine, J.G.; Hopkins, J.R.; McLuckey, S.A.; Zwier, T.S. Gas-Phase Folding of a Prototypical Protonated Pentapeptide: Spectroscopic Evidence for Formation of a Charge-Stabilized β-Hairpin. J. Am. Chem. Soc. 2016, 138, 2849–2857. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Seifert, N.A.; Jäger, W.; Xu, Y.; Jäger, W. A Direct Link from the Gas to the Condensed Phase: A Rotational Spectroscopic Study of 2,2,2-Trifluoroethanol Trimers. Angew. Chem. Int. Ed. 2017, 56, 6289–6293. [Google Scholar] [CrossRef] [PubMed]
- Pérez, C.; Muckle, M.T.; Zaleski, D.P.; Seifert, N.A.; Temelso, B.; Shields, G.C.; Kisiel, Z.; Pate, B.H. Structures of Cage, Prism, and Book Isomers of Water Hexamer from Broadband Rotational Spectroscopy. Science 2012, 336, 897–901. [Google Scholar] [CrossRef] [PubMed]
- Pate, B.H.; Evangelisti, L.; Caminati, W.; Xu, Y.; Thomas, J.; Patterson, D.; Pérez, C.; Schnell, M. Quantitative Chiral Analysis by Molecular Rotational Spectroscopy. In Chiral Analysis: Advances in Spectroscopy, Chromatography and Emerging Methods, 2nd ed.; Polavarapu, P.L., Ed.; Elsevier: Amsterdam, The Netherlands, 2018; pp. 679–729. [Google Scholar]
- Arunan, E.; Desiraju, G.R.; Klein, R.A.; Sadlej, J.; Scheiner, S.; Alkorta, I.; Clary, D.C.; Crabtree, R.H.; Dannenberg, J.J.; Hobza, P.; et al. Definition of the hydrogen bond (IUPAC Recommendations 2011). Pure Appl. Chem. 2011, 83, 1637–1641. [Google Scholar] [CrossRef]
- Steiner, T. The Hydrogen Bond in the Solid State. Angew. Chem. Int. Ed. 2002, 41, 48–76. [Google Scholar] [CrossRef]
- Hutchins, K.M. Functional materials based on molecules with hydrogen-bonding ability: Applications to drug co-crystals and polymer complexes. R. Soc. Open Sci. 2018, 5, 180564. [Google Scholar] [CrossRef]
- Schreiner, P.R. Metal-free organocatalysis through explicit hydrogen bonding interactions. Chem. Soc. Rev. 2003, 32, 289–296. [Google Scholar] [CrossRef]
- Řezáč, J.; Hobza, P. Benchmark Calculations of Interaction Energies in Noncovalent Complexes and Their Applications. Chem. Rev. 2016, 116, 5038–5071. [Google Scholar] [CrossRef]
- Alessandrini, S.; Barone, V.; Puzzarini, C. Extension of the “Cheap” Composite Approach to Noncovalent Interactions: The jun-ChS Scheme. J. Chem. Theory Comput. 2019, 16, 988–1006. [Google Scholar] [CrossRef]
- Scheiner, S. Hydrogen bonding. In A Theoretical Perspective; Oxford University Press: Oxford, UK, 1997. [Google Scholar]
- Nishiyama, Y.; Langan, P.; Chanzy, H. Crystal Structure and Hydrogen-Bonding System in Cellulose Iβ from Synchrotron X-ray and Neutron Fiber Diffraction. J. Am. Chem. Soc. 2002, 124, 9074–9082. [Google Scholar] [CrossRef] [PubMed]
- Gilli, P.; Bertolasi, V.; Ferretti, A.V.; Gilli, G. Evidence for Intramolecular N−H···O Resonance-Assisted Hydrogen Bonding in β-Enaminones and Related Heterodienes. A Combined Crystal-Structural, IR and NMR Spectroscopic, and Quantum-Mechanical Investigation. J. Am. Chem. Soc. 2000, 122, 10405–10417. [Google Scholar] [CrossRef]
- Sharif, S.; Denisov, G.S.; Toney, M.D.; Limbach, H.-H. NMR Studies of Solvent-Assisted Proton Transfer in a Biologically Relevant Schiff Base: Toward a Distinction of Geometric and Equilibrium H-Bond Isotope Effects. J. Am. Chem. Soc. 2006, 128, 3375–3387. [Google Scholar] [CrossRef] [PubMed]
- Nesbitt, D.J. High-resolution infrared spectroscopy of weakly bound molecular complexes. Chem. Rev. 1988, 88, 843–870. [Google Scholar] [CrossRef]
- Larsen, R.W.; Zielke, P.; Suhm, M.A. Hydrogen-bonded OH stretching modes of methanol clusters: A combined IR and Raman isotopomer study. J. Chem. Phys. 2007, 126, 194307. [Google Scholar] [CrossRef] [PubMed]
- Zwier, T.S. The spectroscopy of solvation in hydrogen-bonded aromatic clusters. Annu. Rev. Phys. Chem. 1996, 47, 205–241. [Google Scholar] [CrossRef]
- Dessent, C.E.H.; Müller-Dethlefs, K. Hydrogen-Bonding and van der Waals Complexes Studied by ZEKE and REMPI Spectroscopy. Chem. Rev. 2000, 100, 3999–4022. [Google Scholar] [CrossRef]
- Juanes, M.; Saragi, R.T.; Caminati, W.; Lesarri, A. The Hydrogen Bond and Beyond: Perspectives for Rotational Investigations of Non-Covalent Interactions. Chem. Eur. J. 2019, 25, 11402–11411. [Google Scholar] [CrossRef]
- Caminati, W.; Grabow, J.-U. Microwave Spectroscopy: Molecular Systems. In Frontiers of Molecular Spectroscopy; Laane, J., Ed.; Elsevier: Amsterdam, The Netherlands, 2009; pp. 455–552. [Google Scholar]
- Cametti, M.; Crousse, B.; Metrangolo, P.; Milani, R.; Resnati, G. The fluorous effect in biomolecular applications. Chem. Soc. Rev. 2012, 41, 31–42. [Google Scholar] [CrossRef]
- Suzuki, S.; Green, P.G.; Bumgarner, R.E.; Dasgupta, S.; Goddard, W.A.; Blake, G.A. Benzene Forms Hydrogen Bonds with Water. Science 1992, 257, 942–945. [Google Scholar] [CrossRef]
- Brendel, K.; Mäder, H.; Xu, Y.; Jäger, W. The rotational spectra of the fluorobenzene⋯water and p-difluorobenzene⋯water dimers: Structure and internal dynamics. J. Mol. Spectrosc. 2011, 268, 47–52. [Google Scholar] [CrossRef]
- Evangelisti, L.; Brendel, K.; Mäder, H.; Caminati, W.; Melandri, S. Rotational Spectroscopy Probes Water Flipping by Full Fluorination of Benzene. Angew. Chem. Int. Ed. 2017, 56, 13699–13703. [Google Scholar] [CrossRef] [PubMed]
- Mikami, K.; Itoh, Y.; Yamanaka, M. Fluorinated Carbonyl and Olefinic Compounds: Basic Character and Asymmetric Catalytic Reactions. Chem. Rev. 2004, 104, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Zanda, M. Trifluoromethyl group: An effective xenobiotic function for peptide backbone modification. New J. Chem. 2004, 28, 1401–1411. [Google Scholar] [CrossRef]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef]
- Kang, L.; Novick, S.E.; Gou, Q.; Spada, L.; Vallejo-López, M.; Caminati, W. The shape of trifluoromethoxybenzene. J. Mol. Spectrosc. 2014, 297, 32–34. [Google Scholar] [CrossRef]
- Onda, M.; Toda, A.; Mori, S.; Yamaguchi, I. Microwave spectrum of anisole. J. Mol. Struct. 1986, 144, 47–51. [Google Scholar] [CrossRef]
- Desyatnyk, O.; Pszczółkowski, L.; Thorwirth, S.; Krygowski, T.M.; Kisiel, Z. The rotational spectra, electric dipole moments and molecular structures of anisole and benzaldehyde. Phys. Chem. Chem. Phys. 2005, 7, 1708–1715. [Google Scholar] [CrossRef]
- Jin, Y.; Wang, J.; Gou, Q.; Xia, Z.; Feng, G. Fluorination effect on conformational preferences of trifluorothioanisole. J. Mol. Struct. 2018, 1156, 230–234. [Google Scholar] [CrossRef]
- Velino, B.; Melandri, S.; Caminati, W.; Favero, P.G. Free-jet absorption millimeter spectrum of thioanisole. Gazz. Chim. Ital. 1995, 125, 373–376. [Google Scholar]
- Onda, M.; Kohama, Y.; Suga, K.; Yamaguchi, I. Microwave spectrum and molecular planarity of acetophenone. J. Mol. Struct. 1998, 442, 19–22. [Google Scholar] [CrossRef]
- Lei, J.; Zhang, J.; Feng, G.; Grabow, J.-U.; Gou, Q. Conformational preference determined by inequivalent n-pairs: Rotational studies on acetophenone and its monohydrate. Phys. Chem. Chem. Phys. 2019, 21, 22888–22894. [Google Scholar] [CrossRef] [PubMed]
- Lei, J.; Chen, J.; Feng, G.; Xia, Z.; Gou, Q.; Xia, Z. Rotational spectrum of 2,2,2-trifluoroacetophenone. J. Mol. Spectrosc. 2018, 351, 4–7. [Google Scholar] [CrossRef]
- Pulay, P.; Meyer, W.; Boggs, J.E. Cubic force constants and equilibrium geometry of methane from Hartree–Fock and correlated wavefunctions. J. Chem. Phys. 1978, 68, 5077–5085. [Google Scholar] [CrossRef]
- Pawłowski, F.; Jørgensen, P.; Olsen, J.; Hegelund, F.; Helgaker, T.; Gauss, J.; Bak, K.L.; Stanton, J.F. Molecular equilibrium structures from experimental rotational constants and calculated vibration–rotation interaction constants. J. Chem. Phys. 2002, 116, 6482–6496. [Google Scholar] [CrossRef]
- Piccardo, M.; Penocchio, E.; Puzzarini, C.; Biczysko, M.; Barone, V. Semi-Experimental Equilibrium Structure Determinations by Employing B3LYP/SNSD Anharmonic Force Fields: Validation and Application to Semirigid Organic Molecules. J. Phys. Chem. A 2015, 119, 2058–2082. [Google Scholar] [CrossRef]
- Pickett, H.M. The fitting and prediction of vibration-rotation spectra with spin interactions. J. Mol. Spectrosc. 1991, 148, 371–377. [Google Scholar] [CrossRef]
- Durig, J.R.; Stampf, E.J.; Odom, J.D.; Kalasinsky, V.F. Vibrational spectra and structure of perfluorovinyldifluoroborane and perfluorovinyldichloroborane. Inorg. Chem. 1977, 16, 2895–2900. [Google Scholar] [CrossRef]
- Mendolicchio, M.; Penocchio, E.; Licari, D.; Tasinato, N.; Barone, V. Development and Implementation of Advanced Fitting Methods for the Calculation of Accurate Molecular Structures. J. Chem. Theory Comput. 2017, 13, 3060–3075. [Google Scholar] [CrossRef]
- Balle, T.J.; Flygare, W.H. Fabry–Perot cavity pulsed Fourier transform microwave spectrometer with a pulsed nozzle particle source. Rev. Sci. Instrum. 1981, 52, 33–45. [Google Scholar] [CrossRef]
- Grabow, J.; Stahl, W.; Dreizler, H. A multioctave coaxially oriented beam-resonator arrangement Fourier-transform microwave spectrometer. Rev. Sci. Instrum. 1996, 67, 4072–4084. [Google Scholar] [CrossRef]
- Weak hydrogen bond topology in 1,1-difluoroethane dimer: A rotational study. J. Chem. Phys. 2017, 147, 094301. [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Stephens, P.J.; Devlin, F.J.; Chabalowski, C.F.; Frisch, M.J. Ab Initio Calculation of Vibrational Absorption and Circular Dichroism Spectra Using Density Functional Force Fields. J. Phys. Chem. 1994, 98, 11623–11627. [Google Scholar] [CrossRef]
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef]
- Carnimeo, I.; Puzzarini, C.; Tasinato, N.; Stoppa, P.; Charmet, A.P.; Biczysko, M.; Cappelli, C.; Barone, V. Anharmonic theoretical simulations of infrared spectra of halogenated organic compounds. J. Chem. Phys. 2013, 139, 074310. [Google Scholar] [CrossRef]
- SNSD Basis Set. Available online: https://smart.sns.it (accessed on 20 October 2020).
- Santra, G.; Sylvetsky, N.; Martin, J.M.L. Minimally Empirical Double-Hybrid Functionals Trained against the GMTKN55 Database: revDSD-PBEP86-D4, revDOD-PBE-D4, and DOD-SCAN-D4. J. Phys. Chem. A 2019, 123, 5129–5143. [Google Scholar] [CrossRef]
- Papajak, E.; Zheng, J.; Xu, X.; Leverentz, H.R.; Truhlar, D.G. Perspectives on Basis Sets Beautiful: Seasonal Plantings of Diffuse Basis Functions. J. Chem. Theory Comput. 2011, 7, 3027–3034. [Google Scholar] [CrossRef]
- Boys, S.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies. Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–566. [Google Scholar] [CrossRef]
- Raghavachari, K.; Trucks, G.W.; Pople, J.A.; Head-Gordon, M. A fifth-order perturbation comparison of electron correlation theories. Chem. Phys. Lett. 1989, 157, 479–483. [Google Scholar] [CrossRef]
- Møller, C.; Plesset, M.S. Note on an Approximation Treatment for Many-Electron Systems. Phys. Rev. 1934, 46, 618–622. [Google Scholar] [CrossRef]
- Helgaker, T.; Klopper, W.; Koch, H.; Noga, J. Basis-set convergence of correlated calculations on water. J. Chem. Phys. 1997, 106, 9639–9646. [Google Scholar] [CrossRef]
- Peterson, K.A.; Dunning, T.H. Accurate correlation consistent basis sets for molecular core–valence correlation effects: The second row atoms Al–Ar, and the first row atoms B-Ne revisited. J. Chem. Phys. 2002, 117, 10548–10560. [Google Scholar] [CrossRef]
- Gaussian, 16, release C.01; Gaussian Inc.: Wallingford, CT, USA, 2019.
- Mills, I.M. Vibration-Rotation Structure in Asymmetric- and Symmetric-Top Molecules. In Molecular Spectroscopy: Modern Research; Rao, K.N., Mathews, C.W., Eds.; Academic Press: New York, NY, USA, 1972. [Google Scholar]
- Puzzarini, C.; Bloino, J.; Tasinato, N.; Barone, V. Accuracy and Interpretability: The Devil and the Holy Grail. New Routes across Old Boundaries in Computational Spectroscopy. Chem. Rev. 2019, 119, 8131–8191. [Google Scholar] [CrossRef]
- Penocchio, E.; Piccardo, M.; Barone, V. Semiexperimental Equilibrium Structures for Building Blocks of Organic and Biological Molecules: The B2PLYP Route. J. Chem. Theory Comput. 2015, 11, 4689–4707. [Google Scholar] [CrossRef]
- Glendening, E.D.; Landis, C.R.; Weinhold, F. Natural bond orbital methods. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2011, 2, 1–42. [Google Scholar] [CrossRef]
- Jeziorski, B.; Moszynski, R.; Szalewicz, K. Perturbation Theory Approach to Intermolecular Potential Energy Surfaces of van der Waals Complexes. Chem. Rev. 1994, 94, 1887–1930. [Google Scholar] [CrossRef]
- Laplaza, R.; Peccati, F.; Boto, R.A.; Quan, C.; Carbone, A.; Piquemal, J.; Maday, Y.; Contreras-García, J. NCIPLOT and the analysis of noncovalent interactions using the reduced density gradient. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2020, 1497. [Google Scholar] [CrossRef]
- Bader, R.F.W. A quantum theory of molecular structure and its applications. Chem. Rev. 1991, 91, 893–928. [Google Scholar] [CrossRef]
- Fornaro, T.; Biczysko, M.; Bloino, J.; Barone, V. Reliable vibrational wavenumbers for C [double bond, length as m-dash] O and N–H stretchings of isolated and hydrogen-bonded nucleic acid bases. Phys. Chem. Chem. Phys. 2016, 18, 8479–8490. [Google Scholar] [CrossRef]
- Parrish, R.M.; Burns, L.A.; Smith, D.G.A.; Simmonett, A.C.; DePrince, I.A.E.; Hohenstein, E.G.; Bozkaya, U.; Sokolov, A.Y.; Di Remigio, R.; Richard, R.M.; et al. Psi4 1.1: An Open-Source Electronic Structure Program Emphasizing Automation, Advanced Libraries, and Interoperability. J. Chem. Theory Comput. 2017, 13, 3185–3197. [Google Scholar] [CrossRef]
- Lu, T.; Fei-Wu, C. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2011, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Wang, J.; Spada, L.; Chen, J.; Gao, S.; Alessandrini, S.; Feng, G.; Puzzarini, C.; Gou, Q.; Grabow, J.-U.; Barone, V. The Unexplored World of Cycloalkene-Water Complexes: Primary and Assisting Interactions Unraveled by Experimental and Computational Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 13935–13941. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| I | II | III | IV | |
|---|---|---|---|---|
| Ae | 891.23 | 1277.58 | 1027.75 | 1193.21 |
| Be | 611.55 | 472.14 | 603.12 | 520.70 |
| Ce | 387.60 | 367.15 | 408.87 | 483.30 |
| |μa| | 1.8 | 5.5 | 1.5 | 2.6 |
| |μb| | 1.7 | 2.2 | 2.1 | 0.7 |
| |μc| | 0.0 | 0.0 | 0.5 | 2.5 |
| ΔE | 0.0 | 5.0 | 9.3 | 12.6 |
| ΔE0 | 0.0 | 4.5 | 6.9 | 9.4 |
| De | −22.36 | −17.29 | −13.11 | −9.96 |
| −23.24 | −17.56 | −13.62 | −10.65 |
| Theory 1,2 | TFAP-H2O | TFAP-H218O | TFAP-D2O | TFAP-HOD | TFAP-DOH | |
|---|---|---|---|---|---|---|
| A0/MHz | 879.36 | 878.0858(1) 3 | 833.9848(2) | 830.3953(1) | 845.3957(1) | 861.6862(1) |
| B0/MHz | 605.16 | 609.4679(1) | 607.227(1) | 608.7236(6) | 608.6732(9) | 609.5202(9) |
| C0/MHz | 383.35 | 384.50355(4) | 374.9407(1) | 374.82029(6) | 377.82212(9) | 381.3495(1) |
| DJ/kHz | 0.09 | 0.121(1) | 0.117(2) | 0.1115(9) | 0.115(1) | 0.115(1) |
| DJK/kHz | −0.11 | −0.147(3) | −0.143(3) | −0.138(1) | −0.140(2) | −0.141(1) |
| DK/kHz | 0.03 | 0.035(2) | [0.035] 4 | [0.035] | [0.035] | [0.035] |
| d1/kHz | 0.04 | 0.0557(5) | [0.0557] | [0.0557] | [0.0557] | [0.0557] |
| d2/Hz | −6.6 | −6.3(3) | [−6.3] | [−6.3] | [−6.3] | [−6.3] |
| σ/kHz | 2.4 | 2.7 | 2.1 | 2.4 | 2.8 | |
| N5 | 152 | 62 | 110 | 80 | 87 | |
| Pcc/uÅ2 | 45.75 | 45.1961(1) | 45.1821(7) | 45.2522(4) | 45.2435(6) | 45.2020(6) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lei, J.; Alessandrini, S.; Chen, J.; Zheng, Y.; Spada, L.; Gou, Q.; Puzzarini, C.; Barone, V. Rotational Spectroscopy Meets Quantum Chemistry for Analyzing Substituent Effects on Non-Covalent Interactions: The Case of the Trifluoroacetophenone-Water Complex. Molecules 2020, 25, 4899. https://doi.org/10.3390/molecules25214899
Lei J, Alessandrini S, Chen J, Zheng Y, Spada L, Gou Q, Puzzarini C, Barone V. Rotational Spectroscopy Meets Quantum Chemistry for Analyzing Substituent Effects on Non-Covalent Interactions: The Case of the Trifluoroacetophenone-Water Complex. Molecules. 2020; 25(21):4899. https://doi.org/10.3390/molecules25214899
Chicago/Turabian StyleLei, Juncheng, Silvia Alessandrini, Junhua Chen, Yang Zheng, Lorenzo Spada, Qian Gou, Cristina Puzzarini, and Vincenzo Barone. 2020. "Rotational Spectroscopy Meets Quantum Chemistry for Analyzing Substituent Effects on Non-Covalent Interactions: The Case of the Trifluoroacetophenone-Water Complex" Molecules 25, no. 21: 4899. https://doi.org/10.3390/molecules25214899
APA StyleLei, J., Alessandrini, S., Chen, J., Zheng, Y., Spada, L., Gou, Q., Puzzarini, C., & Barone, V. (2020). Rotational Spectroscopy Meets Quantum Chemistry for Analyzing Substituent Effects on Non-Covalent Interactions: The Case of the Trifluoroacetophenone-Water Complex. Molecules, 25(21), 4899. https://doi.org/10.3390/molecules25214899